

1 Introduction
A fascinating find is the widespread occurrence of aminoacid, particularly tyrosine, radicals [1] in metalloproteins involved in oxygen-dependent enzymatic radical catalysis. This observation has broadly influenced the interests of coordination chemists in choosing phenol-containing ligands to synthesize bioinspired model compounds that contain coordinated phenolate and its one-electron oxidized phenoxyl radical [2]. Additionally, in the field of molecular magnetism [3] redox-active paramagnetic ligands are gaining increasing attention for the generation of molecular high-spin materials based on the hybridization of organic–inorganic molecules in which paramagnetic metal ions are coordinated to organic open-shell radical ligands [4]. This surge of interest has also been due partly to the relevance of such molecules to biological electron-transfer processes. Thus, investigations of radical-containing transition metal complexes are relevant to and important for deepening our knowledge of radicals in metalloproteins [5].
Copper-containing amine oxidases [6] (CAO; EC 1.4.3.6) are widespread in nature, occurring in bacteria, yeast, plants, and mammals. These enzymes catalyse the oxidative deamination of primary amines to form the corresponding aldehyde and ammonia, with subsequent reduction of dioxygen to hydrogen peroxide (Eq. (1)).
| RCH2NH2 + O2 + H2O → RCHO + NH3 + H2O2 | (1) |
CAOs belong to the class of ‘type-2’ or “nonblue” copper proteins that utilize topaquinone (TPQ) (2,4,5-trihydroxyphenylalanine quinone), a modified tyrosine side chain, as a redox cofactor to achieve deamination via an amino transferase mechanism. This new type of metalloenzymes is characterized by the probable participation in catalytic turnover of a semiquinone radical, Cu(I)-TPQ. Recent results, however, seem to question the involvement of copper as a redox-active participant in the oxidative half-reaction. Instead, it has been proposed that it may only serve an electrostatic role. Regardless of the detailed mechanism, a rigorous requirement for active-site bound copper has been attributed to two different but equally critical roles: “to aid assembly of the active site and to act as part of the final catalytic machinery”.
X-ray crystal structures [7] are available for amine oxidases in the resting (oxidized) state from different sources at different levels of resolution (2, 2.2 and 2.4 Å). The coordination geometry of Cu(II), irrespective of the sources, is distorted square pyramidal with three-coordinated histidine imidazoles and two equatorial and axial water ligands, together with the topaquinone residue. Phenoxazinone synthase (PHS) [8], an oligomeric multicopper oxidase, catalyses the oxidative coupling of two molecules of an aminophenol to form the phenoxazinone chromophore. This reaction constitutes the final step in the biosynthesis of actinomycin and is a complex six-electron oxidative condensation (Eq. (2)).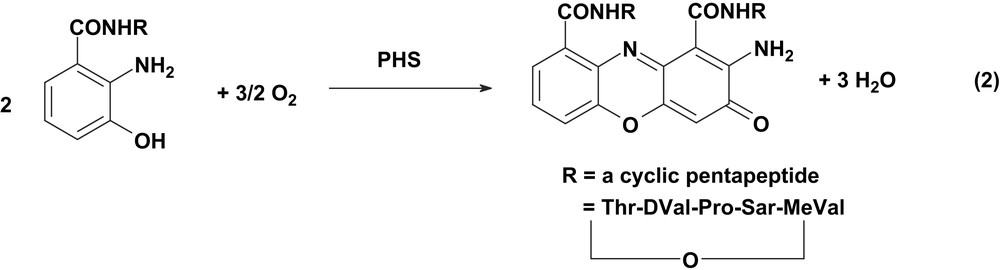
PHS exists in two oligomeric forms: a dimeric and a hexameric form. Recently, the structure of hexameric PHS has been determined using X-ray diffraction to a resolution limit of 2.30 Å [9]. Each subunit of the hexamer contains five copper atoms as detected earlier spectroscopically. The structure confirms the presence of all three copper-binding motifs, as usually known for multicopper oxidases: one type 1 (blue), two type-2 (normal) and one binuclear type-3 centers. The location of the fifth copper center, a type-2 copper, at a distance of ∼25 Å from the blue copper and the other normal type 2 copper, and the requirement of five copper atoms for maximum activity suggest that the fifth copper atom is not merely advantageously bound but has a structural role as well.
With extensive physico-chemical spectroscopic and crystallographic data as a back-up, the above-mentioned two oxidase systems are ‘ripe’ for an in-depth examination of the interaction of aerial dioxygen with a phenolate-oxygen bound to metal center. In a synergistic approach, we have started investigations of radical-containing metal complexes and their oxidative catalytic activities towards different organic substrates in the presence of molecular oxygen as the sole oxidant. The aim is twofold: (i) to gain deeper insight into the electronic structure of the metal core and the ligand surrounding of these complexes and, thereby, to better understand such metal–radical interactions in natural systems; (ii) to explore the aerial-oxidation chemistry of well-characterized metal complexes, which is expected to provide the basis for new catalytic oxidation systems for synthetic and industrial processes.
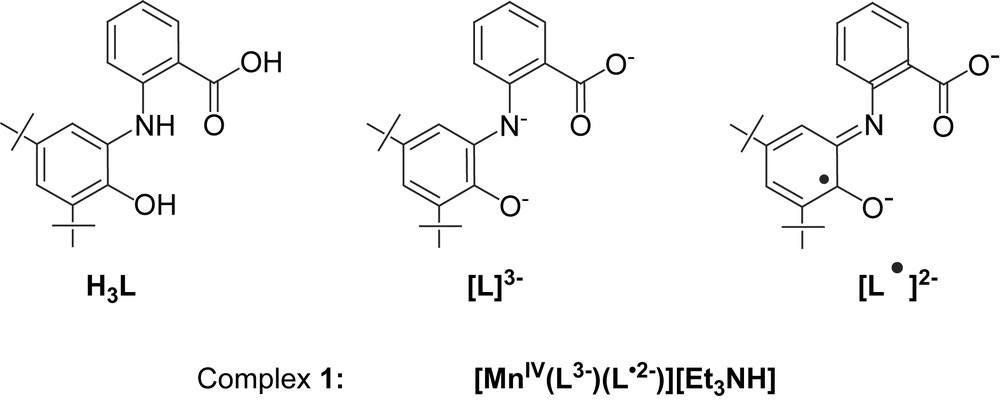
Recently, we have reported several non-innocent ligands [10], based on N-phenyl-2-aminophenol, which can coordinate not only in their different deprotonated forms but also in their oxidized o-iminobenzosemiquinonate radical forms. As a natural progression of using radical-generating non-innocent ligands, we have synthesized the tridentate ligand, H3L, N-(2-hydroxy-3,5-di-tert-butylphenyl)anthranilic acid and its manganese(IV) complex, 1. The crystal structure and magnetism of 1 show unambiguously the presence of Mn(IV) coordinated to two different forms of the ligand, viz. (i) a monoradical dianionic, and (ii) a non-radical trianionic form, as shown together with the ligand, H3L, in Scheme 1.
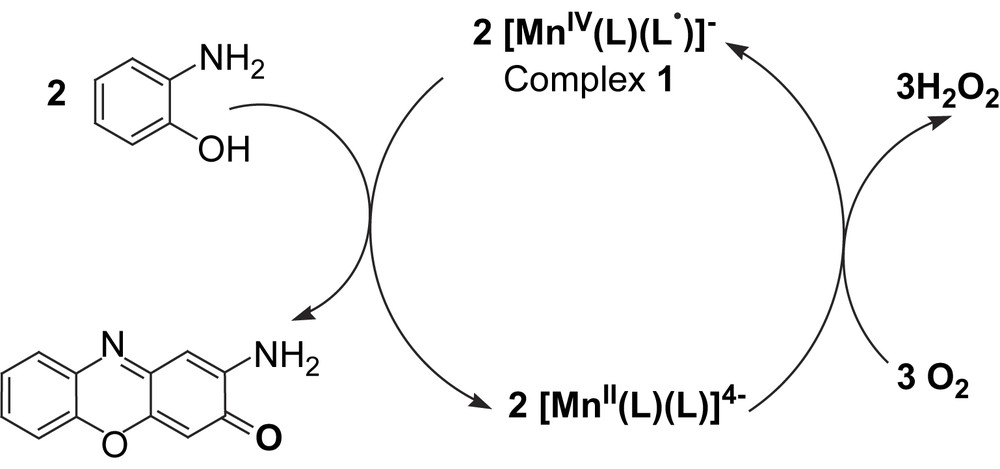
In this paper we describe the spectroscopic and structural characterization of a Mn(IV) complex, 1, which is also capable of catalytic oxidative deamination of primary amines with air. Additionally, 1 is an efficient catalyst for the generation of phenoxazinone chromophore, thus models the function of PHS and also of the enzymes copper amine oxidases (CAO). Although, PHS and CAO contain copper, a manganese(IV)-monoradical complex, 1, models the catalytic functions of the said enzymes, thus exemplification of opening the way for other enzyme mimics being developed that contain metals other than copper.
