

1 Introduction
Nitroalkanes are widely used as chemical intermediates, industrial solvents, and components of inks, paints, varnishes and other coatings and are released in large quantities into the environment. However, many are anticipated to be toxic and some have been demonstrated to be carcinogenic [1–3]. 2-Nitropropane has been shown to cause the formation of both 8-hydroxy- and 8-amino-guanine in the DNA and RNA of liver cells in Sprague Dawley rats [4]. The occurrence of leukemia and non-Hodgkin's lymphoma increased among farmers exposed to solvents containing 2-nitropropane [5]. So the conversion of nitroalkanes into less harmful materials is very useful in industry as well as in environmental conservation.
Primary and secondary but not tertiary aliphatic nitro compounds can be transformed into aldehydes or ketones by treatment of their conjugate bases with sulfuric acid. This is called the Nef reaction and involves hydrolysis of the CN double bond. (Eq. (1)) [6,7].
| (1) |
There are also a fair number of stoichiometric reagents for this conversion. Such methods are the reaction of aliphatic nitro compounds with aqueous TiCl3 [8], cetyltrimethylammonium permanganate [9], tin complexes and NaHSO3 [10], activated dry silica gel [11], or 30% H2O2–K2CO3 [12]. Treatment of the conjugate base of the nitro compound with KMnO4 [13–15], ceric ammonium nitrate [16], MoO5–pyridine–HMPA [17], ozone [18] or singlet oxygen [19] leads to oxo compounds.
Biological organisms can also transform aliphatic nitro compounds into the corresponding oxo species. Nitroalkane oxidase from Fusarium oxysporum catalyzes the oxidation of nitroalkanes to aldehydes, transferring the electrons to dioxygen to form hydrogen peroxide [20,21]. The mechanism proposed for the reaction suggests the presence of a cationic imine as a catalytic intermediate [22,23]. The reaction is initiated by the abstraction of the α-proton from the neutral nitroalkane substrate by aspartate 402 to form the nucleophilic nitroalkane anion. Comparison of enzymatic and non-enzymatic nitroethane anion formation suggests that there are important electrostatic and hydrogen-bonding interactions in the transition state of enzymatic reaction [24]. Similar reaction is the oxidation of nitroalkanes catalyzed by the flavoprotein d-amino acid oxidase [25,26], glucose oxidase [27], and the heme protein horseradish peroxidase [28]. 2-Nitropropane dioxygenase from Neurospora crassa is a flavin-dependent enzyme, which catalyzes the oxidative denitrification of nitroalkanes to their corresponding carbonyl compounds and nitrite [29]. The enzyme was first characterized by Little [30], in 1951 as an oxidase, but was later classified as a dioxygenase, on the basis of the observation that the oxygen atom of the organic product formed upon oxidation of propyl-2-nitrate originates from molecular oxygen and not from water (Eq. (2)) [31].
| (2) |
The enzyme from Streptomyces ansochromogenes is also a nitroalkane dioxygenase-like enzyme rather than a nitroalkane oxidase [32]. 2-Nitropropane dioxygenase has been also isolated from a cell-free extract of Hansenula mrakii [33–35]. From the results obtained with 18O2 and 2-nitropropane, it is evident that two atoms of the dioxygen molecule are incorporated into the carbonyl groups of two molecules of acetone formed, indicating that 2-nitropropane dioxygenase is an intermolecular dioxygenase. Whereas the two enzymes have similar molecular weights of about 40,000, they differ in their prosthetic group content. The enzyme from H. mrakii is a non-heme iron flavoprotein containing FAD (flavin adenine dinucleotide) as prosthetic groups [36], while FMN (flavin mononucleotide) is found in the N. crassa enzyme [31]. Both enzymes are suggested to have similar mechanism because of similarities in several aspects. Kinetic analysis of the oxidation of neutral nitroalkanes and anionic nitronates catalyzed by 2-nitropropane dioxygenase from N. crassa indicates that the enzymatic oxidation occurs through an oxidase-like catalytic mechanism, in which the flavin-mediated oxidation of substrate and the subsequent oxidation of the enzyme-bound flavin occur in two independent steps [29]. In the oxidation of the neutral nitroalkane, a catalytic base is required to initiate the reaction by abstracting the proton from the substrate α-carbon to yield an enzyme-bound nitronate species (Fig. 1). Evidence was obtained for the formation of an anionic flavin semiquinone species as a catalytic intermediate, which then reacts with dioxygen via a rapid one-electron transfer, resulting in the oxidation of the flavin and formation of superoxide. The participation of an acid in the formation of a neutral superoxide species is suggested. The superoxide species would rapidly react with the nitroalkane radical within the enzyme active site, resulting in the formation of an α-peroxynitroalkane intermediate. The final elimination of nitrite from the peroxynitroalkane to yield the carbonyl product would then likely occur through a non-enzymatic attack by a nucleophile. The generation of superoxide anion in the oxidation of anionic 2-nitropropane by 2-nitropropane dioxygenase was revealed by ESR spectroscopy [37]. Recently, the crystal structure of 2-nitropropane dioxygenase from Pseudomonas aeruginosa was determined [38]. The cofactor FMN is bound non-covalently to the active site, and the structure suggests that strictly conserved His152 likely functions as the catalytic base that initiates oxidation of neutral substrates by abstracting a proton from the α-carbon. However, the role of the iron ion in the active site of the enzyme is still obscure. The question is put forward as how the reaction is accelerated and what is the role of the redox metal ion in terms of activation of either dioxygen or the substrate.
