

1 Introduction
Les résultats décrits dans cet article ont trait au magnétisme moléculaire et supramoléculaire. Depuis la découverte de nouvelles familles d'aimants moléculaires (homométalliques) se comportant comme des particules magnétiques monodomaines [1], l'intérêt du chimiste pour élaborer de nouvelles molécules contenant peu de centres paramagnétiques n'a cessé de croître au cours de ces dernières années.
Un des objectifs de notre travail de recherche est de mettre au point de nouveaux systèmes magnétiques, dans lesquels des molécules à couplage magnétique contrôlé puissent s'associer coopérativement avec leurs voisins [2], au travers, par exemple, de molécules magnétiques greffées sur des surfaces (ou agrégats métalliques) adéquates ou s'associant dans des réseaux supramoléculaires spécifiques.
Notre choix s'est porté sur des ligands soufrés (pour le dépôt potentiel sur surface d'or) polydendates, de synthèse aisée et présentant de nombreuses possibilités de contacts « faibles », de type liaison hydrogène ou contacts de van der Waals. Nous avons ainsi élaboré un premier ligand (H2L_2), l'acide 2-hydroxy-1,3-dithiolan-2-ylidenehydrazide benzoïque [3], qui permet l'obtention quantitative de nouveaux complexes mono- et binucléaires de manganèse et fer [3,4]. Certains de ces complexes binucléaires présentent des couplages ferromagnétiques record [4]. L'analyse détaillée (par radiocristallographie, mesures magnétiques et calculs DFT) de ces objets moléculaires asymétriques indique une voix nouvelle en magnétisme moléculaire [5] basée sur les interactions de type CH-π, imposant l'asymétrie au niveau du métal et pouvant aboutir à un contrôle potentiel de l'interaction magnétique par une modification mineure du ligand. Le concept est présenté sur la Fig. 1a. La configuration asymétrique des deux centres métalliques (provenant d'une double connexion CH-π de part et d'autre des métaux) implique une orthogonalité des orbitales dz2, forçant ainsi le système à un couplage ferromagnétique (ici très fort, avec une valeur de constante de couplage proche de 20 cm−1).
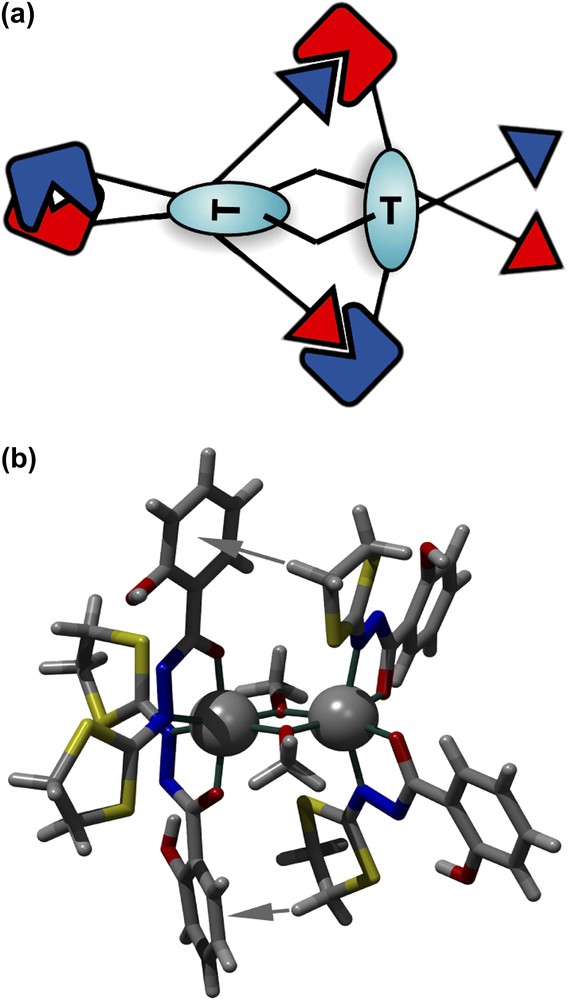
(a) Représentation schématique du principe de la double interaction CH-π fixant l'asymétrie du système binucléaire. (b) Structure moléculaire des nouveaux complexes binucléaires. Les flèches grises indiquent les interactions CH-π.