2 Results and discussion
The ligand, N-(2-hydroxy-3,5-di-tert-butylphenyl)anthranilic acid, H3L, is readily available in moderate yields from the reaction of 3,5-di-tert-butylcatechol and anthranilic acid in n-hexane in the presence of air and the base triethylamine. Excess triethylamine lowers the yield of the product remarkably and thus, is to be avoided. In the IR, the ligand shows characteristic strong peaks at 3456, 3336 and 2959–2904 cm−1 attributable to ν(OH), ν(NH) and ν(CH) of the tert-butyl groups, respectively. Moreover, a strong peak at 1667 cm−1 clearly shows the presence of the COOH group. Selected IR peaks are listed in Section 4.
EI mass spectroscopy shows the molecular ion peak at m/z 341 confirming the composition C21H27NO3. The ligand was also characterized by different other techniques, viz. 1H NMR, together with microanalysis and melting point, and the purity was checked by GC–MS (see Section 4).
When a reaction mixture of the ligand H3L, Et3N and Mn(CH3COO)2·4H2O was stirred under air for 2 h, deep brown microcrystalline solid of complex 1 was obtained in 63% yield. The most salient features observed for complex 1 in the IR are the absence of the frequencies attributable to ν(NH) and ν(OH) stretching. Medium intense to strong sharp bands at 1615, 1594, 1579, 1466, 1443, 1252, 1127, 1101, 1085 and 1023 cm−1 show the existence of the ligand in complex 1 in both iminosemiquinone and amidophenolate forms. ESI-MS in positive and negative modes show molecular ion peaks at m/z 102 (100%) and 735 (100%), respectively, thus confirming the composition [(C2H5)3NH]+[C42H48N2O6Mn]− and indicating that the ligand cannot be present in its “simple” innocent form (Scheme 1).
2.1 Description of the structure
The structure of complex 1 has been determined by single-crystal X-ray crystallography both at 100(2) and 293(1) K. As the crystal-quality for 298 K was better than that for 100 K, we are describing only the structure determined at 298 K; the structural data at 100 K are included as Supplementary material. Fig. 1 shows the structure of the anion in crystals of 1. Selected bond lengths and angles for complex 1 are summarized in Table 1. The result of the crystallographic study, in consonant with the analytical and mass spectroscopic data, shows that a manganese complex is formed in the metal:ligand ratio of 1:2. Each ligand acts as a meridional O,N,O-donor. The geometry around the metal manganese is thus six-coordinate with an N2O4 environment, in which two carboxylate oxygens occupy the cis-positions. In terms of angles the coordination sphere MnN2O4 is close to an ideal octahedron; the dihedral angle between the planes N(8)O(17)N(38)O(1) and O(47)O(17)O(31)O(1) is 88.7°. The largest deviation from linearity is shown by the trans-disposed N(8)–Mn(1)–N(38) angle of 171.0(1)°. The anion in crystals of 1 contains two ligands in different forms: one O,N,O-coordinated carboxylate(1−)/o-iminobenzosemiquinonate(1−) π radical ligand and one N-deprotonated (N8) amidophenolate-carboxylate trianion(3−) ligand. The presence of π radical(2−) ligand is clearly borne out by the observation that (i) N(38) is sp2 hybridized and not protonated; (ii) the six-membered ring of the iminobenzosemiquinonate part displays the typical quinoid distortions, comprising a short, a long, and another short C–C bond followed by three long ones; (iii) the C(32)–O(31) and C(37)–N(38) bond lengths at 1.320(4) and 1.370(4) Å, respectively, are short, approaching double bonds. The second ligand, in contrast, is N-deprotonated: N(8) is sp2 hybridized (the sum of three bond angles C(9)–N(8)–C(7) 124.9(3)°, C(9)–N(8)–Mn(1) 125.6(2)° and C(7)–N(8)–Mn(1) 109.3(2)° is 359.8°), thus providing an amidophenolate-carboxylate trianion(3−) ligand. The six C–C bonds of the amidophenolate ring are equidistant at 1.40 ± 0.02 Å and C(2)–O(1) and C(7)–N(8) distances at 1.339(4) and 1.394(4) Å, respectively, are long. So, it is quite clear that among the two tridentate ligands (O,N,O), one is in iminosemiquinone form with 2(−) charge and the other is in amidophenolate form with 3(−) charge. Thus, MnN2O4 unit being mono-negative, contains the central Mn(1) in +IV (d3) oxidation level. The observed Mn–O and Mn–N bond distances, av. 1.90 (±0.015) and 1.92 (±0.015) Å, respectively, also support this view; they are shorter than the comparable reported values for Mn(III)-complexes and similar to those for Mn(IV) complexes [10,11]. Additionally, the pseudo-octahedral O4N2Mn polyhedron in complex 1 does not show any Jahn–Teller distortion. The crystal structure of complex 1 is in excellent agreement with the charge distribution [MnIV(L)(L)]− and is very similar to, as expected, that for the radical-containing Mn(IV) complexes, reported earlier by us [10,12].
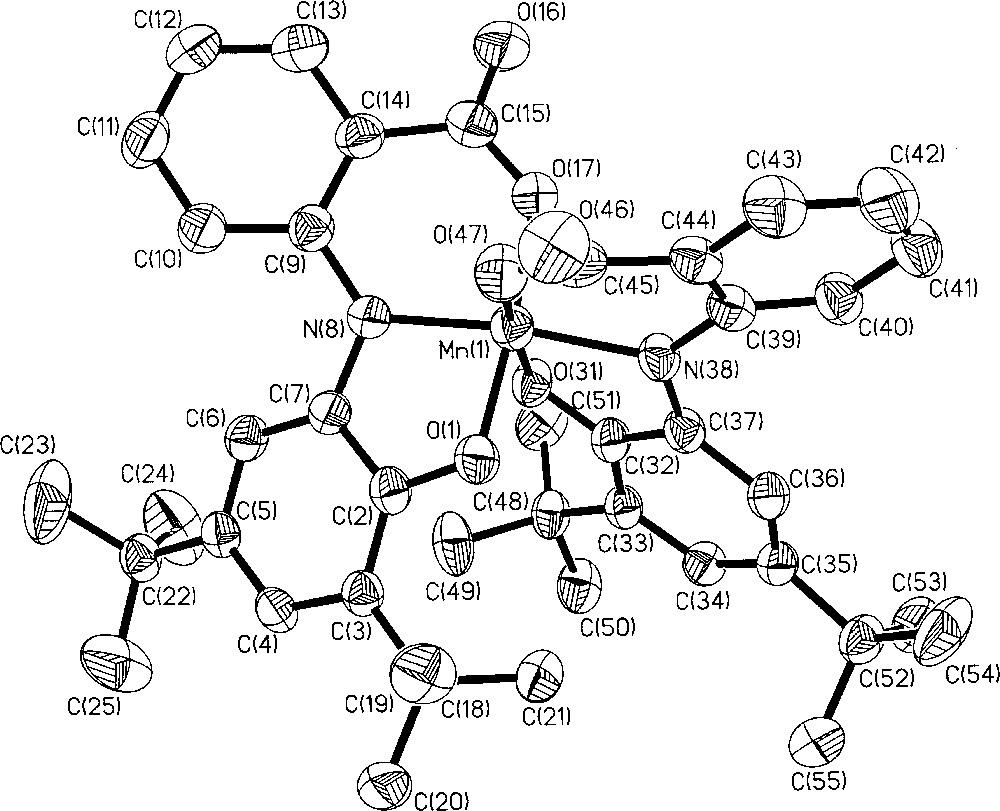
Molecular structure of complex 1.
