

1 Introduction
Enduring goals of catalysis science include the realization of molecules or materials that exhibit high selectivities, rates and stabilities under turnover conditions and a deep, multidisciplinary comprehension of all factors that account for these properties. A few related themes of both fundamental and possible applied importance permeate our catalytic research in recent years and are succinctly addressed in this focused review. One of these themes is to understand and thus combat in new or unconventional ways the decomposition of catalysts that are otherwise attractive (simultaneously selective, rapid, etc.). Formulation of stable and dynamic catalysts (those capable of self-repair during turnover) is one aspect of this effort. Myriad literature studies report catalysts that exhibit rates and/or selectivities that garner attention but because of rapid inactivation during turnover they have little chance ultimately to be used. It is noteworthy that stability is often dismissed and frequently not even mentioned in reports of new catalysts for oxidation processes, an area where catalyst stability, particularly for commodity chemical processes, is a major issue. A second theme of catalytic research from our laboratory is the management of selectivity through systematic control of key intermediates during turnover. A third theme is the use of information from several sources to achieve catalysis of processes that are universally viewed as important but challenging. Two such processes are oxidations that require only the ambient environment (air at room temperature and pressure) to proceed, chemistry with a host of applications, and processes that lead to the controlled oxidative functionalization of hydrocarbons. The latter has been a major focus of the petrochemical and chemical industries for more than 40 years. A fourth theme in our work has been the incorporation of functional complexity into catalysts. This includes the invention of materials that can do multiple tasks such as recognize and sequester a particular substrate, typically a pollutant or a toxic agent, followed by its detection and/or air-based catalytic oxidative removal.
Much of the Hill group research in these areas has used transition metal oxygen-anion clusters (polyoxometalates or ‘POMs’) because they are oxidatively, thermally, and frequently hydrolytically stable while being extremely modifiable [1–3]. A host of precursors (units based on most of the elements in the periodic stable) are compatible with the several synthetic approaches to POMs. The structure of four POMs of considerable ongoing interest and ones addressed in this review is presented in Fig. 1.

Representative polyoxometalates (POMs) in ongoing research addressed in this review. A: [W10O32]4− (decatungstate); B: [X(ML)W11O39]n−, where X, M and L are the central ‘heteroatom’, the d-electron center (more darkly shaded than the 11 WO6 units) and the terminal ligand on that center, respectively. The charge ‘n−’ depends on X, M and L. This is the common α-Keggin structure with one d-electron center substituted in a surface, or ‘addenda’, site; C: [Pt(O)(OH2)(PW9O34)2]16−; D: [Au(O)(OH2)P2W20O70(OH2)2]9−. C and D are late-transition metal–oxo (LTMO) complexes of PtIV and AuIII, respectively, with the linear (H2O)M(O), M = PtIV and AuIII, unit shown in ball and stick notations.
A testimony to the general interest in POM-based catalysts is the commercialization of several homogeneous and heterogeneous acid and oxidation processes based on these complexes [4–8].
2 Catalytic functionalization of alkanes by polyoxometalates
Dioxygen-based oxidation of alkanes catalyzed by insoluble POMs has been known for some time [5], and the thermal and photochemical functionalizations of alkanes catalyzed by soluble POMs were first reported two decades ago [9]. Notable documented reactions of C–H bonds in homogeneous POM-based systems include the recent conversion of methane to methanol [10], and the functionalization of unactivated C–H bonds in positions that are not usually the most thermodynamically reactive (have the strongest C–H bonds) [11–13]. The selective conversion of methane to methanol has been, for 30+ years and will be for the foreseeable future, a target of intense intellectual and practical interest. The intellectual interest stems in large part from the challenge of overcoming the intrinsic selectivity problem in oxidative hydrocarbon functionalization. In short, methanol is almost always more reactive than methane in both homolytic and heterolytic oxidation processes, and as a consequence, it is extremely difficult to avoid overoxidation of the methanol product during conversions of methane that are of economic interest. The practical interest in converting methane selectively to methanol is that the latter is a liquid under ambient conditions that can thus be shipped and stored far more cheaply and safely than methane itself. The interest in functionalization (true functionalization versus stoichiometric activation as remains the rule in organotransition metal alkane reaction systems) of conventionally unreactive bonds in large molecules derives from the fact that such reactions are as potentially useful in synthesis as they are unusual. At present there is no general synthetic methodology to do this effectively. Controlled and selective oxidative functionalization of strong bonds in molecules with many bonds remains a grand challenge in chemistry.
