

1 Introduction
Les fluides organo-silicones constitués d'entités structurales (–(CH3)2SiO–) ont une grande variété d'applications (produits de beauté, élastomères, résines). L'utilisation croissante des polysiloxanes dans la technologie macromoléculaire a conduit à des études portant sur la polymérisation des cyclosiloxanes et de la polycondensation des silanols en présence de catalyseur [1]. Ces composés sont par ailleurs identifiés comme de potentiels substituts aux produits halogénés utilisés en tant qu'inhibiteurs de la propagation des flammes [2]. La compréhension de la cinétique réactionnelle à haute température de ces composés revêt une grande importance pour le développement de modèles de combustion du type proposé par Lipowitz [3]. La cinétique et le mécanisme de thermolyse de l'octaméthylcyclosiloxane (–(CH3)2SiO–)4 désigné par l'appellation D4, ont été étudiés par Davidson et al. [4,5] à de faibles degrés de conversion dans un domaine de température allant de 767 à 925 K. Leurs résultats, en accord avec ceux de Gusel'nikov et al. [6,7], qui ont étudié plusieurs diméthylcyclosiloxane (Dn : n = 3 à 7) ont permis de proposer un mécanisme pour la dégradation thermique des cyclosiloxanes.
Cet article présente un travail complémentaire à celui de Davidson, en étendant le domaine d'étude à des températures plus élevées, à savoir 1058–1197 K.
2 Partie expérimentale
La technique utilisée dans ce travail est celle de réacteur « fast-flow », dont les espèces sont prélevées par formation d'un faisceau moléculaire couplée avec une détection par spectrométrie de masse. Le dispositif expérimental présenté sur la Fig. 1 a été décrit par ailleurs [8,9] ; seulement quelques caractéristiques principales seront données ici.
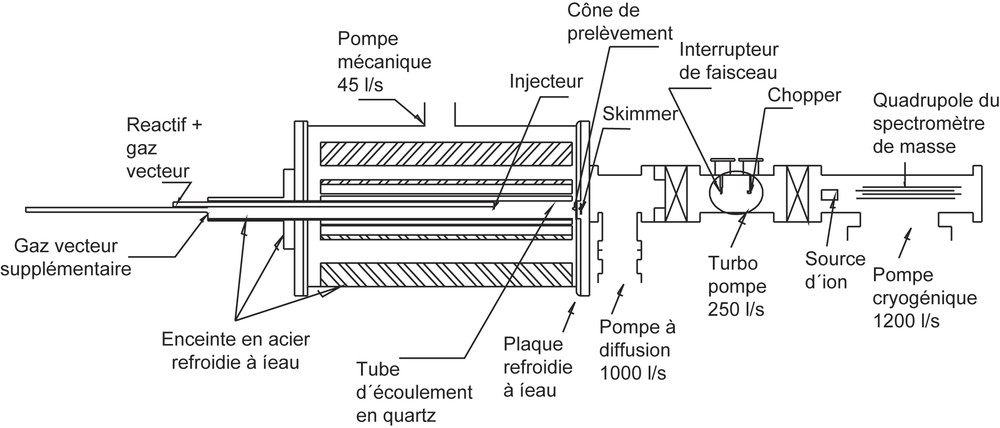
Réacteur à écoulement rapide/système de faisceau moléculaire.
Le tube du réacteur fait en quartz a un diamètre intérieur de 3,75 cm et une longueur de 90 cm. Le réacteur est logé dans une enceinte d'acier inoxydable sous vide ; deux injecteurs en quartz en assurent l'alimentation en réactif et en diluant.
La technique de prélèvement des échantillons gazeux par formation d'un faisceau moléculaire consiste en une sonde conique suivie de trois étages, dans lesquels la pression est abaissée de façon progressive par un pompage adéquat. La troisième enceinte, qui contient le spectromètre de masse est maintenue à une pression de l'ordre de 10−7 Pa. Le cône de prélèvement est muni d'un orifice de 0,04 cm de diamètre. Le jet moléculaire créé par le cône de prélèvement est dirigé vers le premier étage, qui est maintenu à une pression voisine de 2 × 10−4 Pa. Un second cône (écorceur ou skimmer) extrait la partie centrale du faisceau vers le deuxième étage, dans lequel règne une pression d'environ 10−5 Pa. L'écorceur est placé à la distance optimale permettant à la fois d'assurer un pompage efficace et d'éviter la formation d'un disque de Mach en aval du cône de prélèvement.