Selected bond distances (Å) and angles (°) for complex 1
| Mn(1)–O(1) | 1.878(2) | O(1)–Mn(1)–O(31) | 88.25(11) |
| Mn(1)–O(31) | 1.887(2) | O(1)–Mn(1)–O(47) | 89.81(11) |
| Mn(1)–O(47) | 1.900(3) | O(31)–Mn(1)–O(47) | 174.85(11) |
| Mn(1)–N(8) | 1.907(3) | O(1)–Mn(1)–N(8) | 83.82(11) |
| Mn(1)–O(17) | 1.915(3) | O(31)–Mn(1)–N(8) | 89.79(11) |
| Mn(1)–N(38) | 1.934(3) | O(47)–Mn(1)–N(8) | 94.75(12) |
| O(1)–C(2) | 1.339(4) | O(1)–Mn(1)–O(17) | 175.24(11) |
| C(2)–C(3) | 1.401(5) | O(31)–Mn(1)–O(17) | 91.66(11) |
| C(2)–C(7) | 1.416(5) | O(47)–Mn(1)–O(17) | 90.67(12) |
| C(3)–C(4) | 1.394(5) | N(8)–Mn(1)–O(17) | 91.43(12) |
| C(4)–C(5) | 1.407(5) | O(1)–Mn(1)–N(38) | 90.68(11) |
| C(5)–C(6) | 1.372(5) | O(31)–Mn(1)–N(38) | 82.97(11) |
| C(6)–C(7) | 1.405(5) | O(47)–Mn(1)–N(38) | 92.29(12) |
| C(7)–N(8) | 1.394(4) | N(8)–Mn(1)–N(38) | 171.04(12) |
| N(8)–C(9) | 1.389(4) | O(17)–Mn(1)–N(38) | 94.03(12) |
| O(31)–C(32) | 1.320(4) | C(9)–N(8)–C(7) | 124.9(3) |
| C(32)–C(33) | 1.419(5) | C(9)–N(8)–Mn(1) | 125.6(2) |
| C(32)–C(37) | 1.423(5) | C(7)–N(8)–Mn(1) | 109.3(2) |
| C(33)–C(34) | 1.373(5) | C(37)–N(38)–C(39) | 124.6(3) |
| C(34)–C(35) | 1.420(5) | C(37)–N(38)–Mn(1) | 110.2(2) |
| C(35)–C(36) | 1.367(5) | C(39)–N(38)–Mn(1) | 125.2(2) |
| C(36)–C(37) | 1.413(5) | ||
| C(37)–N(38) | 1.370(4) | ||
| N(38)–C(39) | 1.400(4) |
2.2 Magnetic susceptibility and EPR measurements
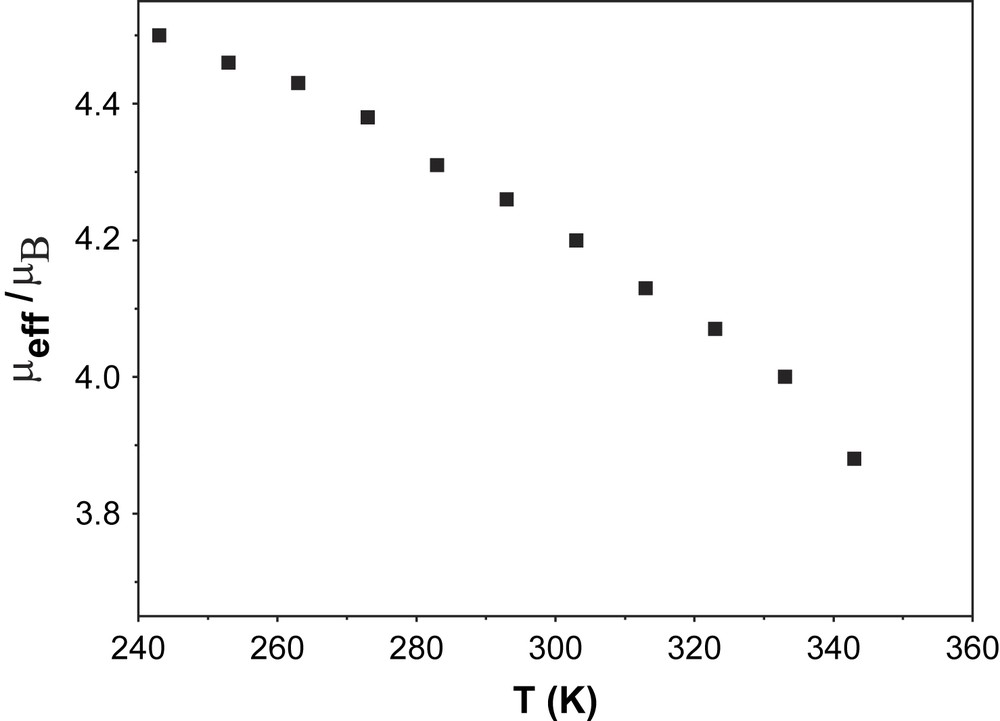
In order to establish the electronic ground state for complex 1, variable-temperature (2–290 K) magnetic susceptibility measurements on powdered sample of 1 were performed by using a SQUID magnetometer in an external magnetic field of 1.0 T. Fig. 2 shows the temperature dependence of the magnetic moment, μeff, for complex 1. The magnetic moment in the temperature range 20–290 K is practically constant with a value of μeff = 2.81 ± 0.03 μB, indicating an St = 1 ground state for 1. A strong antiferromagnetic interaction (J > 200 cm−1) between the two paramagnetic centers, SMn(IV) = 3/2 and SR = 1/2, leads to the ground state St = 1. Below 20 K, μeff starts to decrease steadily reaching a value of 1.59 μB at 2 K, probably due to zero-field splitting of the ground state. The experimental data are simulated by using the following parameters: S = 1 (fixed), gMn = 1.985, gRadical = 2.00 (fixed) and D = |6.3| cm−1 and the simulation is shown as a solid line in Fig. 2. The ground state of St = 1 was also confirmed by the variable-temperature variable-field measurements at 1, 4 and 7 T and these magnetization measurements are shown in Fig. 3. The solid lines in Fig. 3 are theoretical simulations with parameters St = 1, D = |6.3| cm−1. Thus, magnetic measurements unambiguously show that the ground state St = 1 for complex 1 arises from a strong antiferromagnetic interaction between a Mn(IV) (S = 3/2) and a radical ligand (SR = 1/2), thus confirming the monoradical nature of 1, [MnIV(L)(L)]−, as elucidated from the structural data.
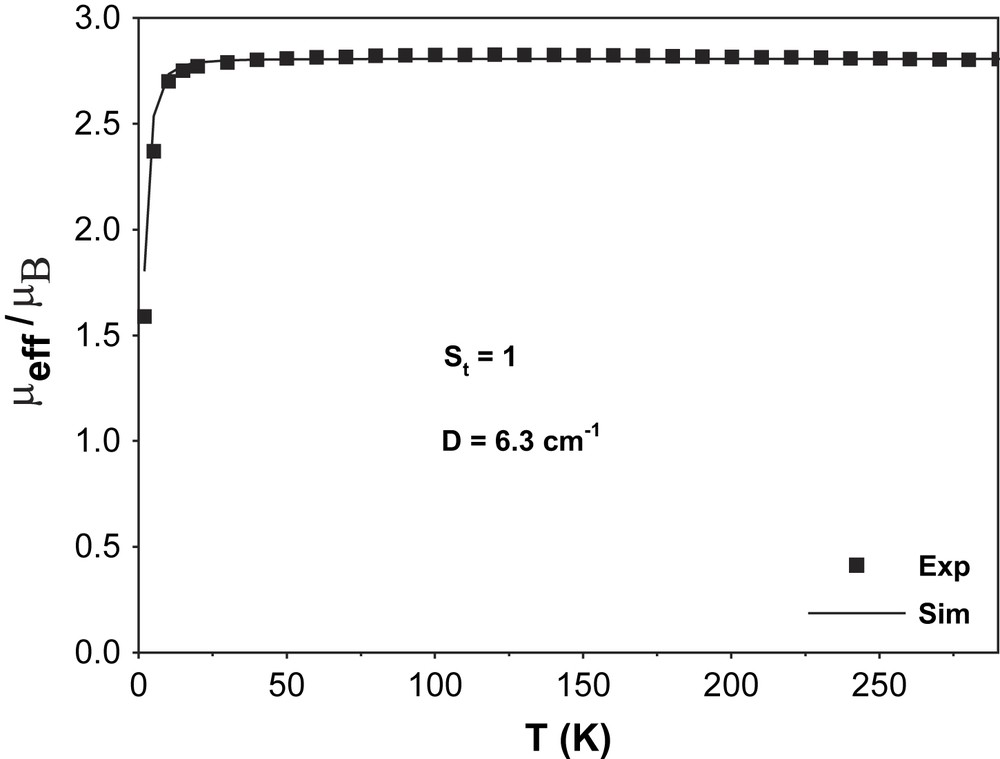
Plot of μeff/μB vs. T (K) for complex 1.