Ce type d'édifice moléculaire (Fig. 1b) présente ainsi un double intérêt:
- (1) un couplage magnétique record, qu'il s'agira d'améliorer encore par une chimie intramoléculaire adaptée (modification des fonctions pontantes, par exemple);
- (2) une partie organique facilement modulable. Nous pourrons ainsi comprendre quels sont les groupements indispensables à la « fixation asymétrique » (dans le solide et en solution), puis la possibilité de rompre cette « fixation » pour permettre la génération contrôlée d'entités ferromagnétiques ou antiferromagnétiques.
Dans cet article, nous présentons nos premiers résultats relatifs à la modulation du ligand et la synthèse d'un nouveau complexe obtenus à partir des ligands H2L_3 et H3L_4 (Schéma 1).

Liste des ligands.
2 Procédures expérimentales
2.1 Procédures générales
Toutes les réactions et manipulations ont été effectuées en utilisant les techniques classiques des tubes de Schlenk (sous atmosphère d'argon dans le cas du complexe de fer). Les solvants ont été séchés et distillés avant leur utilisation. Les analyses élémentaires pour C, H, N, et S ont été effectuées au service de microanalyse de la faculté de chimie de l'université Louis-Pasteur (Strasbourg, France). Les spectres infrarouge ont été collectés (à partir de pastilles de KBr) sur un spectromètre de type PerkinElmer 1600 series FTIR. Les spectres RMN du proton ont été collectés à 300 MHz, ceux du 13C à 75 MHz sur un spectromètre Bruker AC300.
2.2 Synthèse
2.2.1 Acide 2-hydroxy[bis(méthylthio)méthylène]hydrazide benzoïque H2L_3
À une solution agitée de salicylhydrazide (2,52 g; 16,6 mmol) avec NaOH (1,32 g; 33,2 mmol) dans l'éthanol absolu (30 ml) est ajouté goutte à goutte le disulfure de carbone CS2 (1 ml; 16,6 mmol). Le mélange est laissé sous agitation toute la nuit, puis le composé CH3I (2,06 ml; 33,2 mmol) est ajouté. 100 ml d'éther diéthylique froid est ensuite ajouté au mélange réactionnel, le précipité blanc formé est filtré et rincé avec de l'éther diéthylique. La recristallisation dans l'éthanol donne des cristaux incolores H2L_3 (3,44 g, Rdt = 81%) de taille suffisante pour la diffraction sur monocristal.
RMN 1H (DMSO-d6): δ 11,78; 10,50 (br s, br s, 2H, du NH amide et OH phénolique), 7,98–7,95 (m, 1H, H aromatique), 7,44–7,39 (m, 1H, H aromatique), 7,02–6,96 (m, 2H, Hs aromatiques), 3,77–3,73; 2,60 et 2,51 (m, m, 6H, Hs les deux SCH3). 13C NMR (DMSO-d6):δ = 15,19; 15,21 (2C, CH3); 117,24; 117,81; 120,16; 131,04; 133,79 (Carom); 152,03; 156,74; 161,22 (CO, C–OH, S–C–S). Analyse élémentaire. Calc. Pour H2L_3: C10H12N2O2S2: C. 46,85; H. 4,72; N. 10,93%. Exp. C. 47,4; H. 4,1; N. 11,2%. IR (KBr, cm−1): ν = 3240, 3034, 1632, 1527, 753, 593.