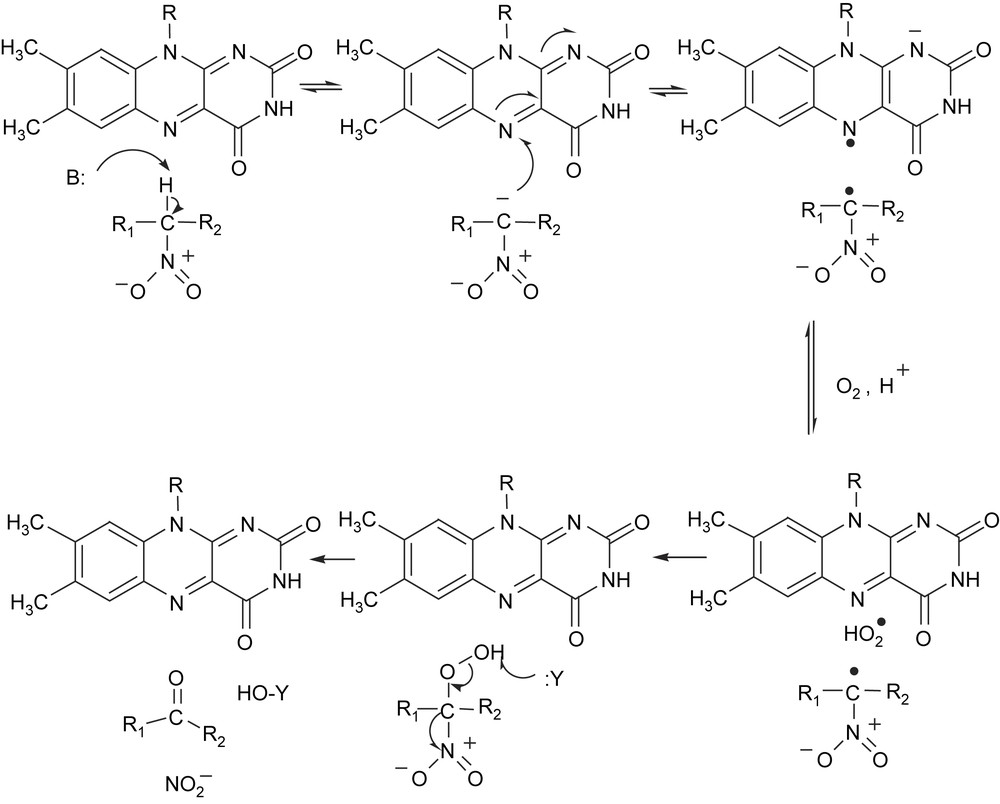
Proposed mechanism of 2-nitropropane dioxygenase.
Cobalt Schiff-base complexes have been found to promote the oxygenolysis of 1-aryl-1-nitroethanes probably via a hydroperoxo intermediate [39]. Since the 2-nitropropane dioxygenase is an iron-containing enzyme, we initiated studies on copper- and iron-mediated oxygenations of nitroalkanes. Recently, we demonstrated the facile copper- and iron-assisted oxygenation of nitroalkanes, and proposed a mechanism for these curious reactions. In order to gain more insight into the mechanism of these reactions, kinetic studies on the oxygenation of potassium alkylnitronates in aprotic solvent were also carried out.