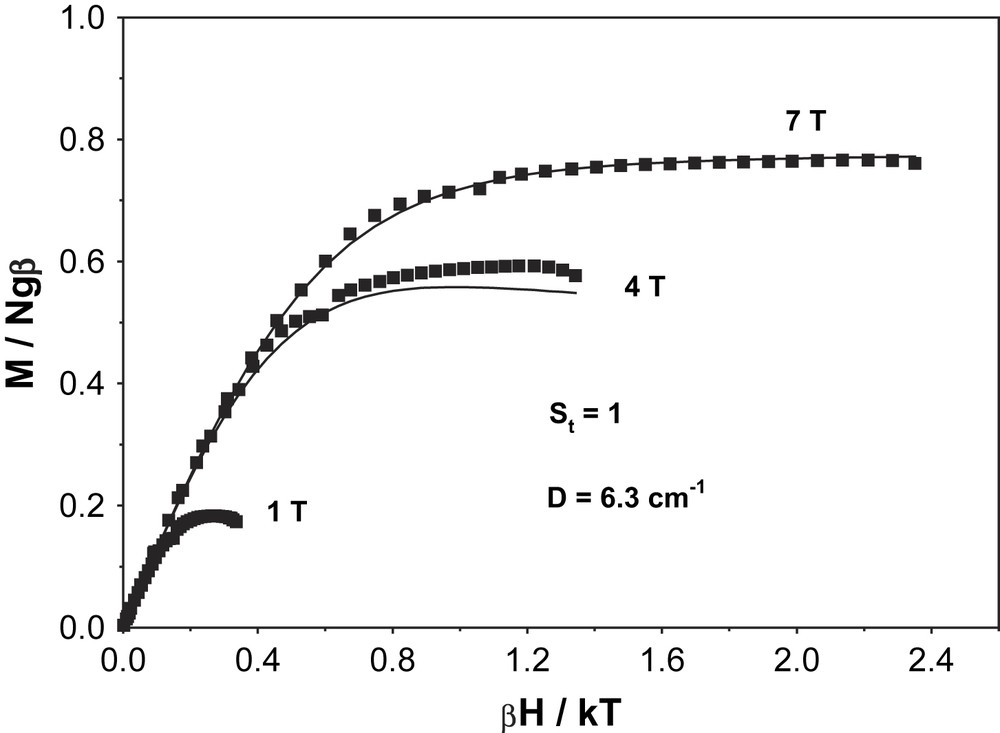
VTVH magnetization measurements at 1, 4 and 7 T for complex 1.
That complex 1 is EPR silent is also in conform with the ground state St = 1, as determined by the susceptibility measurements.
2.3 Identification of a dimeric form in solution
At room temperature the electronic spectrum of 1 in CH2Cl2 and ClCH2CH2Cl solution exhibits a distinct band in the NIR range with a maximum at 945 nm. The intensity of this band (λ = 945) depends on the concentration of 1, being higher at higher concentrations; in Fig. 4 the molar absorption coefficients ɛ are plotted vs. the concentration (c) of 1. Apparently complex 1 dimerizes in solution. It is seen that the limiting ɛ (λ945) value for infinite dilution is zero and the value at high concentrations approaches a value of ɛ (945)lim = 4.8 × 103 M−1 cm−1. The ratio ɛ (945)/ɛ (945)lim should then correspond to dimeric fraction of 1 and (1 − ɛ (945)/ɛ (945)lim) to the degree of dissociation, α. If the α values thus obtained are plotted vs. the concentration of 1 according to Ostwald's law of dilution, the same association constant Ka = 4 × 104 M−1 is obtained for all concentrations and degrees of dissociation (0.12 < α < 0.85), within the experimental error. This clearly shows that dimerisation occurs indeed.
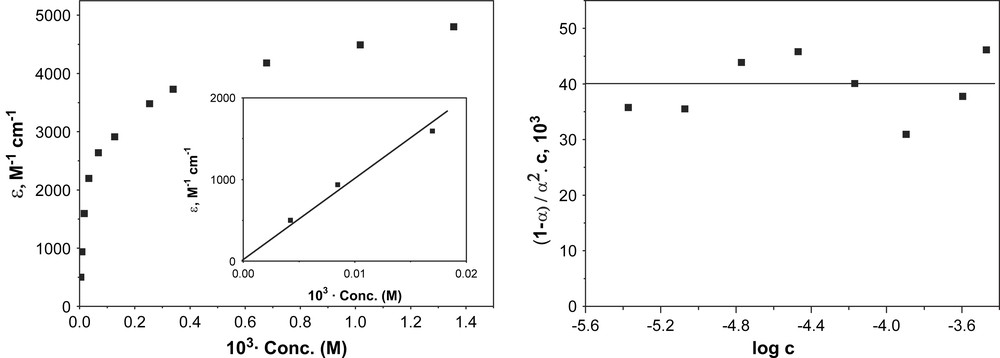
Plots of ɛ at λ = 945 nm vs. concentration (left) and (1 − α)/α2C vs. log c (concentration) (right) for complex 1.
The intensity of the band at 945 nm is also influenced by the temperature: when the temperature is decreased, the intensity of the band increases, i.e. low temperatures favour dimerisation. The spectral changes are observed to be reversible and isosbestic behaviour at 415 nm and 475 nm shows that only two species are present in the solution, which are reversibly interconverted on varying the temperature. Fig. 5 shows the changes of the absorption spectra as a function of temperature of complex 1 (1.8 × 10−4 M) together with the plot of absorption at λ = 945 nm vs. 1/T of a dichloromethane solution.
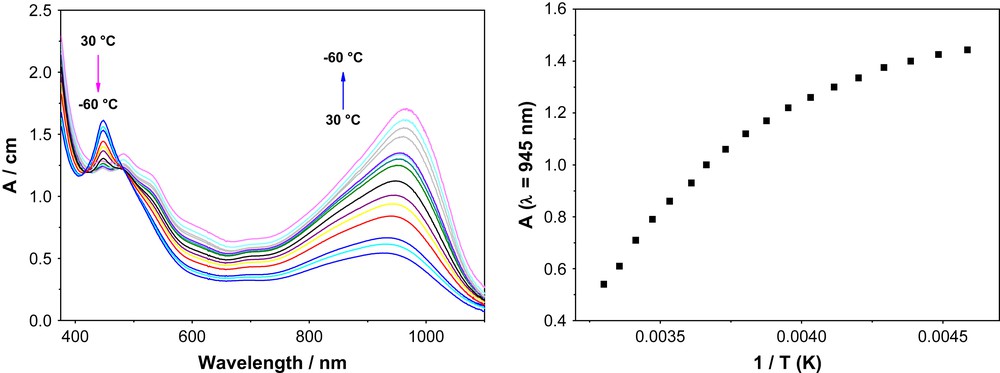
Temperature dependence of the electronic absorption spectrum of a dichloromethane solution (left) and absorption at 945 nm vs. 1/T plot (right) for complex 1.
It is seen that in the accessible temperature range (+30 °C to −60 °C) the limiting values of the high and low temperature forms are not reached. However, the diagram suggests that the limiting OD values of the pure forms are close to zero at high temperatures and around 1.6 for low temperatures. Using these values, equilibrium constants K for the different temperatures could be calculated. If they are plotted in the form ln K vs. 1/T according to van't Hoff's law, a reasonable straight line is indeed obtained (Fig. 6). The ΔH value obtained from the plot is −22 kJ mol−1. Using the equilibrium constant at 300 K, the thermodynamic parameters are calculated to be ΔG300 = +1.2 kJ mol−1 and ΔS300 = −77 J mol−1 T−1. The signs of the thermodynamic data are taken for the direction of cooling, i.e. for formation of {[MnIV(L)(L)]−}2.