2.2.2 Le mélange acide 2-hydroxy [bis(méthylthio)méthylène]-hydrazide benzoïque H2L_3 et acide 2-hydroxy-2-[(méthylthio)thioxo-méthyl]-hydrazide benzoïque H3L_4
À basse température (<10 °C), nous faisons réagir le disulfure de carbone (1 ml; 16,6 mmol) avec le salicylhydrazide (2,52 g; 16,6 mmol) en présence d'une base de type hydroxyde de potassium (0,9 g; 16,6 mmol) dans l'éthanol (20 ml). Nous rajoutons ensuite l'iodure de méthyle (1 ml; 16,6 mmol). 50 ml d'eau froide sent rajoutés, conduisant à un précipité blanc. La recristallisation du solide obtenu dans la diméthylformamide avec l'eau utilisée comme non-solvant permet d'obtenir des cristaux incolores (de taille suffisante pour la diffraction sur monocristal), avec un rendement de 42%.
RMN 1H (DMSO-d6): δ 11,89; 10,72 (br s, br s, 4H, des NH amide et OH phénolique), 7,97–7,81 (m, 2H, H aromatique), 7,47–7,41 (m, 2H, H aromatique), 7,00–6,83 (m, 4H, Hs aromatiques), 2,60, 2,51 et 2,48 (m, m, m, 9H, Hs les trois SCH3). NB: les analyses élémentaires et les spectres IR n'ont pas été collectés sur ce composé, qui est un mélange de H2L_3 et H3L_4 (voir 2.3).
2.2.3 Acide 2-hydroxy-2-[(méthylthio)thioxométhyl]hydrazide benzoïque [Na(HL_4)(EtOH)]
Ce ligand a été synthétisé au laboratoire de la même façon que le ligand H2L_3, en utilisant un équivalent d'iodure de méthyle (1 ml; 16,6 mmol). Le solide blanc obtenu est recristallisé dans le méthanol avec un rendement de 35%.
1H NMR (DMSO-d6): δ 12,41; 11,67 (br s, br s, 2H, du NH amide et OH phénolique), 7,71–7,68 (m, 1H, H aromatique), 7,39–7,33 (m, 1H, H aromatique), 6,93–6,88 (m, 2H, Hs aromatiques).
Analyse élémentaire. Calc. Pour Na(HL_4)(EtOH): C11H12N2O3S2Na: C. 43,0; H. 3,9; N. 9,1%. Exp. C. 43,1; H. 4,0; N. 9,6%. IR (KBr, cm−1): ν = 3428, 3169, 3136, 1635, 1599, 1485, 1365, 891, 741, 644, 539.
2.2.4 β-Mn(O2CMe)2
Sous atmosphère d'argon, des essais de complexation du ligand H2L_3 avec l'acétate de manganèse (II) dans la DMF ont conduit au complexe β-Mn(O2CMe)2 [6], de formule C12H18Mn3O12, qui cristallise dans le système cristallin orthorhombique, selon le groupe d'espace P212121 et les paramètres de maille a = 11,310(5) Å, b = 11,548(5) Å, c = 15,265(5) Å; V = 1993,7(1) Å3. Ce complexe est signalé dans la littérature et il a été synthétisé par une autre méthode [6]. Nous avons alors choisi un autre métal de transition (le fer) pour tenter de synthétiser un complexe avec ce ligand.
2.2.5 Complexe de fer(III): FeIII(HL_3′)3
(L_3′: Phénol,2-[5-(méthylthio)-1,3,4-oxadiazol-2-yl])
Le complexe FeIII(HL_3′)3 est synthétisé sous atmosphère d'azote. Nous ajoutons, à une solution de 0,128 g de H2L_3 dans 2 ml de DMF, 0,024 g d'hydrure de sodium. Après réaction, nous ajoutons 0,063 g de chlorure de fer (II) anhydre dissous dans 1 ml de DMF. Après quelques heures àĺair, des cristaux bruns se forment dans le milieu réactionnel (rendement de 52%).
Analyse élémentaire. Calc. pour FeIII(HL_3′)3: C27H21N6O6S3Fe: C. 47,8; H. 3,1; N. 12,4%. Exp: C. 46,7; H. 3,3; N. 12,2%. IR (KBr, cm−1): ν = 1604, 1563, 1499, 1474, 1331, 862, 760, 519, 441.