À l'entrée du spectromètre de masse, un système de modulation du faisceau moléculaire (chopper), constitué d'un disque alternant des pales évidées et pleines, occulte périodiquement le jet. La modulation permet de faire des mesures alternées du signal correspondant au faisceau moléculaire et au résiduel, d'une part, et du signal correspondant au résiduel seul, d'autre part. Par différence, la contribution du résiduel se trouve éliminée.
La pression typique dans le réacteur en fonctionnement est de 263 Pa. Tous les gaz ont été débités par des débitmètres massiques pré-calibrés. L'argon est utilisé comme diluant. Les puretés sont celles données par le fabricant : Ar, 99,999%, D4 – 98%. D4 est stocké dans un barboteur dans lequel des traces de l'éthanol sont piégées par des filtres moléculaires de 4 Å. Avant utilisation, D4 est soumis à un dégazage. Le flux d'argon dans le barboteur à pression et température constantes a pour fonction de débiter la quantité désirée de D4 dans le réacteur. La pression de vapeur de D4 à la température ambiante est de 131 Pa et, typiquement, sa pression partielle dans le réacteur est de 2,6 Pa. La variation du temps de séjour est obtenue par la variation du débit global dans le réacteur, l'injecteur étant maintenu à une position fixe par rapport au cône de prélèvement, dont la position est arbitrairement utilisée comme origine. Les températures réelles sont référencées à la température de décomposition du cyclohexène, dont la constante de vitesse, déterminée par Tsang [10] selon l'expression suivante :
3 Résultats et discussions
Pour vérifier la cohérence de la méthode appliquée à la réaction de référence (décomposition de cyclohexène), nous avons mesuré la constante de vitesse d'une autre réaction, à savoir la décomposition du tétraéthoxysilane (TEOS), dont la constante de réaction a été mesurée par plusieurs auteurs [8,11,12]. Les mesures effectuées pour la décomposition de TEOS dans le domaine de température 1018–1083 K sont présentées sur la Fig. 2.
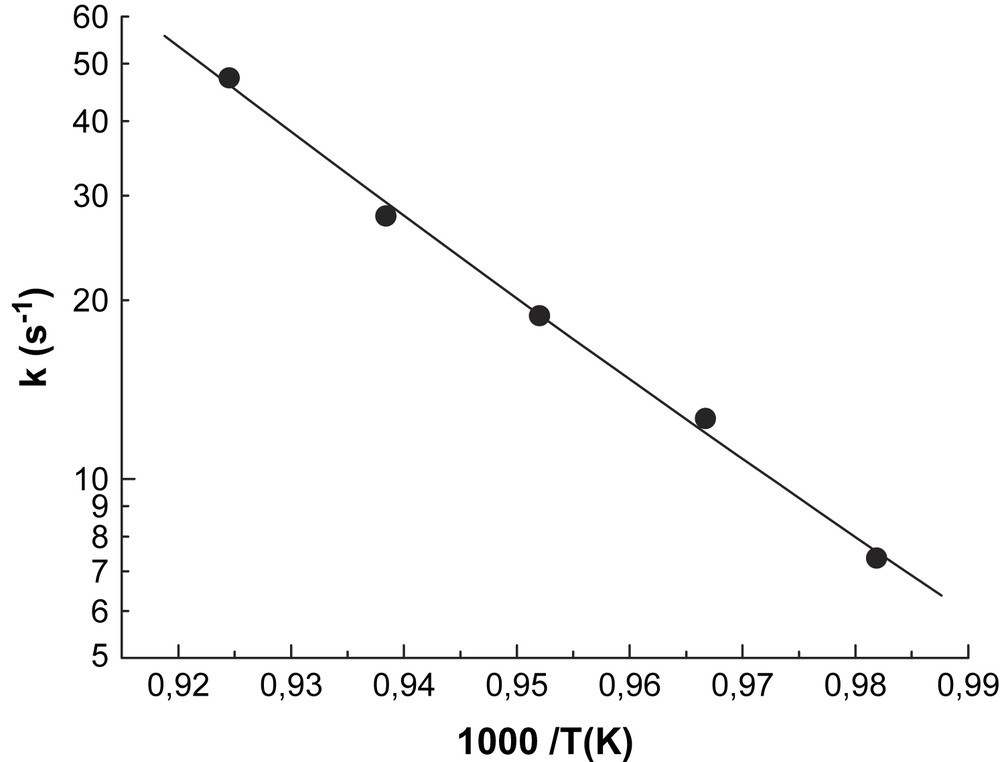
Constante de vitesse de la décomposition de TEOS.