The catalytic photochemical functionalization of alkanes by polytungstates under anaerobic conditions facilitates high yield, high selectivity routes to several useful alkyl derivatives and H2 via Eqs. (1)–(3) [14–19]
| POM + hν → POM∗ | (1) |
| POM∗ + SubH2 → POMred + Sub + 2H+ | (2) |
| POMred + 2H+ → POM + H2 | (3) |
Net reaction from Eqs. (1)–(3):
| SubH2 + hν → Sub + H2 | (4) |
The thermodynamic (most substituted) alkenes are formed by abstraction of hydrogen from the weakest C–H bonds (ultimately generating the most stable carbocation intermediate) followed by deprotonation. However, if decatungstate, [W10O32]4−, is used as the POM photocatalyst, and the reaction is conducted under conditions where most of the decatungstate is reduced, then intermediate alkyl radicals are reduced to carbanions which quickly react with several electrophilic functional groups including alkyl nitriles [15,16,19,20]. Reaction with the latter forms, after hydrolysis, yields ketones [19]. This work leads to the realization that substrate photooxidation processes (with POM reduction) such as hydrogen abstraction from unactivated C–H bonds, could be coupled with substrate reduction by the reduced POM to complete a catalytic cycle (reoxidation of the POM photocatalyst) [21]. The reduced forms of the POMs with more negative reduction potentials, including [W10O32]6− (the two-electron-reduced form of decatungstate) are sufficiently nucleophilic to cleave C–X, X = Cl, Br, SH(R) bonds rapidly at ambient temperature in the dark. Thus photoreduction of POMs like decatungstate by compounds with C–H bonds along with the simultaneous presence of substrates with cleavable C–X bonds results in catalytic cycles for the light-driven oxidation of the C–H substrate with associated dehalogenation (or desulfurization) of the C–X substrate [21–23].
If reaction conditions are chosen that lead to alkyl radical generation in the presence of other alkenes, CO or methyl cyanoformate (MCF) then homologated alkanes, aldehydes and nitriles/α-iminoesters, respectively, are readily formed [17,18]. These conditions include an anaerobic atmosphere to avoid the extremely facile trapping of alkyl radicals by dioxygen, and shorter irradiation times to minimize build-up of two-electron-photoreduced POMs.
Significantly, two photochemical reactions of unactivated C–H bonds catalyzed by POMs lead to the functionalization of homolytically stronger bonds and not the conventionally weaker more reactive bonds. As such, these reactions address the general challenge noted above in the catalytic functionalization of hydrocarbon domains. First, a few hydrogen atom abstraction processes by photoexcited POMs including some reactions in complex molecules [11,12], involve the stronger bonds because these are more sterically accessible and thus more reactive kinetically than the weaker bonds. This is consistent with the considerable steric bulk immediately proximal to many of the surface oxygen atoms in POMs which are the hydrogen abstracting atoms. Second, under conditions where the intermediate radicals resulting from C–H bond cleavage by photoexcited POMs are not reduced, oxidized or trapped in bimolecular processes with non-substrate molecules such as CO, alkene, alkynes, etc. the concentration of these alkyl radicals builds up and radical–radical disproportionation and combination processes start to take place. The key here is that the disproportionation of alkyl radicals leads to the less stable (less substituted) alkene product (and alkane, which is the substrate) [13,15]. The synthetic utility of these reactions, and in particular catalytic generation of alpha olefins from alkanes, could be significant. Some of the alkane functionalization processes photocatalyzed by polytungstates are summarized in Fig. 2.

Functionalization of unactivated C–H bonds photocatalyzed by polyoxometalates (POMs), illustrated using 2,3-dimethylbutane. All reactions proceed with high to very high selectivity. Acetonitrile is one solvent compatible with all these reactions; conversions of alkane are usually, but not always, kept moderate.