2.3 Résolution structurale par diffraction des rayons X sur monocristal
Les monocristaux des composés H2L_3, H3L_4/H2L_3, Na(HL_4)(EtOH) et FeIII(L_3′)3 ont été analysés au moyen d'un diffractomètre automatique de type Nonius Kappa-CCD (Mo Kα λ = 0,71073 Å). Les intensités diffractées ont été intégrées à l'aide de la suite logicielle Denzo [7]. Les paramètres de maille ont été déterminés à partir de réflexions diffractées collectées sur 10 images (pas de 1,0° en rotation Phi) exposées 20 s chacune. Les structures cristallines ont été résolues par les méthodes directes (SHELXS97), puis affinées sur la base de F2 à l'aide du logiciel SHELXL97 [8]. L'absorption n'a pas été corrigée. Tous les atomes (sauf les atomes d'hydrogène) ont été affinés anisotropiquement. Les positions des atomes d'hydrogène ont été calculées en accord avec la stéréochimie et affinées en modèle rigide avec SHELXL97. Les données radiocristallographiques (à l'exclusion des facteurs de structure) ont été déposées au Cambridge Crystallographic Data Centre comme matériel supplémentaire. Les numéros de dépôt correspondants sont: n° CCDC 613175-613178. Ces données peuvent être obtenues gratuitement à l'adresse suivante: CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44)1223-336-033; e-mail: deposit@ccdc.cam.ac.uk). Le Tableau 1 résume les résultats des analyses radiocristallographiques.
Données cristallines and paramètres des affinements radiocristallographiques pour les composés H2L_3, (H2L_3)(H3L_4), Na(HL_4)(EtOH) et Fe(L_3′)3
| Composés | H2L_3 | (H2L_3)(H3L_4) | Na(HL_4)(EtOH) | Fe(L_3′)3 |
| Formule | C10H12N2O2S2 | C19H22N4O4S4 | C11H15N2NaO3S2 | C27H21FeN6O6S3 |
| Masse molaire (g mol−1) | 256,34 | 498,65 | 310,36 | 677,53 |
| Système cristallin | orthorhombique | triclinique | triclinique | cubique |
| Groupe spatial | P21cn | P | P | I |
| a [Å] | 11,7350(4) | 9,745(1) | 6,5890(10) | 22,574(5) |
| b [Å] | 12,7900(5) | 11,540(2) | 6,9700(10) | 22,574(5) |
| c [Å] | 15,7880(6) | 12,828(2) | 16,586(5) | 22,574(5) |
| α [°] | 90,00 | 63,702(5) | 99,60(5) | 90,00 |
| β [°] | 90,00 | 69,265(5) | 96,75(5) | 90,00 |
| γ [°] | 90,00 | 67,966(5) | 100,12(5) | 90,00 |
| V [Å3] | 2369,63(15) | 1167,8(3) | 730,8(3) | 11503(4) |
| Z | 8 | 2 | 2 | 16 |
| Densité (calc.) [g cm−3] | 1,437 | 1,418 | 1,410 | 1,565 |