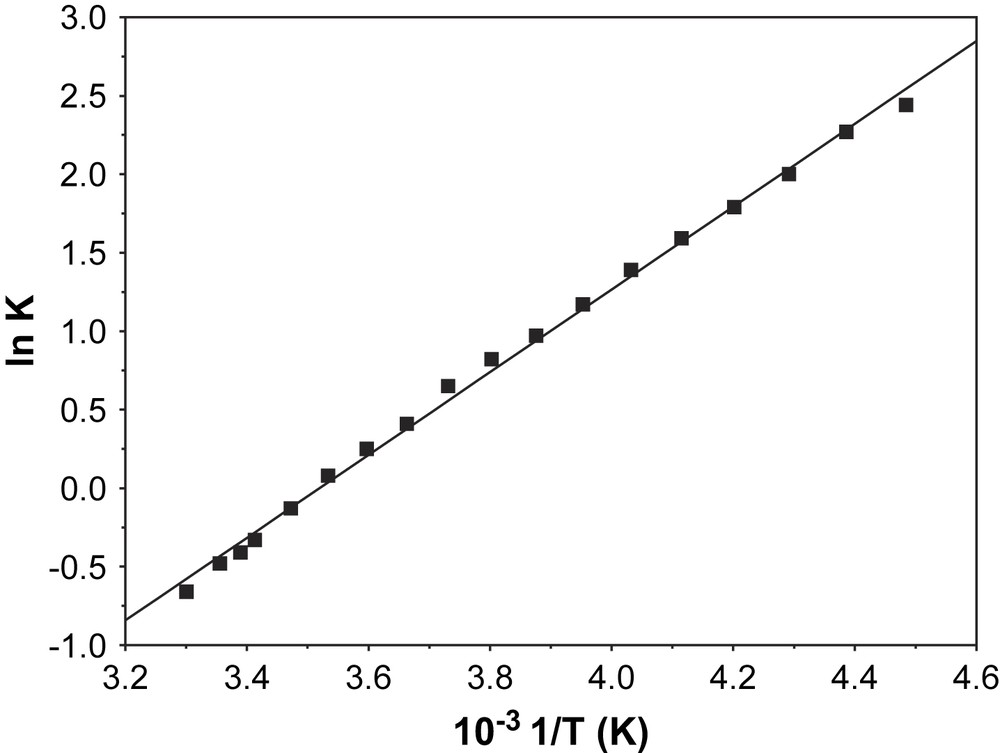
Plot of ln K vs. 1/T for complex 1.
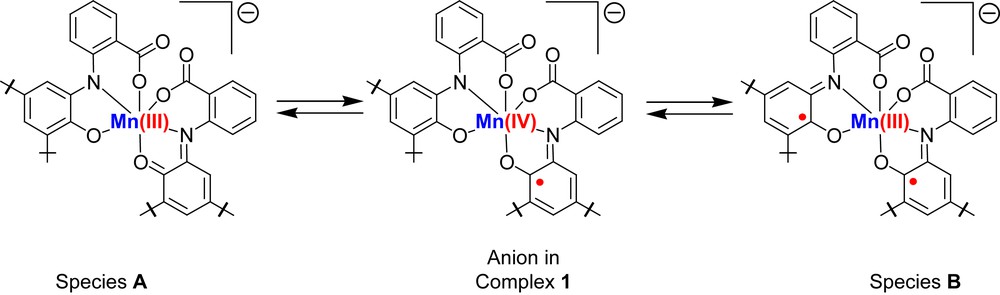
The other alternative that complex 1 undergoes thermally driven intramolecular electron transfer (valence tautomerism) can be ruled out. If the process was a valence tautomerism then complex 1 would be in equilibrium with species A or species B as shown in Scheme 2. The stabilization of species A is quite unlikely as it contains ligands in the strong oxidizing iminoquinone form and the strong reducing amidophenolate form. Hence, disproportionation will be the result. Moreover, entropy of the system decreases on cooling. On the other hand, if complex 1 shifts to species A or species B then entropy should increase as oxidation state of the metal decreases from +IV to either +III or +II (for a 2e transfer process). Therefore, the thermodynamic data support again dimerisation as shown below and not valence tautomerism [13,4b].
Further support for the formation of dimer on cooling the solution was obtained from 1H NMR (ClCD2CD2Cl) spectra measured in the temperature range +70 °C to −40 °C. The monomeric form has an St = 1 ground state whereas the dimeric form could have St = 2, 0 or two degenerate St = 1 ground states. Fig. 7 shows the μeff vs. T plot. The μeff values were calculated using Evan's method [14]. At −40 °C μeff of the 9 mM solution of 1 in dichloroethane-d4 is 4.5 μB. On increasing the temperature of the solution the μeff value decreases gradually and reaches 3.82 μB at +70 °C. The decrease in μeff value with increase in temperature of the solution indicates splitting of the dimeric species to monomeric species and at +70 °C both dimeric and monomeric species exist in the solution. Measurements of 1H NMR spectra above +70 °C were not possible as the complex decomposes above the temperature.
Change in μeff of a solution in dichloroethane-d4 with temperature for complex 1.
2.4 Catalytic aerial oxidation of primary amines: mimicking the function of amine oxidases
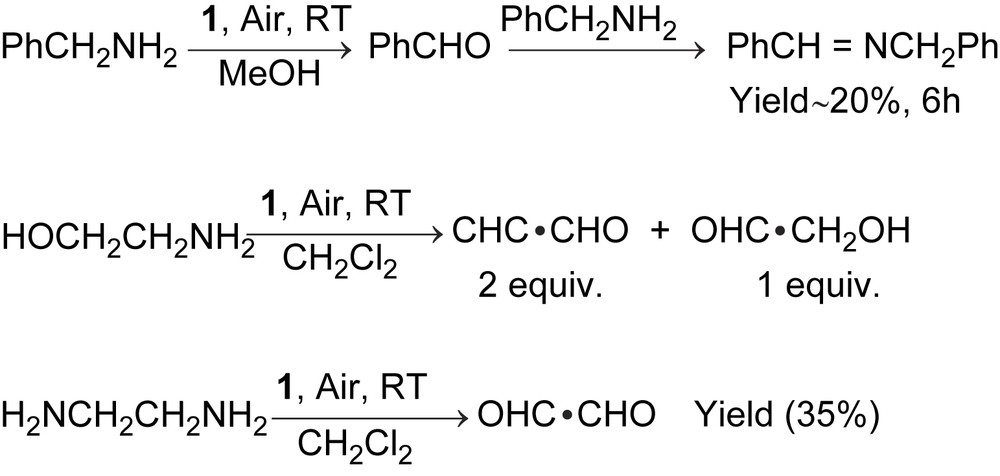
The Mn(IV)-monoradical complex, 1, catalyses the aerial oxidation of amines with two α-H atoms, like benzylamine, ethylenediamine, 2-aminoethanol, as shown in Scheme 3. No oxidation of secondary amines or amines with one α-H atom was observed.
The kinetic of the deamination of benzylamine was investigated in detail.
When a catalytic amount of complex 1 (10−5 mol) was introduced for the aerial oxidation of a 100-fold excess of benzylamine (10−3 mol) and 20% of benzylidinebenzylamine was determined in 6 h by GC using n-hexadecane as an internal standard. No more conversion was noted after 6 h because of the decomposition of complex 1 after that period. Though decomposition of hydrogen peroxide occurred due to the catalase activity of the manganese complex at room temperature, hydrogen peroxide is detected at −25 °C. Ammonia is also detected qualitatively. The kinetics for this catalytic reaction were studied by varying the total concentration of benzylamine (10−2–2.5 × 10−6 mol) keeping the concentration of complex 1 as constant and vice versa. The rate law deduced from the kinetic data is
| rate = k[complex][amine] |
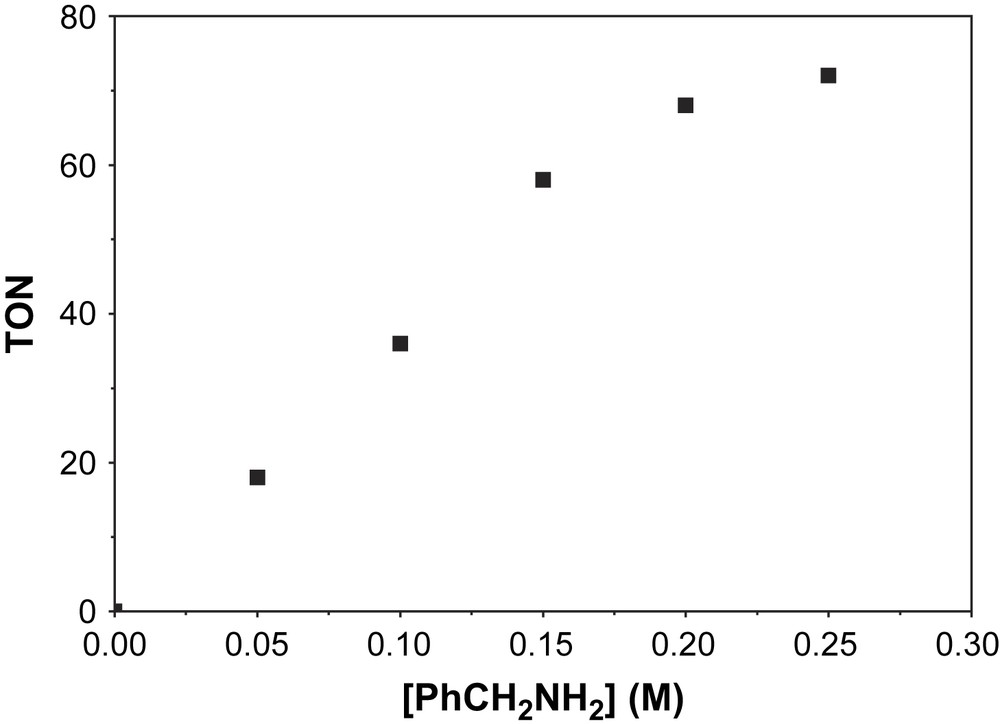
The maximum turnover number TON (the ratio between the concentration of product and catalyst) that has been achieved for the above catalytic process is 72 and is shown in Fig. 8. With the selectively deuterated substrate at the α-C atom, PhCD2NH2, a kinetic isotope effect (KIE = kH/kD) of about 1 was evaluated and is depicted in Fig. 9. This indicates clearly that the H-atom abstraction from the α-C atom of the substrate is not the rate-determining step for the catalysis.