Quantum yields for all these POM photocatalyzed processes are high using blue or UV light (λ < 430 nm). Work in collaboration with J. Winkler and M. Kozik (then at Brookhaven National Laboratory) and T. Netzel (Georgia State University) established that the initial charge-transfer excited state of decatungstate is shorter lived than the 30 ps rise time of the laser flash photolysis equipment used (frequency tripled YAG) [24–26]. The initial excited state decays into a second excited state with a lifetime of tens of nanoseconds which is the one actually responsible for the photooxidation of organic substrates (frequently cleavage of C–H bonds) [26,27].
Subsequent work by Giannotti et al. [28,29] on shorter time-scales than tens of picoseconds and thorough work by Tanielian et al. [30–32] have established the rates and quantum yields of other fundamental processes involving decatungstate.
While some attempts to shift the photoredox chemistry of POMs from the blue and ultraviolet to the visible regions of the spectrum have been taken, energy limitations have kept the quantum yields for visible-light-driven processes low. On the positive side, the quantum yields for production of the photoredox active excited state for decatungstate exceed those for commercially used TiO2 in the high energy regions of the terrestrial solar spectrum [33]. Based on this, some light-driven organic degradation and transformation processes could well be useful in context with environmental remediation and decontamination.
3 Self-repairing catalysts
The fact that organic structure and O2 (and most oxidants) are unstable with respect to CO2 and H2O made the development of oxidatively resistant ligands for homogeneous oxidation catalysts a cottage industry for years [34–41]. Making organic ligands for oxidation catalysts, including porphyrins and Schiff base ligands, more resistant to oxidative degradation by replacing C–H bonds with C–X, X = Cl, F, and NO2 groups was and still is a major theme in this area of research [39]. After six years of developing and investigating metalloporphyrin catalysts ourselves, we decided to replace the organic ligand environment of the catalytic metal center entirely by using multidentate defect POMs as ligands [42]. Since POMs are composed solely of fully oxidized transition metal centers, most commonly d0 W(VI), Mo(VI) and V(V) centers, bridged by oxide ions, these totally inorganic ligands are completely resistant to oxidation by all reagents used in organic oxidation processes including O2.
After our initial report of hydrocarbon oxidation catalyzed by soluble d-electron-metal-substituted POMs (‘inorganic metalloporphyrins’) [42], many groups, including those of Neumann [43–52], Mizuno [53,54], Finke [55,56], Pope [57–59], Lyons [60], and Shannon [61], reported related studies. Some promising catalysts have been developed and informative mechanistic studies have appeared. Activity in this area, and in making more stable homogenous catalysis of all kinds for organic oxidations continues and doubtlessly will continue for some time as new substrates and challenges, including integrated fully green operation, arise.
However, there is more to designing a superior oxidation catalyst than making it thermodynamically stable to oxidative degradation. An ideal catalyst would not only be stable to oxidative, hydrolytic or any other means of potential degradation but also have an engineered ability to reverse any damage that does occur during reaction. It occurred to us that the successful catalysts in Nature do not follow the somewhat simplistic paradigm of having a single optimally stable static structure. Instead, biological catalysis takes places in the larger context of cells and extensive DNA-coded molecular machinery that not only makes and often modifies catalysts for optimal operation but also repairs the damage that biological catalysts (and for that matter nearly all catalysts) inevitably suffer during reaction. For example if a particular amino acid in an enzyme is modified during catalytic turnover rendering the protein (or multi-protein active assembly) inactive, or worse, active but non-selective (hydroxylation of a residue in oxidase active sites is a classic example), then such damage is noted and the compromised enzyme is removed. After disassembly, removal of the altered amino acid and synthesis of fresh undamaged enzyme, effective catalysis continues. The catalytic system in this case is not simply the molecule or multi-unit assembly but rather the cell or multi-cellular structure and its considerable stored information and associated capabilities.