2 Copper-mediated oxygenation of nitroalkanes
2.1 Synthesis and oxidation of copper(I) nitronate complexes [40]
Our studies on the activation of dioxygen [41] and on model systems mimicking dioxygenases [42], and the fact that nitronate salts of nonredox metals can be converted into nitro- and carbonyl compounds with singlet dioxygen inspired us to prepare (nitronato)copper complexes possessing a redox-active metal ion, and oxygenate them with triplet dioxygen. Nitroalkanes are known to exhibit tautomerism to yield nitronic acids and equilibria lay well on the side of the latter [43]. Deprotonation of both tautomers leads to nitronate ions. Mesitylcopper(I) [44] reacts with 2-nitropropane and triphenylphosphine in anhydrous acetonitrile under an atmosphere of argon at room temperature; the copper(I) aci-2-nitropropanate complex [Cu(NP)(PPh3)2] (4) is formed (Eq. (3)).
| (3) |
This complex is moderately soluble in polar organic solvents, and its solutions are not very sensitive toward dioxygen, due to the facts that the PPh3 stabilizes the copper in the oxidation state one and the coordinated nitronato ligand is not susceptible to oxidation. Characteristic NMR features of the complex in CDCl3 solution include a single CH3 resonance downfield shifted by 0.4 ppm from the free 2-nitropropane and a multiplet resonance of the aromatic protons of the phosphine ligands in the 1H NMR, downfield shifts of coordinated nitronato carbons in the 13C NMR together with a single peak in the 31P NMR spectrum, in the IR spectrum, intense sharp bands are assigned to ν(CN) at 1602, νas(NO2) at 1145 and 1134, and νs(NO2) at 938 cm−1. Single-crystal X-ray analysis of compound 4 revealed a distorted tetrahedral geometry around copper(I). The oxygen atoms form strong bonds with the copper atom having short bond distances (2.139(3) Å). The aci-2-nitropropanate ion is planar and the observed C–N distance lies between those for oximes and for nitro compounds. It seems likely that these bonds have appreciable double-bond character.
Motivated by our original goal to oxygenate a (nitronato)redox-metal complex, we investigated the reactivity of 4 toward dioxygen. A slurry of [Cu(NP)(PPh3)2] in acetonitrile reacts with dioxygen slowly in a 2:1 stoichiometry with the concomitant formation of [Cu(NO2)(PPh3)2] (5) as colorless crystals and acetone (Eq. (4)).
| (4) |
The diamagnetic compound exhibits simple NMR spectroscopic features, showing only signals characteristic for PPh3 in the 1H, 13C, and 31P NMR spectra and ν(NO2) bands in the IR due to coordinated nitrito ligand. Single-crystal X-ray analysis data of complex 5 were identical to that found in Ref. [45]. 18O2 labeling experiments of the oxygenation of 4 were carried out with a mixture of 18O2 and 16O2. The GC–MS analysis of the solution showed the presence of 18O- and 16O-acetone, the relative abundance of base peaks (m/z 45 and 43) parallels the 18O2 enrichment used. In the IR spectra, peaks assigned to ν(C16O) (1714 cm−1) and ν(C18O) (1684 cm−1) of acetone were observed, whereas no new bands of 5 due to ν(N18O16O) or ν(N18O18O) was detected showing that 18O2 has been incorporated only into acetone. Kinetic measurements on the oxygenation of 4 in pyridine solution resulted in a first-order dependence with respect to 4 and dioxygen with a second-order rate constant of k = 2.48 × 10−2 mol−1 dm3 s−1. On the basis of the stoichiometry, kinetics, and labeling experiments on reaction (4), the mechanism shown in Eq. (5) can be proposed.
| (5) |
The aci-2-nitropropanate anion is inactive against 3O2 [46], but the Cu(I) center is redox-active in complex 4, even in the presence of a strong Cu(I)-stabilizing ligand such as PPh3 [47]. The rate-limiting reaction step seems to be the interaction of dioxygen with 4, leading to a (superoxo)copper(II) complex 6, as found in the case of many other copper(I) complexes [48], and finally to an intermolecular peroxidic species 7. A fast breakdown of 7 may result in the copper(I)nitrite complex (5) and acetone.