Ces données ont permis d'établir l'expression de la constante de vitesse sous la forme :

Comparaison des constantes de vitesse de la décomposition de TEOS mesurées par divers auteurs.
Les quatre travaux présentés sont conformes à un facteur 2 près, et une expression unique peut être déduite pour couvrir l'intervalle de température 750–1285 K sous la forme :
À l'aide de cette méthode de référence, la constante de vitesse de réaction de la thermolyse de D4 a été mesurée en suivant sa décomposition dans le temps. Le pic mesuré est celui correspondant au rapport masse sur charge m/z = 281, qui est le pic parent ayant perdu le fragment –CH3.
La Fig. 4 présente le rapport du signal de D4 à celui de l'argon, qui demeure le signal de référence. La normalisation de tous les signaux à celui de l'argon permet de prendre en considération les variations de réglage du dispositif de mesure. La Fig. 4 est typique de la décomposition de premier ordre de D4 en fonction du temps de séjour.
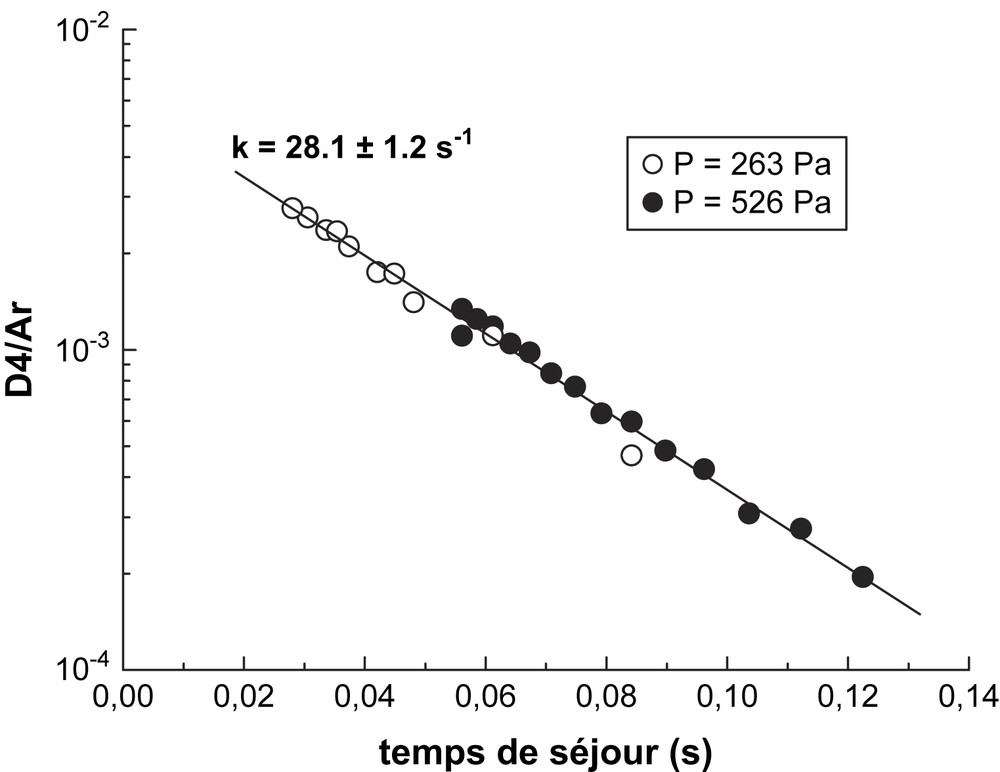
Décomposition de premier ordre de D4 à 263 et 526 Pa.
Pour cette expérience, les mesures effectuées à 263 et 526 Pa n'ont montré aucune dépendance par rapport à la pression. L'ordre de la réaction a été vérifié comme étant unimoléculaire sur une variation d'un ordre 10 de la concentration initiale de D4.