We reasoned that if a catalyst is formulated to be both thermodynamically stable (for example a metal oxide solid or a POM for an oxidation reaction) and equilibrating with its components or possible fragments under turnover conditions, then a special and powerfully protective situation exists [62–66]. In such circumstances, any damage or disassembly of the catalyst during reaction would be reversed and the active form of the catalyst would be reconstituted. Damaged forms of such a catalyst system would experience a thermodynamic driving force and a kinetic ability to re-form the active catalyst [66]. Effectively one would have a self-repairing catalyst system [63,66]. We further realized that catalytically useful POMs, such as [AlVVW11O40]6− can be prepared in water by heating the right ratio of metal oxide or oxometalate salt precursors of this polytungstate in water at the right pH and ionic strength. In other words, one equivalent of aluminum(III) (as in Al2O3), one equivalent of vanadium(V) (as in V2O5) and 11 equivalents of tungsten(VI) (as in WO42−) produce [AlVVW11O40]6− when heated in water in the presence of O2 (air) [64,66]. In contrast to conventional (kinetic, serial and multi-step) syntheses which afford a single pure product (in this instance only [AlVVW11O40]6− itself), the above non-conventional one-pot equilibration synthesis produces a thermodynamically stable distribution (ensemble) of POMs that can be equilibrating at appropriate temperatures, such as the temperature of catalysis. During one-pot thermodynamic generation of [AlVVW11O40]6−, the polytungstates [Al3W10O38]7−, [V2W4O19]4− and [W7O24]6− are formed simultaneously [64,66]. This ensemble of equilibrating POMs can then be used as a stable yet dynamic self-repairing oxidant system as outlined in Fig. 3 below.
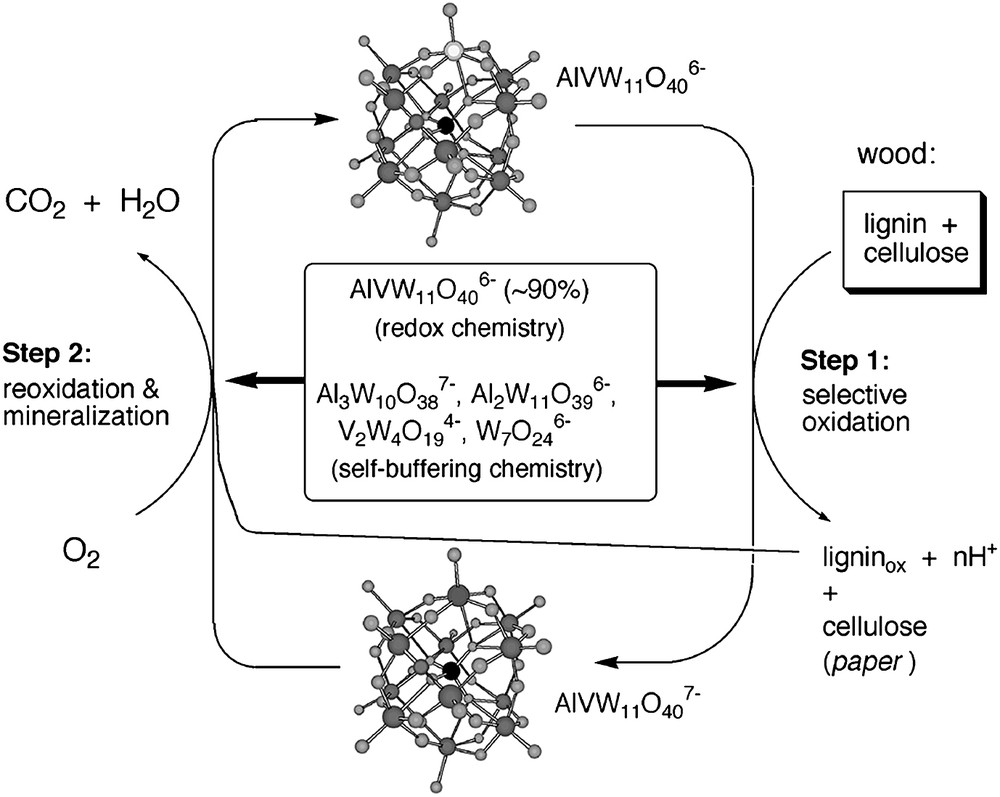
Thermodynamically stable, self-buffering system for selective use with O2 as the oxidant in H2O as the solvent, illustrated in this case by the delignification of wood. Step 1 involves selective oxidation of the lignin in wood or wood pulp by the oxidizing POM, [AlVVW11O40]6−. The protons that are generated by oxidations in water are consumed by the more basic POMs present in the equilibrating POM ensemble, [Al3W10O38]7−, [Al2W11O39]6−, [V2W4O19]4− and [W7O24]6−. The oxidized lignin fragments are soluble and easily removed from the cellulose, which can be further processed to generate paper products. Step 2 involves reoxidation of the reduced POM, [AlVIVW11O40]7−, while the hydroxide generated by coupled O2 reduction is again consumed by the equilibrating POM ensemble. The equilibrating ensemble consumes, alternatively, both H+ and OH− keeping the pH approximately neutral as the cycle is repeated many times.