The copper-assisted intermolecular oxygenation of the CN double bond of the coordinated aci-2-nitropropanato ligand with triplet dioxygen may indicate that the presence of copper(I) is essential in the activation of triplet dioxygen, and the reduced oxygen species is ready for oxidative splitting of the CN double bond.
2.2 Copper-catalyzed oxygenation of nitroalkanes in the presence of N-ligands [49]
Primary and secondary nitro compounds are easily transformed into aldehydes or ketones and nitrous acid in rather good yields by dioxygen catalyzed in the presence of metallic copper and N-ligands, such as N,N,N′,N′-tetramethylethylenediamine (tmeda), 2,2′-bipyridine (bpy), and 1,10-phenanthroline (phen) in various solvents (DMF, CH3CN, py) (Eq. (6)). If methanol is used as solvent, the methyl esters are also formed to a large extent, along with minor byproducts.
| (6) |
In these reactions, the copper metal reacts with the aci form of the nitro compounds to give copper nitronate complexes. The (nitronato)copper compounds are then further oxygenated to give the carbonyl compounds and nitrous acid. The solvent has a profound effect on the effectivity of the reaction. More coordinating solvents as well as donor N-ligands accelerate the reaction remarkably.
2.3 Stoichiometric oxygenation of 2-nitropropane [50]
Stoichiometric oxygenation of 2-nitropropane (NPH) in DMF in the presence of copper and N-ligands results in acetone and a copper(II) complex. According to parallel gas-volumetric (O2), titrimetric (NO3), spectrophotometric (NO2) and GC (acetone) measurements, the stoichiometry of the oxygenation reaction corresponds to Eq. (7).
| Cu0 + L + 2 NPH +2 O2 = [CuII(L)(NO2)(NO3)] + 2(CH3)2CO + H2O | (7) |
The presence of the intermediate H2O2 in the reaction mixture could be demonstrated either by iodometry or oxidizing triphenylphosphine to triphenylphosphine oxide. We believe that in the first step copper metal, in the presence of N-ligands, reacts with the aci form of the secondary nitro compounds to give copper nitronate complexes and H2O2 (Eq. (8)).
| Cu0 + L + 2 NPH +O2 = [CuII(L)(NP2)] + H2O2 | (8) |
Similar reactions were reported for the reaction of copper metal with acidic compounds in air [51–54]. In the presence of molecular oxygen, both copper(I) and copper(II) nitronates are probably first formed, the former being then transformed into [CuII(L)(NP)2], which can be oxygenated into the desired products (Eq. (9)).
| CuII(L)(NP2)] + O2 = [CuII(L)(NO2)2] + 2(CH3)2CO | (9) |
This is then followed by the fast consecutive formation of [Cu(L)(NO2)(NO3)], which is assumed to be obtained as a product of the oxidative process between [Cu(L)(NO2)2] and H2O2 (Eq. (10)).
| [CuII(L)(NO2)2] + H2O2 = [CuII(L)(NO2)(NO3)] + H2O | (10) |
2.4 Copper-catalyzed oxygenation of 2-nitropropane
The oxygenolysis of 2-nitropropane in the presence of copper and tmeda in DMF results in acetone and acetone oxime. The amount of tmeda influences the chemoselectivity, higher tmeda concentrations preferentially leading to the formation of the oxime. We believe that excess tmeda acts as a base and, in the presence of HNO2, the salt tmeda·HNO2 is formed, which reacts with 2-nitropropane, resulting in the oxime (Eq. (11)).
| (11) |
On addition of ether to the reaction mixture, a green solid precipitated, from which, after recrystallization from ethanol, green crystals of [Cu(NO2)2(tmeda)] (11) were obtained. The presence of [Cu(NO2)2(tmeda)] in the reaction mixture clearly suggests that the first step in the catalytic oxygenation of 2-nitropropane is the formation of the fairly stable [Cu(NP)2(tmeda)] (12) complex from copper powder, 2-nitropropane, tmeda and dioxygen, which reacts further rapidly with dioxygen to form the more stable nitrito species 11.