Turnover numbers (TON) as a function of benzylamine concentration for aerial oxidation of the amine catalysed by complex 1.
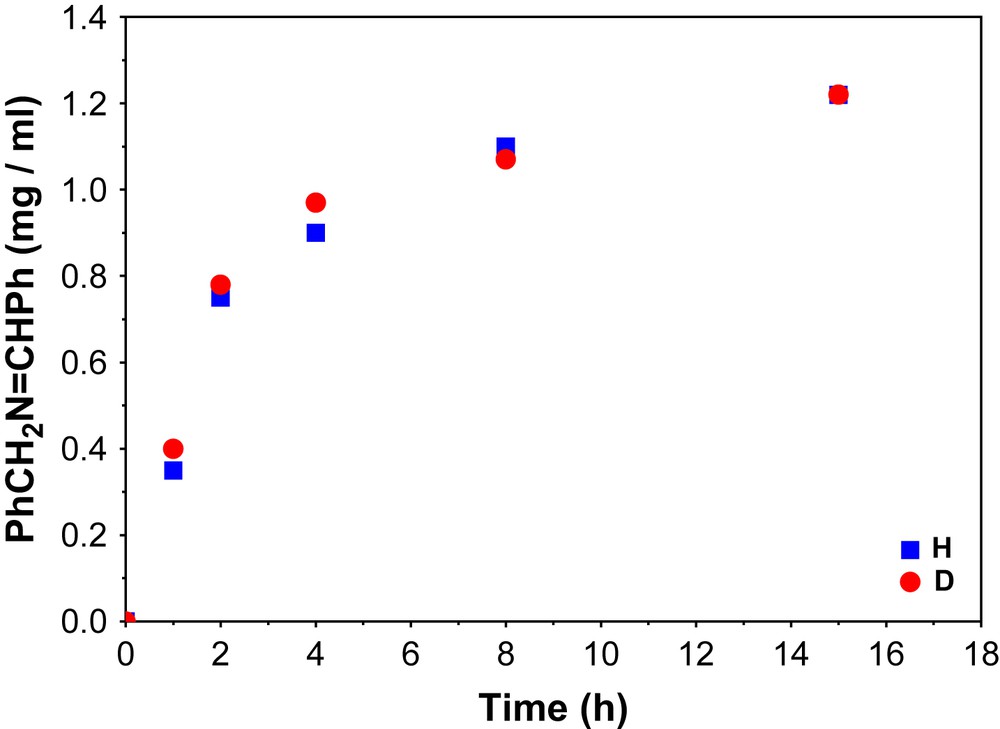
Kinetic isotope effect for the aerial oxidation of benzylamine.
2.4.1 Reaction in absence of aerial oxygen
When 12.5 equivalents of benzylamine were allowed to react with 1 equivalent of complex 1 under anaerobic condition, about 1.5 equivalent benzylidinebenzylamine was found by GC analysis. The X-band EPR spectrum at 10 K and UV–vis/NIR spectrum at room temperature of the solution were measured and are shown in Fig. 10. The X-band EPR spectrum shows a signal indicative of a Mn(II) species with six major hyperfine lines centered around g = 2 which originate from the hyperfine splitting of the transition Ms = 1/2 to Ms = −1/2 of S = 5/2 paramagnet with I = 5/2 of the 55Mn nucleus. Thus EPR spectrum unambiguously exhibits the presence of a d5 hs species, generated through the reduction by the substrate.
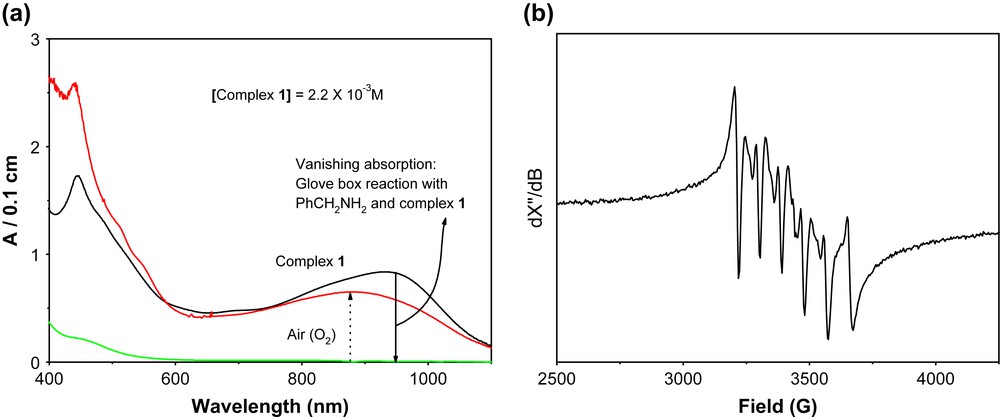
(a) UV–vis/NIR spectral changes of a reaction solution containing 1 and the substrate under Ar-atmosphere in a glove-box and the effect of air on it (left). (b) X-band EPR spectrum of a reaction solution, containing 1 and the substrate, obtained through the glove-box reaction (right). Experimental conditions: temperature 11.5 K; solvent methanol; microwave power 200 μW; microwave frequency 9.632 GHz; modulation amplitude 7.5 G.
Hence, from the X-band EPR spectrum it is quite clear that the Mn(IV)-monoradical center gets reduced to Mn(II) in presence of benzylamine under anaerobic condition. The UV–vis/NIR spectrum in absence of air shows clearly that there is no absorption band at around 930 nm that is present in complex 1. This band may be assigned to the intervalence ligand-to-ligand charge transfer band (between iminosemiquinone and amidophenolate forms of the ligand) [15]. The absence of that charge transfer band in the presence of substrate under anaerobic conditions is an indication that the radical involved in the catalysis, i.e. the iminosemiquinone form of the ligand has been reduced to its amidophenolate form. When the solution is exposed to air the absorption spectrum feature changes and shows the regeneration of radical-containing species by reproducing the band at around 930 nm.
From the above experiments a mechanism for the formation of benzilidinebenzylamine can be proposed, as shown in Scheme 4.
2.5 Catalytic aerial oxidation of 2-aminophenol; mimicking the function of phenoxazinone synthase (PHS)
Actinomycin D is a member of an interesting class of natural products in which the yellow-red 2-aminophenoxazinone chromophore is linked to two cyclic pentapeptides. These compounds are among the most potent antineoplastic agents known [8].
When complex 1 was used as a catalyst for the oxidation of 2-aminophenol to 2-aminophenoxazine-3-one by aerial oxygen at room temperature (RT) in methanol or a 1:1 dichloromethane/methanol solvent mixture, 84% conversion of 2-aminophenol to 2-aminophenoxazine-3-one was observed within 2 h. The reaction was monitored spectrophotometrically at 435 nm (ɛ435 = 2.4 × 104 M−1 cm−1) (Fig. 11).