Neumann and co-workers also reported a catalyst that self assembles under turnover conditions [67,68]. An additional feature of an equilibrating POM ensemble catalyst is that the acid that is produced during oxidation (Fig. 3, step 1) as well as the hydroxide produced during reoxidation of the reduced POM is consumed by the other polytungstates present, namely [Al3W10O38]7−, [V2W4O19]4− and [W7O24]6− and thus such an equilibrating recycling system is self buffering [64,66]. The formulation of self-repairing and self-buffering catalysts for several green (air-based and water-compatible) processes would offer advantages. As a consequence, further research in this area would certainly appear to be warranted.
4 Catalysts for air-based oxidations under ambient conditions
The pollutants most dangerous to human health and those most odorous (aldehydes, sulfur compounds, nitrogen compounds) in indoor air (workplace, home, family car, hospital, etc.) are amenable, in principle, to oxidative decontamination by reaction with O2 [69,70]. So too are some of the principal chemical warfare agents, including mustard (HD) and VX [71]. While all these organic compounds are readily removed using potent stoichiometric oxidants [71,72], including bleach (aqueous hypochlorite), or by more environmentally attractive catalyzed peroxide-based decontamination processes [73], all such processes require reagents, usually solvents, and often heat and/or light to be effective. These logistical requirements render them undesirable if not unworkable for many needs. Clearly the development of catalytic molecules or materials that would require only the air itself (O2 at ambient pressure and temperature) to catalyze oxidations of pollutants and toxic agents could be of significant value. The availability of solid forms of such catalysts should facilitate the development of coatings, fabrics (clothing, upholstery, carpeting), devices for confined indoor areas, and protective cosmetics that would remove these toxic and/or odorous compounds without energy or media requirement – only contact of the contaminant with the catalyst-containing material in the air would lead to effective decontamination. Further, if catalytic turnover is rapid and/or the surface area of the catalyst material is high, only tiny quantities of the catalyst itself would be required. These points are well and good, but there is a major barrier to such materials being realized.
The central problem is the reactivity of O2 itself. Dioxygen either reacts violently with organic materials, namely combustion which requires high temperatures, or extremely slowly, the usual case under ambient conditions. The challenge is to develop catalysts for O2-based oxidations that are so active that only ambient air is needed for rapid turnover. Ideally such catalysts would work as solids and not require dissolution because most of the above and other applications require active solids, not solutions.
Our group identified some highly active catalysts from diversity-based screening methods, including libraries comprising POMs of greatly varying composition and reduction potential, with combinations of counter cations and likely co-catalysts. A few active catalysts for air-based oxidation of both aldehydes and sulfur compounds (sulfide mustard simulants and thiol odorants) were discovered or developed which included Au(III)Cl2NO3(thioether) complexes [74–76], and some heterogeneous systems. The latter included Ag5[PV2Mo10O40] [77], and H5[PV2Mo10O40], a highly effective and thus much studied catalyst for O2-based oxidation in solution [7,78,79], when deposited on cotton cloth, polyacrylic fiber, nylon fiber, carbon powder (Ambersorb 572), and the Japanese ‘self-deodorizing’ fabric Smoklin® [80]. However, these early developed systems (research conducted in 1999–2002) were not sufficiently active with ambient air to be of compelling interest. Two more active solid catalysts for air-based oxidation in the dark were developed, a coordination network polymer involving the redox active polyanion, [PV2Mo10O40]5−, and bridging –CuII(OH2)4– units [81], and cationic silica nanoparticles with the POM, [(FeIII(OH2)2)3(A-α-PW9O34)2]9− electrostatically bound to the particle surfaces [82,83]. However, the most active catalysts of this category, including FeIII[H(ONO2)2]PW11O395−, were identified in the last two years [84]. Proprietary solid forms of related species are in development with two companies. Unlike nearly all the other catalysts noted elsewhere in this account-like review, we have yet to unequivocally characterize the geometric and electronic structures of these extremely active catalysts for dark air-based oxidations despite considerable effort in this direction. This has also rendered elucidation of the mechanism very difficult to date. However, given the potential significance of these systems, efforts in these directions will and must continue.