2.5 Characterization of the [Cu(NO2)2(tmeda)] (11) complex
[Cu(NO2)2(tmeda)] displays three bands at 1360, 1286 and 808 cm−1 in its IR spectrum assigned to the asymmetric and symmetric stretching and bending frequencies of the nitro group. There are three additional bands at 953, 845 and 593 cm−1 due to deformation and bending frequencies of the nitro group. Three bands at 2983, 2941 and 2899 cm−1 can be assigned to the ligand tmeda coordinated to the metallic copper. The electronic absorption spectrum in acetonitrile showed two intense bands at 271 and 222 nm, which were assigned to LMCT bands. The d–d transition of Cu(II) was found at 642 nm. Further, the LMCT bands for the nitro complex at 271 nm may include contributions from the n → π* transition as well [55]. Crystallographic characterization of the complex shows that the molecule is monomeric; the copper(II) center has an elongated rhombic octahedral geometry with a CuO2N2O2 chromophore and a (4 + 2∗)-type coordination involving two off-the-z-axis bonds. Two N atoms of the bidentate tmeda ligand and two O atoms of the NO2 occupy basal positions, the other two O atoms of NO2 ligand being in apical positions.
2.6 Synthesis and characterization of the [Cu(NP)2(tmeda)] (12) complex
The complex [Cu(NP)2(tmeda)] was prepared by the stoichiometric reaction of [Cu(OMe)2], 2-nitropropane and tmeda in THF under Ar. The IR spectrum of the complex shows intense sharp bands corresponding to the coordinated aci-2-nitropropanate ligands at 1612 ν(CN), 1162 and 1123 νas(NO2) and 943 νs(NO2) cm−1. The electronic spectrum of the complex shows an absorption band at 727 nm due to d–d transitions. A higher energy band at 314 nm is associated with a charge-transfer transition. Room-temperature magnetic measurements yielded a value of μeff = 1.92 BM for the complex, consistent with a copper(II) ion. The geometry around the copper(II) center is similar to that of the [Cu(NO2)2(tmeda)] complex, with an elongated rhombic octahedral geometry. Two N atoms of the bidentate tmeda ligand and two O atoms of the nitronate ligand occupy basal positions, while the other two O atoms of nitronate ligand are in apical positions. The carbon–nitrogen bond [1.290 Å] in the nitronate ligand is essentially double bond in character (C–N 1.47 Å, CN 1.27 Å) [56].
2.7 Kinetic measurements
Kinetic measurements of the oxygenation of 2-nitropropane in the presence of catalytic amount of copper and tmeda were performed in DMF in the temperature range 363.16–373.16 K. The kinetics of the reaction, measured at 90 °C, resulted in a rate equation of first-order dependence on copper and dioxygen and second-order dependence on 2-nitropropane. The rate law −d[NPH]/dt = d[acetone]/dt = kcat[NPH]2[Cu][O2] describes the kinetic data (kcat = 5.37 × 10−2 mol−3 dm−9 s−1, Ea = 131 kJ mol−1, ΔH≠ = 127 kJ mol−1 and ΔS≠ = 80 J mol−1 K−1). The catalytic oxygenation of 2-nitropropane by [Cu(NO2)2(tmeda)] and [Cu(NP)2(tmeda)] complexes as catalysts was also carried out. It could be seen that the mean values of the kinetic constant for [Cu(NO2)2(tmeda)] and [Cu(NP)2(tmeda)] complexes are very similar to those found in the reactions, where the catalyst was prepared ‘in situ’, suggesting that the two complexes above were involved in the catalytic process when copper metal was used as the catalyst precursor. The kinetics of the stoichiometric oxygenation of [Cu(NP)2(tmeda)] into [Cu(NO2)2(tmeda)] and acetone resulted in the overall second-order rate equation with a rate constant, activation enthalpy, and entropy at 313.16 K of ks = 0.46 mol−1 dm3 s−1, Ea = 38 kJ mol−1, ΔH≠ = 35 kJ mol−1 and ΔS≠ = −142 J mol−1 K−1, respectively. Values of the activation parameters indicate that the oxidation of [Cu(NP)2(tmeda)] is a bimolecular reaction involving the associative transition state of the complex and dioxygen.
2.8 Copper-mediated acetylation of alcohols by 2-nitropropane [57]
The catalytic oxidation reactions of 2-nitropropane by metallic copper were carried out in various alcohols in order to study the effect of the alcoholic media for the catalytic activities and selectivities, with emphasis on the ester formation. The oxygenolysis was performed in alcoholic solvents (methanol, ethanol, 2-propanol, 1-butanol, 2-butanol) in the presence of metallic copper and tmeda or [Cu(NO2)2(tmeda)]. The results show that the formation of acetone is the main reaction route and the corresponding esters are formed only in a maximum of 30% yield. We have found that the solvent has a profound effect on the selectivity of this reaction. In the case of simple alcohols such as ethanol, the amount of ester was increased. It was shown that [Cu(NO2)2(tmeda)] is an even more efficient catalyst than that formed from metallic copper, however, in the case of methanol, ethanol and 2-propanol, the conversion is lower.