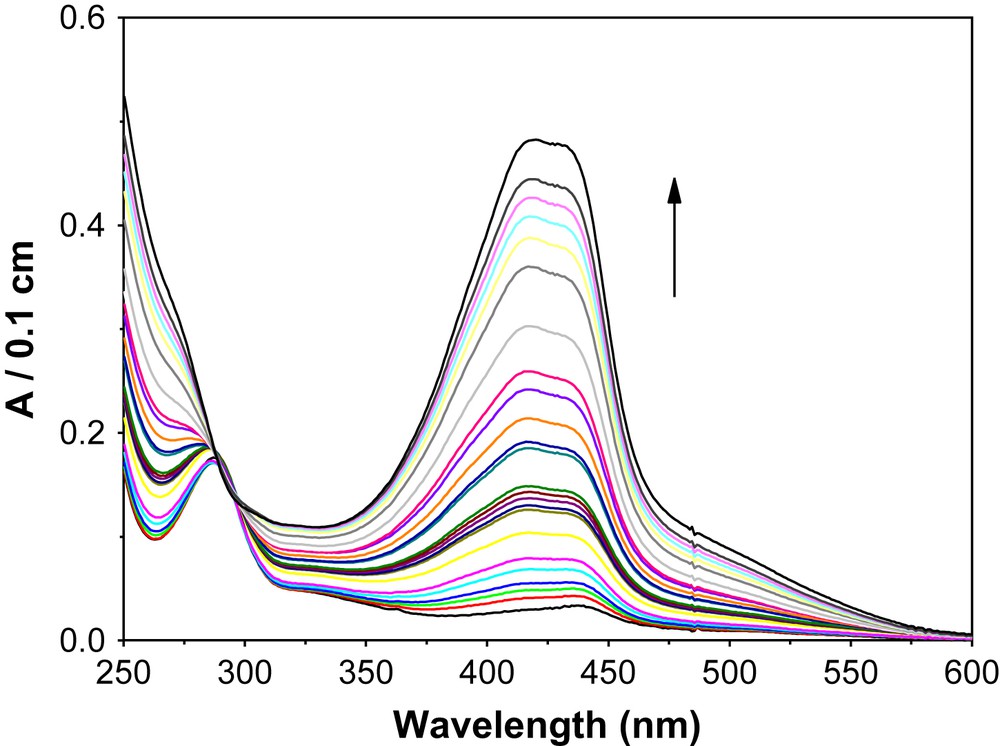
Time resolved formation of phenoxazinone chromophore.
Moreover, after 2 h of the catalytic reaction, the solvent was removed and the solid was examined by mass spectrometry. The mass spectrum shows a peak that has a composition similar to the 2-aminophenoxazine-3-one compound [16]. GC and GC–MS analysis were performed after passing the solution through a neutral Al2O3 column (0.5 g neutral Al2O3 was taken) in order to find out the number of products, percent of yield and the composition. GC and GC–MS results showed the formation of 2-aminophenoxazine-3-one by aerial oxidation of 2-aminophenol as a sole product.
Catalase activity by manganese complexes is quite common. Complex 1 is not exceptional as confirmed by allowing methanolic solution of the complex to react with 30% H2O2. Moreover, in the catalytic aerial oxidation of 2-aminophenol by complex 1, no H2O2 was detected at room temperature.
Oxygen uptake measurements using complex 1 as catalyst at 23 °C and −25 °C were carried out. At 23 °C, the ambient temperature, there is negligible amount of oxygen uptake by the system. This is probably due to generation of oxygen in the system through the decomposition of hydrogen peroxide owing to the catalase activity of complex 1. Moreover, no hydrogen peroxide is detected using TiO2+.
When the reaction was monitored at −25 °C in a 39:1 dichloromethane:methanol solvent mixture the volume of oxygen uptake by the system increased with time linearly and then saturation appeared. The volume of O2 taken-up by the system at −25 °C vs. time plot is illustrated in Fig. 12. The produced H2O2 was extracted into an aqueous phase from the reaction solution and the concentration of hydrogen peroxide was determined spectrophotometrically (λ = 415 nm, ɛ = 675 M−1 cm−1) by titanyl sulfate-reagent (Fig. 12). The concentration of H2O2 corresponds to 3/2 times the concentration of the substrate and to the amount of oxygen taken-up by the system. Hence, the stoichiometry at −25 °C is represented by the equation: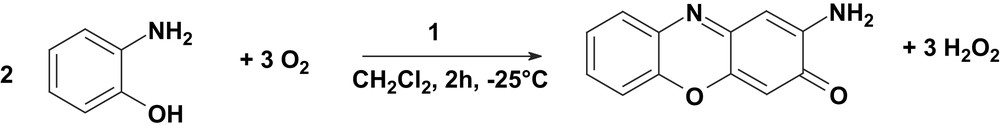
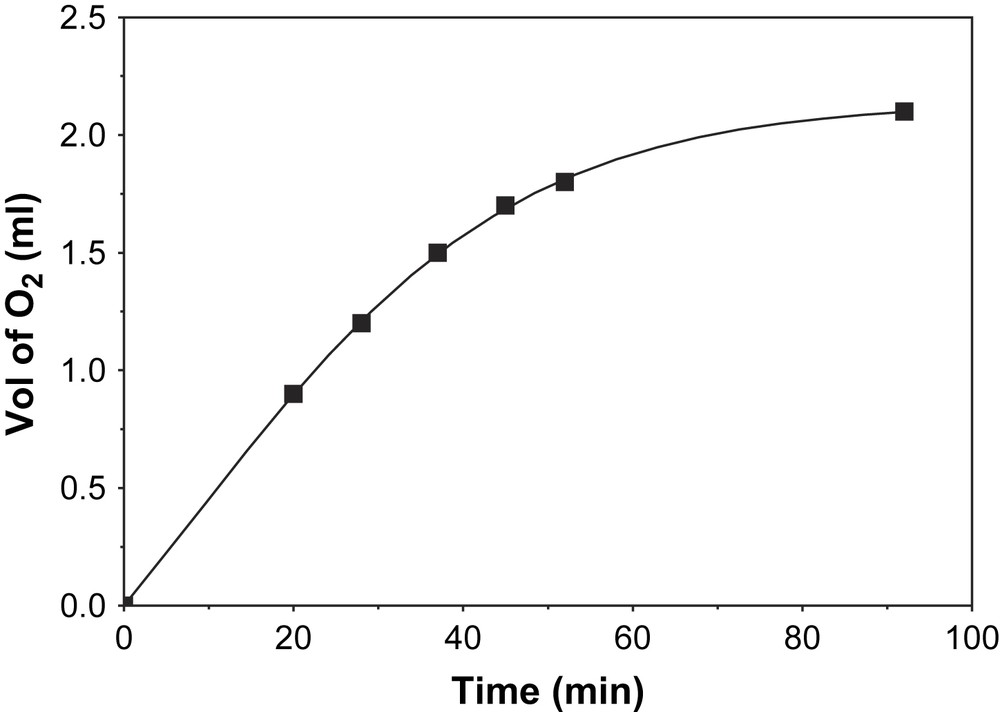
Plot of volume of O2 vs. time at −25 °C.
When 1 equivalent of 1 was allowed to react with approximately 10 equivalents of 2-aminophenol under an argon atmosphere in a glove-box at room temperature in MeOH for 2 h, 0.5 equivalent of phenoxazinone chromophore to the concentration of complex 1 was found to form spectrophotometrically at 435 nm. The X-band EPR spectrum feature of the solution at 10 K is same as shown in Fig. 10, i.e. the formation of Mn(II) species. Exposure of the solution to air increases the concentration of phenoxazinone chromophore. Hence, the process is an aerial oxidation process.
A few other model complexes with cobalt, iron and copper are also known in the literature for the oxidative coupling reaction of aminophenols [17,18]. A mechanism can be proposed from the above experimental results as shown in Scheme 5.
3 Concluding remarks
To conclude, the following points of this study deserve particular attention: An ortho-substituted aniline, anthranilic acid, has been used to prepare a tridentate non-innocent ligand, N-(2-hydroxy-3,5-di-tert-butylphenyl)anthranilic acid, H3L, which forms a Mn(IV)-monoradical complex, 1. As expected, this Mn(IV) compound exhibits strong antiferromagnetic coupling between the radical ligand (SR = 1/2) and the Mn(IV) ion (SMn = 3/2) yielding an St = 1 ground state.