Pour la plupart des expériences, aucun piège de radicaux n'a été utilisé, mais nous avons éliminé la possibilité d'une réaction radicalaire en mesurant la constante de vitesse à la température la plus élevée, en utilisant le toluène comme piège de radicaux. La différence des constantes de vitesse mesurées en présence et en l'absence de toluène est inférieure à 3%, ce qui est conforme avec l'erreur globale de nos mesures, estimée à 10%. Les constantes de vitesse établies dans le domaine de température 1058–1197 K sont récapitulées sur la Fig. 5.
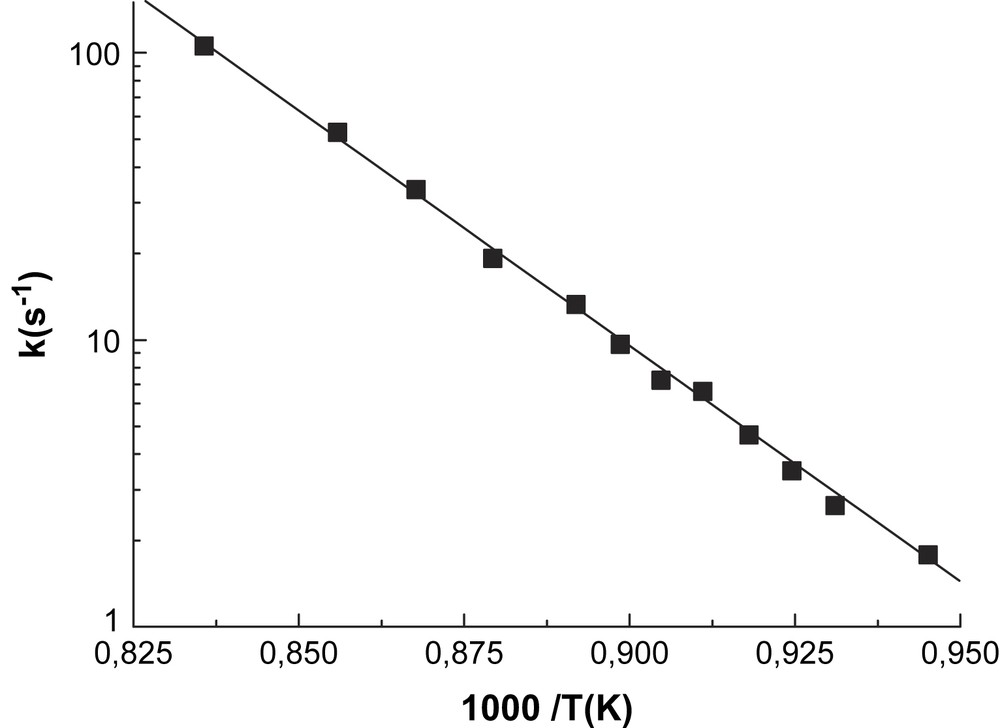
Constante de vitesse de la décomposition de D4.
Pour la réaction D4 → produits, l'expression d'Arrhenius obtenue avec un coefficient de régression de 99,79% s' écrit :
- – les mesures effectuées dans le cadre de ce travail, extrapolées dans le domaine de température des mesures de Davidson et al. (767–925 K), sous-estiment leurs résultats de 15% au plus ;
- – l'extrapolation de l'expression obtenue par Davidson et al. dans le domaine de température étudié ici donne des valeurs de constante de réaction deux fois plus faibles que nos mesures.