2.9 Mechanistic pathways in the oxygenation of nitroalkanes mediated by copper complexes
Based on the stoichiometric and kinetic data on the copper-catalyzed oxygenation of 2-nitropropane into acetone and acetylation of alcohols by 2-nitropropane in the presence of tmeda, a probable mechanism is proposed as shown in Fig. 2.
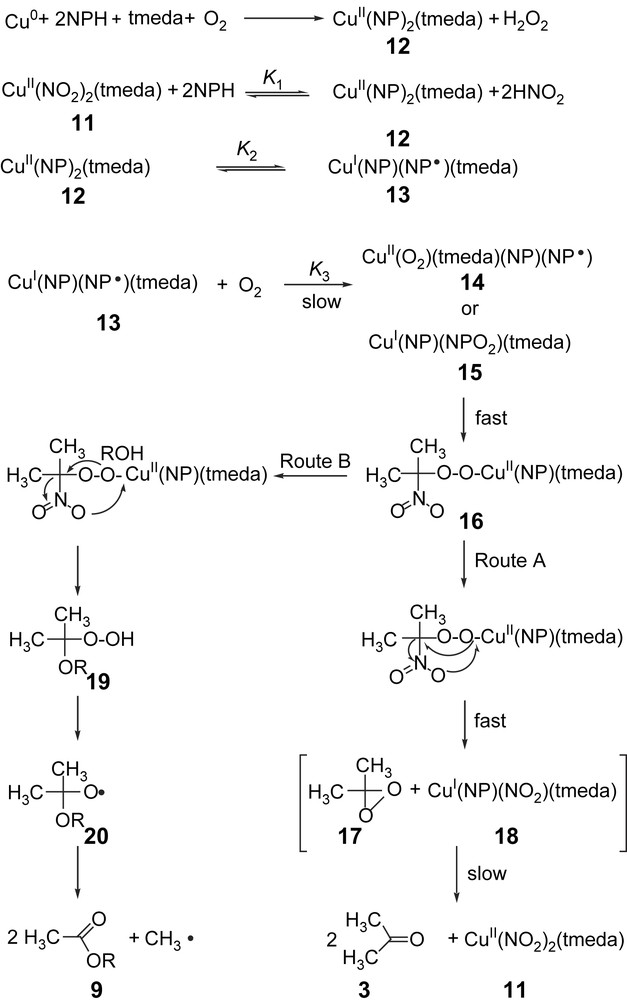
Mechanism of the copper-mediated oxygenolysis of 2-nitropropane and acetylation of alcohols by 2-nitropropane.
According to that, the first step is the formation of the complex [Cu(NP)2(tmeda)] from copper powder, 2-nitropropane, tmeda, and dioxygen or from [Cu(NO2)2(tmeda)] through substitution of NO2− by NP− in a pre-equilibrium in the catalytic cycle. This is then followed by an intramolecular electron transfer from the ligand 2-nitropropanate to the copper(II) ion, resulting in a (2-nitropropyl radical)copper(I) complex (13). This equilibrium is largely shifted to the left (K2 is rather small). At the intradiol cleavage of catechols, either by iron- [58] or copper-containing [42] catechol 1,2-dioxygenases along with model studies [47], it has been proven that valence isomerism plays an important role. The molecular orbitals involved in this process are close to each other in energy and so the electron transfer is easily feasible. The equilibrium depends, of course, on auxiliary ligands, as shown in the case of copper catecholate complexes, whereas soft ligands prefer the lower oxidation state of the metal ion [59]. The 2-nitropropanate anion does not react with 3O2 [46]. The absence of the electron transfer from [Cu(NP)2(tmeda)] to dioxygen could also be proved by the test for free O2− with nitroblue tetrazolium (NBT), where no reduction of the added dye to the blue formazan took place, excluding the formation of free O2− [60]. In the complex [Cu(NP)(NP)(tmeda)], there are two redox-active centers, namely the nitropropanyl radical NP and the copper(I) ion. The biradical dioxygen may react at both redox-active sites in the complex 13, in a radical–radical reaction with the coordinated radical ligand NP, leading to the peroxo intermediate 15, or in an oxidative addition of O2 to the copper(I) ion, giving the (superoxo)copper(II) complex 14, as found in the case of many other copper(I) complexes (Fig. 2) [48]. Both mechanistic assumptions satisfy the kinetic data, but we believe that the reaction of the copper(I) center of 13 with O2 is more probable. We have observed earlier that radical ligands bound to metal ions that cannot be oxidized (e.g. Cu(II), Fe(III), etc.) do not react with O2 at all, or undergo very slow reactions only [61]. The (superoxo)copper(II) complex 14 then undergoes an intramolecular radical–radical reaction of the coordinated ligands O2− and NP, resulting in the peroxide species 16. This then decomposes to the mixed-ligand copper(II) complex 18 and 1,1-dimethyl-dioxirane 17 (Fig. 2, route A). The dioxirane 17 then oxygenates the coordinated NP− in 18, resulting in [Cu(NO2)2(tmeda)] and acetone. Previous studies have shown that dimethyldioxirane oxidation of nitronate anions affords the corresponding carbonyl product [62]. On the other hand, the mechanism involves the decomposition of the peroxide species 16 intermediate by the addition of the alcohol, giving rise to (1-alkoxy-1-methylethylperoxo)copper(II), which would release [Cu(NP)(NO2)(tmeda)] and a hydroperoxide intermediate that undergoes Baeyer–Villiger decomposition into the corresponding esters (Fig. 2, route B).
3 Iron-mediated oxygenation of nitroalkanes
3.1 Synthesis of iron nitronate complexes [63]
In order to understand the coordination of nitroalkanes to iron and their conversion to oxo compounds, we prepared and characterized 2-propanenitronato-O,O-Fe(III) complexes and investigate their oxygenation reaction.
Iron(III) chloride reacts with potassium 2-propanenitronate to give the homoleptic tris(2-propanenitronate-O,O)iron(III) complex [Fe(NP)3] (21). In ethanol, complex 21 undergoes ligand exchange: ethanol displaces the nitronate ligand and bridges the two iron(III) ions to form [Fe2(NP)4(C2H5O)2] (22) in an equilibrium (Eq. (12)).
| (12) |
The IR spectra of 21 and 22, obtained as KBr pellets, reveal a particularly diagnostic absorption ν(CN) at 1639 and 1644 cm−1, respectively. The relative intensity of this absorption suggests a similar mode of coordination for both complexes and implies considerable carbon–nitrogen double-bond character. Magnetic measurements gave values of μB = 5.87 per Fe(III) for 21 and 7.29 per 2Fe(III) for 22 in accord with high spin Fe(III) centers, with a slight antiferromagnetic interaction in complex 22. The Mössbauer spectra reflect the different coordination of Fe(III) in complexes 21 and 22. Although the identical isomer shift values (δ1 = 0.48 mm s−1 and δ2 = 0.48 mm s−1) reveal the same electron density of the iron in both compounds, the quadrupole splittings (Δ1 = 0.90 mm s−1 and Δ2 = 0.53 mm s−1) differ significantly, owing to different ligand contributions to the electric-field gradient (EFG). The trigonal distortion of complex 21 makes the EFG rather large; however, this is considerably reduced upon dimerisation, when the local environment of the iron becomes closer to octahedral. The crystal structure of [Fe(NP)3] shows a distorted octahedral geometry around the iron atom, with all coordination sites being occupied by the bidentate 2-nitropropanate ligands. The complex exhibits a propeller-like structure with the iron atom being 0.007 Å out of the plane of the three N atoms of the three ligands. The crystal structure of 22 shows that there are two very similar ethoxy-bridged diiron molecules, with slightly different bond distances and angles. The geometry around the iron atoms is approximately octahedral, the six coordination sites being occupied by two ethoxy groups and two bidentate 2-propanenitronate ligands. The carbon–nitrogen bonds in both complexes (1.283–1.291 Å) are essentially double bond.