| μ(MoKα) [mm−1] | 0,436 | 0,440 | 0,398 | 0,796 |
| F(000) | 1072 | 520 | 324 | 5552 |
| Dimension du cristal [mm] | 0,15 × 0,15 × 0,10 | 0,12 × 0,11 × 0,10 | 0,10 × 0,10 × 0,10 | 0,10 × 0,10 × 0,10 |
| Données collectées | ||||
| Theta min–max | 2,05–30,04 | 1,82–27,44 | 1,26–29,98 | 2,21–30,02 |
| Dataset[h, k, l] | 0/16, 0/17, −17/22 | −11/12, −12/14, 0/16 | −9/9, −9/9, −23/18 | −31/12, −19/24, –27/21 |
| Tot., Uniq. Data, R(int) | 6626, 3618, 0,0460 | 5285, 5284, 0,0435 | 8212, 4186, 0,0476 | 11 829, 2785, 0,0817 |
| Obs. data [I > 2σ(I)] | 2278 | 3241 | 2764 | 2026 |
| Affinement | ||||
| Nrefl., Nparam. | 3618, 313 | 5284, 298 | 4186, 184 | 2785, 128 |
| R2, R1, wR2,wR1, Goof | 0,1259, 0,0796, 0,2183, 0,1941, 1,024 | 0,1497, 0,0850, 0,2034, 0,1628, 1,040 | 0,1253, 0,0737, 0,1941, 0,1637, 1,081 | 0,0960, 0,0597, 0,1351, 0,1109, 1,112 |
| Max. and av. shift/error | 0,008, 0,002 | 0,001, 0,000 | 0,000, 0,000 | 0,000, 0,000 |
| Min,max. resd. dens. [e/Å3] | −0,427, 0,967 | −0,358, 0,408 | −0,490, 1,032 | −0,397, 0,501 |
3 Résultats et discussion
Nous présentons dans cet article nos premiers résultats relatifs à la synthèse de nouveaux ligands pouvant conduire à l'élaboration de systèmes magnétiques moléculaires comparables à ceux décrits récemment [3,4] et qui présentent à la fois des couplages magnétiques forts et des possibilités de modification de ces couplages par voie chimique ou physique. Nous avons ainsi synthétisé les ligands H2L_3 et H3L_4 (voir Schéma 2) et fait des tests de métallation à partir du chlorure ferrique et l'acétate manganeux tétrahydrate. Comme le montre l'analyse structurale intra- et supramoléculaire des ligands à l'état solide (voir ci-après), l'existence de nombreux contacts de type liaison hydrogène justifie à lui seul les essais de métallation que nous avons effectué. Un nouveau complexe, de structure originale, a ainsi été mis en évidence. En revanche, aucun complexe binucléaire n'a pu être stabilisé avec ces ligands, semblant indiquer que l'ouverture du cycle à cinq de type dithiolane n'est pas favorable à la formation d'un complexe binucléaire tel qu'il est présenté sur la Fig. 1.
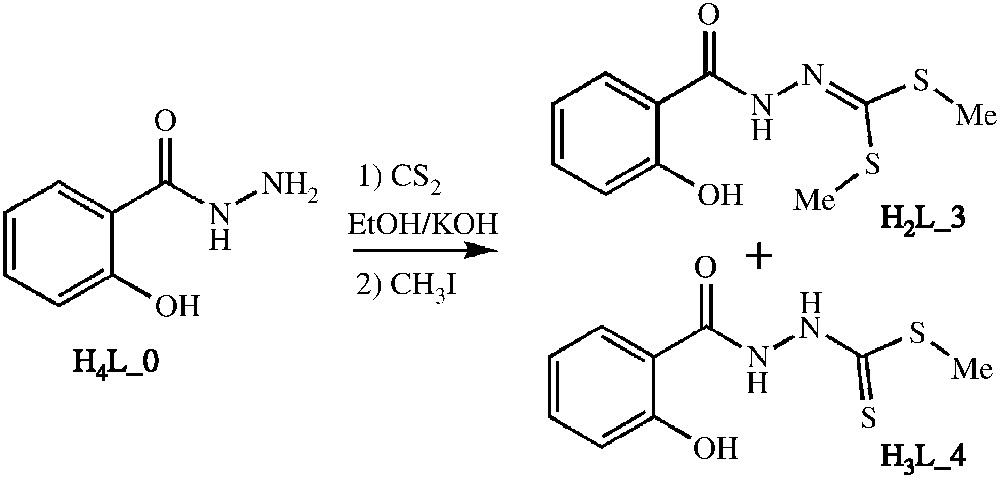
Synthèse des ligands H2L_3 et H3L_4 à partir de l'acide salicylhydrazide.