L'écart entre les deux expressions apparaît dans les différences sur le terme pré-exponentiel (un ordre de grandeur), et l'énergie d'activation (écart de 17 kJ mol−1). Ces différences semblent beaucoup plus liées aux contraintes du meilleur procédé de lissage des données et aux erreurs systématiques internes des deux expériences. Néanmoins, quand l'ensemble des deux travaux est considéré sur le domaine de température de 767 à 1197 K, elles sont bien corrélées (R2 = 99,97%) par la constante de vitesse suivante :
Le facteur pré-exponentiel et l'énergie d'activation obtenus avec ces expériences sont indicatifs d'une rupture de liaison. La liaison la plus faible dans la molécule D4 est vraisemblablement Si–CH3, typiquement dans la gamme de 377 kJ mol−1 [14], tandis que l'énergie de a liaison Si–O est de 540 kJ mol−1. Si la réaction était un pur processus de rupture de liaison, on s'attendrait à ce que l'énergie d'activation soit au moins égale à l'endothermicité. L'énergie d'activation mesurée est de 318 kJ mol−1 ; elle est de 60 kJ/mol plus basse que l'énergie de la liaison Si–CH3. Par ailleurs, les expériences conduites en présence ou en absence de toluène à haute température n'ont pas donné de variations significatives sur les vitesses de réaction. Ces observations sont cohérentes avec le mécanisme suggéré par Davidson et al. [4,5], qui a proposé une élimination transannulaire du diméthylsilanone, (CH3)2SiO avec les réactions subséquentes suivantes :
| D4 ⇋ D3 + D1 | (1,−1) |
| D4 + D1 ⇋ D5 | (2,−2) |
| D5 + D1 ⇋ D6 | (3,−3) |
| D6 → 2 D3 | (4,−4) |
Ce mécanisme était cohérent avec les produits observés, notamment D3 et D5, durant la décomposition de D4. L'addition de l'éthylène utilisée pour piéger D1 permet de n'avoir que D3 comme produit majoritaire de la décomposition tandis que la concentration D5 est fortement réduite. Nos mesures des produits de réaction sont récapitulées sur les Figs. 6 et 7, correspondant respectivement à deux températures différentes, 1089 et 1113 K.
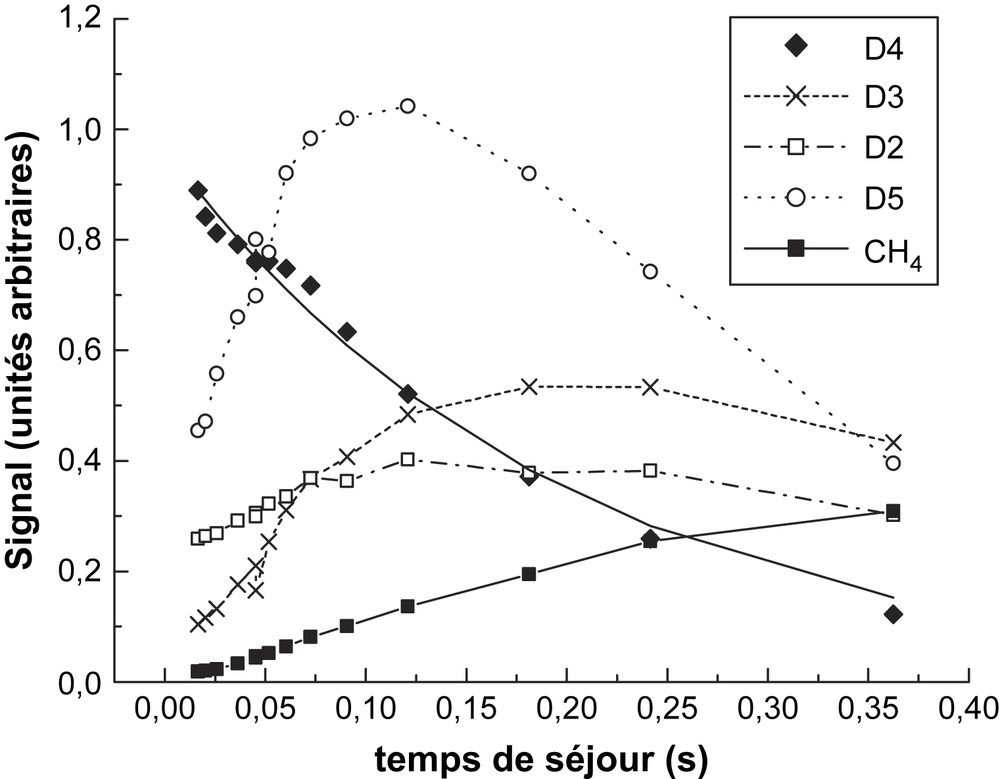
Produits de décomposition de D4 à 1089 K.