The investigation emphasizes that the Mn(IV)-monoradical complex 1 can catalyse the oxidation of primary amine with molecular oxygen as the sole oxidant to afford aldehyde under mild conditions to mimic the function of the copper-containing enzyme amine oxidases (CAO):
Complex 1 has been found to be a good catalyst for the oxidative coupling of 2-aminophenol to phenoxazinone chromophore. Thus, 2-aminophenol was oxidized by aerial oxygen quantitatively to 2-aminophenoxazine-3-one by using complex 1 as a catalyst.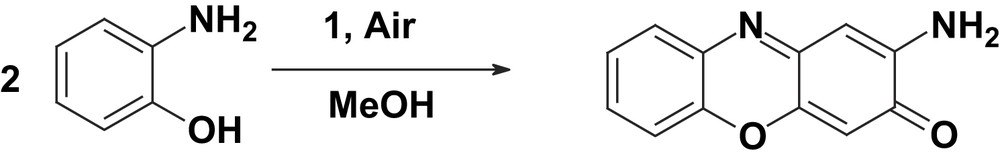
Thus, complex 1 is an efficient catalyst for the generation of phenoxazinone chromophore, hence modelling the function of the copper-containing enzyme phenoxazinone synthase (PHS).
Although PHS and CAO contain copper, a manganese(IV)-monoradical complex 1 models the catalytic functions of the said enzymes, thus exemplification of opening the way for other enzyme mimics being developed that contain metals other than copper.
4 Experimental section
4.1 Materials and physical measurements
Commercial grade chemicals were used for the synthetic purposes and solvents were distilled and dried before use. Fourier transform IR spectroscopy on KBr pellets was performed with a Perkin–Elmer 2000 FT-IR instrument. Solution UV–vis spectra were measured with a Perkin–Elmer Lambda 19 spectrophotometer. Mass spectra were recorded either in the EI or ESI (in CH2Cl2) mode with a Finnigan MAT 95 or 8200 spectrometer. Magnetic susceptibilities of the polycrystalline samples were recorded with a SQUID magnetometer (MPMS, Quantum Design) in the temperature range 2–290 K with an applied field of 1 T. Diamagnetic contributions were estimated by using Pascal's constants. X-band EPR spectra were recorded with a Bruker ESP 300E spectrometer. GC of the organic products during catalysis were performed either on HP 5890 II or HP 6890 instruments using RTX-1701 15 m S-41 or RTX-5 Amine 13.5 S-63 columns, respectively. GC–MS was performed using the above columns coupled with a HP 5973 mass spectrometer with a mass selective detector. NMR spectra were measured using a DRX 400 spectrometer.
4.2 X-ray crystallographic data collection and refinement of the structure
The crystallographic data for 1 are summarized in Table 2. Graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) was used for 1. A dark red-brown crystal of 1 was fixed with perfluoropolyether onto glass fibers and mounted on a Nonius Kappa-CCD diffractometer equipped with a cryogenic nitrogen cold stream, and intensity data were collected both at 100(2) and 293(1) K. Intensity data were corrected for Lorentz and polarization effects. The data set was not corrected for absorption. The Siemens ShelXTL software package (G. M. Sheldrick, Universität Göttingen) was used for solution, refinement, and drawing of the structure; the neutral atom scattering factors of the program were used.
Crystal data and structure refinement for complex 1
| Empirical formula | C48H64MnN3O6 |
| Formula weight | 833.96 |
| Temperature | 293(2) K |
| Wavelength | 0.71073 Å |
| Crystal system, space group | Monoclinic, P21/c, no. 14 |
| Unit cell dimensions | a = 9.706(2) Å, α = 90° |
| b = 10.323(2) Å, β = 92.03(2)° | |
| c = 45.132(6) Å, γ = 90° | |
| Volume | 4519.2(14) Å3 |
| Z, Calculated density | 4, 1.226 Mg/m3 |
| Absorption coefficient | 0.342 mm−1 |
| F(000) | 1784 |
| Crystal size | 0.09 × 0.05 × 0.03 mm |
| Theta range for data collection | 2.91 to 22.50° |
| Reflections collected/unique | 20 414/5834 [R(int) = 0.0562] |
| Completeness to theta = 22.50 | 98.6% |
| Absorption correction | None |
| Refinement method | Full-matrix least squares on F2 |
| Data/restraints/parameters | 5834/145/563 |
| Goodness-of-fit on F2 | 1.047 |
| Final R indices [I > 2σ(I)] | R1 = 0.0537, wR2 = 0.1254 |
| R Indices (all data) | R1 = 0.0773, wR2 = 0.1389 |
| Largest diff. peak and hole | 0.366 and −0.299 e Å−3 |
CCDC reference numbers are 613530 and 613531 for 1 at 100 and 293 K, respectively. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (internat.) +44 1223 336 033. E-mail: deposit@ccdc.cam.ac.uk].
4.3 Preparation
4.3.1 Synthesis of the ligand, H3L
Anthranilic acid (1.45 g; 10 mmol), 3,5-di-tert-butylcatechol (2.25 g; 10 mmol) and 0.01 ml Et3N were taken in 50 ml of n-hexane. The solution was heated to reflux for 5 h and then stirred under air for 2 days and filtered, washed with water and then n-hexane to procure a white color solid. Yield: 1.9 g (56%). Mp > 220 °C. IR (KBr, cm−1): 3456s, 3336s, 2959–2868s, 1667s, 1595s, 1577s, 1420s, 1255m. Mass (EI): m/z 341. 1H NMR (CD2Cl2, ppm): 1.27 (s, 9-tBu H), 1.44 (s, 9-tBu H), 6.12 (s, 1-NH H) 6.5–8.4 (6H from aromatic ring), 8.75 (s, 1-COOH H). Anal. calcd. for C21H27NO3: C, 73.90; H, 8.0; N, 4.10. Found: C, 74.5; H, 7.7; N, 3.9%.
4.3.2 Complex 1
MnII(CH3COO)2·4H2O (0.49 g; 2 mmol) was dissolved in 30 ml of MeOH, and H3L (0.68 g; 2 mmol) was added. To the solution 0.3 ml of Et3N was added and stirred under air for 2 h. Microcrystalline solid precipitated which was filtered, washed with MeOH. Single crystals suitable for X-ray analysis were grown from a 1:1 CH2Cl2–CH3CN solution mixture. Yield: 1.05 g (63%). IR (KBr, cm−1): 2950–2870s, 1619s, 1574s, 1467s, 1255m, 1104s. m/z (ESI(+) and ESI(−), CH2Cl2): 102 [HNEt3]+, 731 [M]−. Anal. calcd. for C48H64Mn1N3O6: C, 69.13; H, 7.73; N, 5.04; Mn, 6.60. Found: C, 69.0; H, 7.75; N, 5.0; Mn, 6.6%.
4.4 General procedure for the oxidation of benzylamine and determination of the concentration of benzilidinebenzylamine
In 20 ml methanol, 5 × 10−4 M complex 1 and 5 × 10−2 M benzylamine were added. The resulting solution was stirred under air for 15 h and then 1 ml of the catalytic solution was passed through a neutral alumina (0.5 g) and the column was washed with 9 ml methanol. To the resulting 10 ml solution 5 μl n-hexadecane was added as standard. GC and GC–MS were measured. Concentration of benzilidinebenzylamine was calculated against the standard.
4.5 General procedure for the oxidation of ethylenediamine and 2-aminoethanol and determination of the concentration of glyoxal and 2-hydroxy-acetaldehyde
In 20 ml methanol, 5 × 10−4 M complex 1 and 5 × 10−2 M ethylenediamine or 2-aminoethanol were added. The resulting solution was stirred under air for 15 h and then 0.5 ml of the catalytic solution was extracted by 10 ml water. To the 0.5 ml water part, 0.5 ml 1% aqueous 3-methyl-2-benzothiazolone hydrazine hydrochloride [19] was added and the mixture was allowed to stand for 30 min. Then 2.5 ml 2% freshly prepared aqueous FeCl3 solution was added. Following a 5 min waiting period, the mixture is diluted to 10 ml with acetone. The absorbance was then determined at 634 nm and 664 nm against a blank.
4.6 General procedure for the oxidation of 2-aminophenol and determination of the concentration of phenoxazinone chromophore
In 20 ml methanol or dichloromethane, 2.5 × 10−4 M complex 1 and 2.5 × 10−2 M 2-aminophenol were added. The resulting solution was stirred under air for 2 h. The solution was diluted as required and UV–vis spectrum at 435 nm (ɛ = 24 000 M−1 cm−1) was measured against blank.
Acknowledgements
Financial support from the German Research Council (DFG) is gratefully acknowledged (Project: Priority Program “Radicals in enzymatic catalysis”, Ch111/2-3). Skilful technical assistance of Mrs. H. Schucht, Mrs. R. Wagner, Mrs. U. Westhoff, Mr. A. Göbels, Mr. J. Bitter and Mr. U. Pieper is thankfully acknowledged.