Dans le cas du complexe de fer(III) de symétrie cubique, la chélation par le ligand HL_3′ est originale, puisque l'on observe que l'oxygène du groupement phénol participe à la complexation sur l'atome de fer central (Schéma 3). Ceci n'avait pas encore été observé avec ce type de ligand. L'anion L_3′- est probablement le produit d'une attaque nucléophile de l'ion énolate de l'hydrazide sur le disulfure de carbone au sein du ligand H2L_3 en donnant le phénol,2-[5-(méthylthio)-1,3,4-oxadiazol-2-yl. Ce phénomène de cyclisation est décrit dans la littérature [10]. Une recherche sur la base de données CSD associant l'atome de fer et la symétrie cubique conduit aux 65 composés moléculaires, de symétrie cubique ou équivalente, connus à ce jour. En revanche, pour une configuration octaédrique de type « FeO3N3 », seuls deux autres complexes de symétrie cubique ont été signalés dans la littérature [11,12].

Synthèse du complexe FeIII(HL_3′)3.
3.1 Description des structures cristallines
Le Tableau 1 rassemble l'ensemble des données radiocristallographiques relatives aux 4 structures décrites ci-après. Les interactions faibles de type liaisons hydrogène sont détectées et analysées à l'aide du logiciel PLATON [9].
3.1.1 Acide 2-hydroxy [bis(méthylthio)méthylène]hydrazide benzoïque (H2L_3)
Ce ligand cristallise dans le système orthorhombique, selon le groupe d'espace P21cn.
Dans le solide cristallin, la partie hydrazide est quasi planaire (Fig. 2), avec présence d'une liaison hydrogène intramoléculaire entre l'azote N1 (ou N3) de l'hydrazide et l'oxygène O1 (ou O3) du groupement phénol N1–H1N⋯O1: dN1–O1: 2,573(7) Å, avec un angle N1–H1N–O1 de 133(1)° (ou N3–H3N…O3: dN3–O3: 2,598(8) Å, avec un angle N3–H3N–O3 de 133(1)°).
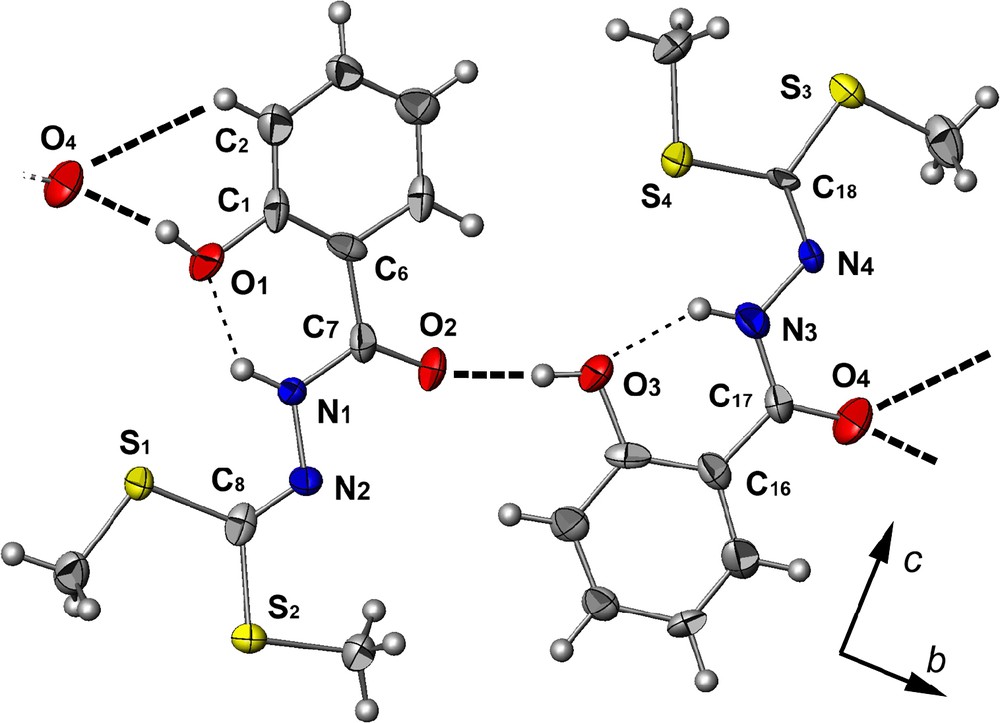
Vue supramoléculaire (ATOMS) du ligand H2L_3 (projection sur le plan b,c) avec une labellisation partielle. Les ellipsoïdes d'agitation thermique englobent 50% de la densité électronique. Les liaisons hydrogène sont représentées en pointillés (fin pour les liaisons H intramoléculaires and gras pour les liaisons intermoléculaires).