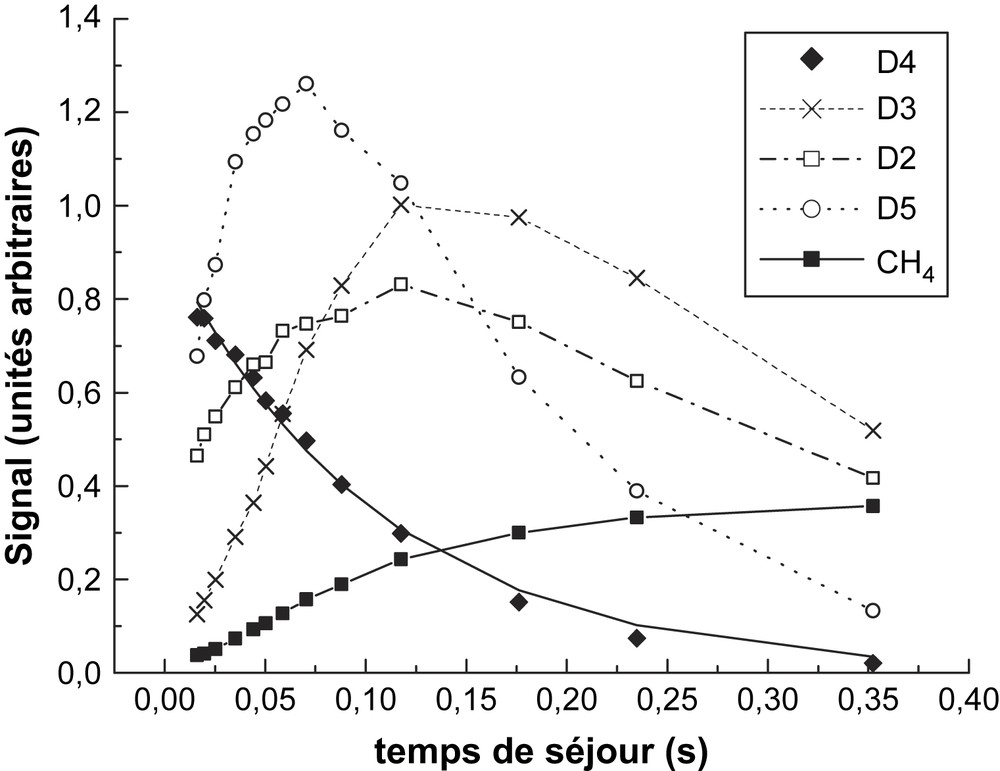
Produits de décomposition de D4 à 1113 K.
Les pics mesurés sont m/z = 281 pour D4, m/z = 355 pour D5, m/z = 207 pour D3, m/z = 118 pour D2 et m/z = 16 pour CH4 ; les signaux présentés sur les Figs. 6 et 7 sont rapportés à la concentration D4 initiale, afin de fournir des données comparables. Ces signaux ne représentent pas des concentrations absolues en raison des facteurs d'étalonnage, inconnus pour les espèces détectées. Toutefois, les signaux relatifs mesurés fournissent des informations qualitatives sur le profil temporel des pics mesurés et sur leur évolution en fonction de la température. Les résultats plus marquants sont observés sur l'instabilité temporelle de D5 lorsque la température augmente. À partir du profil de D5 présenté sur les figures, on peut estimer sa vitesse de consommation à 4,2 et 8,4 s−1, respectivement à 1089 et 1113 K. Cette même approche appliquée à 1058 K fournit une vitesse de 1,7 s−1. Ces trois données fournissent une estimation de la constante de vitesse à
| D5 → D4 + D1 | (−2) |
| D5 → D3 + D2 | (5) |
À l'opposé des cyclosiloxanes, la formation de méthane se produit très tôt, ce qui est en parfait accord avec l'analyse de Davidson et al. [4], qui ont pu observer le méthane et (HMeSiO) (Me2SIO)2, comme les produits finaux de la rupture de la liaison Si–CH3 lors de la thermolyse de D3 :
| D3 → CH3 + MeSIO(Me2SIO)2 | (6) |
4 Conclusion
La pyrolyse de D4 en phase gazeuse a été étudiée à des températures relativement élevées, à l'aide d'un dispositif de type fast-flow, à de hauts degrés de conversion. La constante de vitesse mesurée pour la réaction D4 → produits donne l'expression d'Arrhenius suivante :
Remerciements
Ce travail a été réalisé grâce au soutien du National Institute of Standards and Technologies (NIST), à qui nous exprimons nos sincères remerciements.