5 Terminal metal–oxo complexes of the noble metals
A final and actively ongoing effort in our laboratory that addresses oxygen chemistry and catalysis involves the generation of late-transition metal–oxo (LTMO) complexes. Terminal transition metal–oxo compounds are some of the most important oxidants in industry and biology. The stability of these inorganic units decreases and reactivity increases as d electrons are added to the metal. Thus, while terminal metal–oxo species of d0 Ti(IV), Nb(V), V(V), Mo(VI), W(VI), etc. are well known, stable and not very reactive, the terminal metal–oxo species of Mn, Fe (typically d3 and d4 in various spin states) are unstable and highly reactive oxidants [85–92]. Despite much effort by organometallic and inorganic chemists for years, only one terminal oxo complex of an element to the right of the column 8 (Fe, Ru and Os) in the periodic table had been successfully prepared and characterized: (trimesityl)IrV–oxo by Wilkinson and co-workers [93].
POMs share geometric and electronic structural features as well as chemical properties in common with redox active metal oxides such as TiO2 and CeO2 that are heavily used as supports in the fairly ubiquitous noble metal–metal oxide catalysts. In addition, POMs are strong (multidentate and chelating) and electron withdrawing ligands. As such, POMs are a logical choice to stabilize high-d-electron-count transition metal centers.
Indeed, reaction of the polytungstate, [A-PW9O34]9− with Pt(II) salts in water in the presence of O2 forms [PtIV(O)(OH2)(PW9O34)2]16−, PtIV–oxo complex [94], while reaction under similar conditions but with a buffer to keep the pH in a particular range, forms [PdIV(O)(OH)WO(OH2)(PW9O34)2]13−, PdIV–oxo complex [95]. Terminal oxo complexes of Au have also been prepared and characterized [96]. The unprecedented nature of these compounds required the use of a large number of complementary techniques (14 in the case of the Au complexes) to make a defensible case that these complexes are real and their oxidation states and electronic structures can be described. Multiple very-low temperature X-ray and neutron diffraction studies revealed the geometric structures of these complexes and neutron diffraction confirmed that no hydrogen was proximal to the terminal oxo oxygen atoms in the Pt and Au complexes. In other words, these compounds do contain the metal–oxo unit and not metal–hydroxo or metal–aqua units. Pd EXAFS confirmed the distances in that complex and XAS, redox titration, coulometry, ultra-low-temperature optical spectra, variable-temperature magnetism, and other data confirmed the oxidation state assignments.
All these complexes can be solubilized in organic solvents by exchanging the hydrophilic (usually potassium) counterions for hydrophobic counterions including tetra-n-butylammonium (TBA) ion [96]. 17O NMR established that the some of the LMTO complexes transfer the oxo oxygen on the noble metal, and this oxygen only (none of the oxygens in the polytungstate ligands) to a range of substrates including allylic C–H bonds in aprotic solvents. The LTMO complexes also mediate O2-based oxidations of some organic substrates. However, this finding is very recent so virtually no details of this chemistry are known yet.
Given the possible importance of Pt–, Pd– and Au–oxo intermediates in a host of significant technologies involving these noble metals and dioxygen ranging from nearly all vehicle catalytic converters to fuel cell cathodes to highly reactive catalysts for air-based oxidation, these recently characterized LTMO complexes and their dioxygen and oxidation chemistry will be carefully investigated.
Acknowledgments
CLH thanks many co-workers in addition to his co-authors of this perspective review article and long time funding by the National Science Foundation (for fundamental mechanistic and structural work), the Army Research Office (for fast and air-based decontamination systems and detector-catalyst materials) and the Department of Energy (for probing geometric and electronic structures of catalyst active sites in a close integration of experimental and computational work).