La connexion dans le cristal est assurée principalement par une interaction hydrogène intermoléculaire bifurquée (impliquant deux atomes donneurs et un accepteur, voir, par exemple, la référence [13]) entre l'oxygène O4 du groupement carbonyle et l'oxygène O1 du phénol et le carbone C2 du phénol de la molécule adjacente (1/2 + x, –1/2 + y, 5/2 − z), [dO4–O1: 2,578(8) Å avec un angle O1–H1O–O4 de 161(1)° et dC2–O4: 3,246(11) Å avec un angle C2–H2–O4 de 127°]. Compte tenu d'une autre liaison hydrogène entre l'oxygène O2 du groupement carbonyle et l'oxygène O3 du cycle aromatique d'une deuxième molécule (–1/2 + x, –1/2 + y, 5/2 – z), [dO2–O3: 2,588(8) Å, avec un angle O2–H3O–O3 de 160(1)°], l'empilement des molécules dans le cristal peut être décrit par la succession de pseudo-dimères (générés par la liaison hydrogène bifurquée) formant une chaine en zigzag le long de l'axe b (la Fig. 2 montre une chaîne de molécules liées par la liaison hydrogène la plus courte).
3.1.2 Co-cristallisation de l'acide 2-hydroxy [bis(méthylthio)méthylène]-hydrazide benzoïque H2L_3 et de l'acide 2-hydroxy-2-[(méthylthio)thioxo-méthyl]-hydrazide benzoïque H3L_4
L'analyse radiocristallographique de ces cristaux montre que le composé obtenu cristallise dans le système cristallin triclinique, selon le groupe d'espace centrosymétrique P
La résolution de la structure cristalline indique sans ambiguïté que l'unité asymétrique est constituée d'un mélange stœchiométrique des deux ligands H2L_3 (acide 2-hydroxy [bis(méthylthio)méthylène]-hydrazide benzoïque) et H3L_4 (acide 2-hydroxy-2-[(méthylthio)thioxométhyl]-hydrazide benzoïque) (Fig.3).
Dans l'unité asymétrique, des liaisons hydrogène du type O–H⋯O et O–H⋯N sont observées entre les deux ligands. L'oxygène O1 du groupement phénol du ligand H3L_4 fait une liaison bifurquée [13] avec l'oxygène O4 du carbonyle et l'azote N4 du ligand H2L_3 [dO1–O4: 2,643(5) Å et dO1–N4: 3,207(5) Å]. L'oxygène O3 du carbonyle du ligand H3L_4 est connecté par une autre liaison hydrogène à l'oxygène O2 du groupement phénol du ligand H2L_3 [dO5–C31: 3,31(1) Å]. L'arrangement supramoléculaire ainsi formé peut être décrit comme une succession de doublets (formés par deux molécules identiques tête bèche) perpendiculaires à deux autres doublets constitués par l'autre ligand, le long de l'axe c du réseau cristallin (voir Fig. 3).

(Partie supérieure) Vue supramoléculaire partielle (ATOMS) du ligand L_3_4 avec une labellisation partielle. Les ellipsoïdes d'agitation thermique englobent 50% de la densité électronique. Les liaisons hydrogène sont représentées en pointillés (fins pour les liaisons H intra-moléculaires and gras pour les liaisons intermoléculaires). (Partie inférieure) Vue complète des connexions supramoléculaires le long de l'axe c de la structure cristalline.
3.1.3 Acide 2-hydroxy-2-[(méthylthio)thioxo-méthyl]hydrazide benzoïque [Na(HL_4)(EtOH)]
L'analyse radiocristallographique de ces cristaux montre que le composé obtenu cristallise dans le système cristallin triclinique, selon le groupe d'espace P
La résolution de la structure indique que le composé cristallise sous forme d'un sel de sodium [Na(HL_4)(EtOH)] (Fig. 4). Le sodium est pentacoordiné et son environnement est de type pyramidal à base carrée. Une molécule d'éthanol (O3– dNa–O3 = 2,330(3) Å) et trois ligands H2L_4– (O1, O2, N1 et S1 avec les distances respectives: dNa–O1 = 2,381(3) Å, dNa–O2 = 2,369(3) Å, dNa–N1 = 2,499(3) Å et dNa–S1 = 2,98(1) Å) participent à la coordination de l'ion sodium. La structure forme un réseau polymérique suivant l'axe a du réseau. On notera enfin la présence de deux liaisons hydrogène intramoléculaires entre O2 et O1 (dO2–O1: 2.552(3) Å avec un angle O2–H2O–O1 de 154(4)°) et N2 et S1 (N2–H2N S1 2.891(3) Å avec un angle de 118(3)°).

(Gauche) Vue de type ATOMS de l'unité asymétrique du composé NaH2L_4(EtOH). Les ellipsoïdes d'agitation thermique englobent 50% de la densité électronique. Les liaisons hydrogène intramoléculaires sont représentées en pointillés. (Droite) Représentation de l'empilement moléculaire dans la structure cristalline (les sphères représentent les atomes de sodium).
3.1.4 Complexe de fer(III) de symétrie C3: FeIII(HL_3′)3. (HL_3′: phénol,2-[5-(méthylthio)-1,3,4-oxadiazol-2-yl])
Ce complexe cristallise dans le système cristallin cubique, selon le groupe d'espace I
La structure de ce complexe ferrique Fe(HL_3′)3 montre que l'ion FeIII est chélaté par trois anions bidentates (L_3′–) (Fig. 5), formant ainsi un octaèdre déformé, puisque les longueurs de liaison Fe–N et Fe–O ne sont pas équivalentes (dFe–O ≈ 1.92 Å et dFe–N ≈ 2.16 Å).

Vue de type ATOMS du complexe Fe(L_3′)3 avec une labellisation partielle. Les ellipsoïdes d'agitation thermique englobent 50% de la densité électronique. Les atomes d'hydrogène sont volontairement omis par souci de clarté, à l'exception de H7, qui montre une interaction hydrogène faible vers le centroïde du groupement phénylique (C2_C7) des molécules adjacentes.
On notera enfin que les interactions supramoléculaires prépondérantes sont probablement les interactions de type CH-π présentes entre l'hydrogène H7 et le centroïde du groupement phénol (dH7-centroïde = 2,76 Å avec un angle de 167° – voir Fig. 5).
4 Conclusions
Dans cet article, nous avons présenté les structures cristallines de deux nouveaux ligands ainsi que la synthèse et la structure d'un nouveau complexe associant un métal de transition, le fer au degré d'oxydation III. Ces résultats entrent dans le cadre d'une recherche systématique de ligands pouvant conduire à des complexes paramagnétiques et asymétriques à couplages magnétiques contrôlés par des liaisons hydrogènes. La présente étude nous apprend ainsi que l'ouverture du cycle à cinq de type dithiolane n'est pas favorable à la synthèse des systèmes souhaités, mais montre néanmoins que les ligands dérivant de l'acide salicylhydrazique conduisent, en réaction avec les métaux de transition, à des complexes inattendus et dont les propriétés physiques peuvent être intéressantes à étudier plus avant.
Remerciements
Les résultats présentés dans cet article sont une partie du travail de thèse de Chahrazed Beghidja et Nouri Bouslimani. Nous tenons à remercier M. André DeCian pour sont aide lors de la sélection des monocristaux ainsi que pour l'enregistrement des données issues de la diffraction sur monocristaux.