Complex [Fe(NP)3] is very sensitive to dioxygen and moisture, while [Fe2(NP)4(C2H5O)2] is reasonably stable to both in the solid state. Preliminary oxygenation reactions of 2-nitropropane in the presence of 21 and 22 in pyridine at 90 °C resulted in the formation of acetone and HNO2. Conversion of ca. 80% could be achieved at substrate:catalyst ratios of 20–40:1 during 20 h. The conversion-time profiles of the catalytic reactions were very similar for complexes 21 and 22, indicating that 22 in transformed, into 21 if an excess of 2-nitropropane is present. To our knowledge, these reactions represent the first examples of iron-catalyzed oxygenation of 2-nitropropane resembling enzyme action, and indicate the possible role of iron ion as a cofactor.
3.2 Ferroxime(II)-catalyzed oxygenation of nitroalkanes [64]
The complex [Fe(Hdmg)2(py)2] (Hdmg = dimethylglyoxime), ferroxime(II) was found to be a selective catalyst for the oxidative denitrification of nitroalkanes such as 1-nitroethane, 1-nitropropane, 2-nitropropane, 1-nitropentane and nitrocyclohexane into the corresponding aldehydes or ketones (Eq. (13)).
| (13) |
The oxidation was performed in DMF and EtOH under Ar in the presence of different oxidants (tBuO2H, H2O2 and O2). Catalytic oxidations were effective only with TBHP and H2O2. The best conversions have been obtained with TBHP in EtOH solution (31%). During the oxidation of 2-nitropropane by TBHP in the presence of ferroxime(II), the characteristic band of the complex at 509 nm immediately disappeared, due to loss of coordinated py; a new species was formed, signalled by the appearance of a broad absorption band at around 610 nm. The spectroscopic characteristics of the species absorbing at 610 nm are identical to those of the mononuclear alkylperoxo-iron complex, [Fe(III)(OOR)] as reported earlier [65]. From the analysis of the results obtained for the oxidation of 2-nitropropane with TBHP, the following mechanism is proposed (Fig. 3).
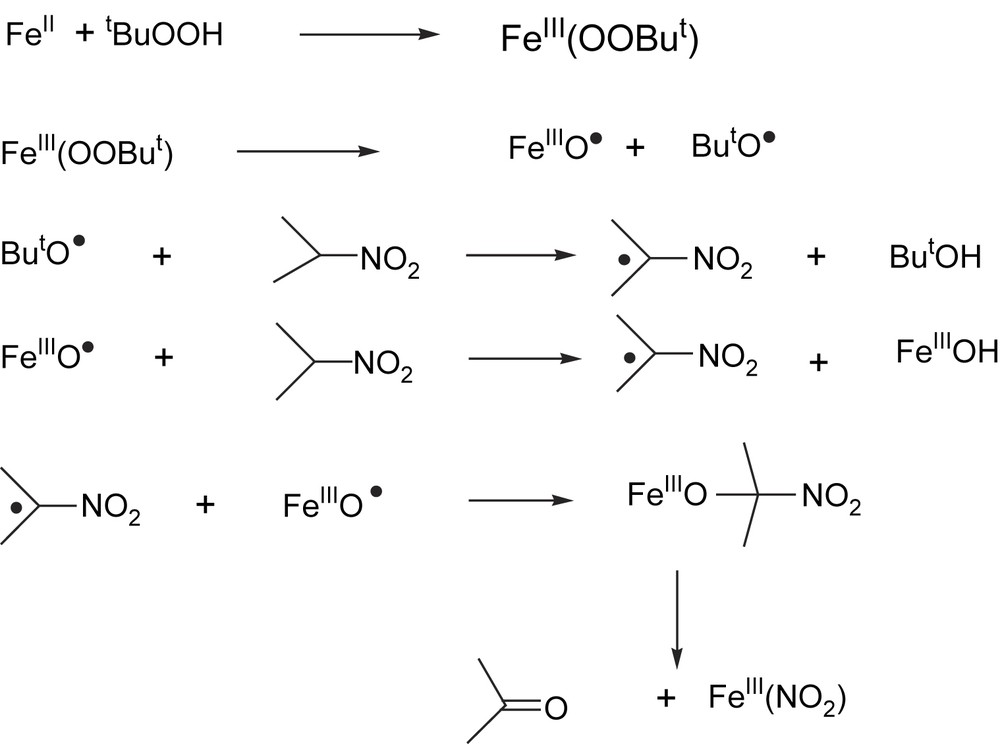
Proposed mechanism of ferroxime(II)-catalyzed oxygenation of 2-nitropropane.
The first step of the reaction is the binding of the oxidant to the iron catalyst, forming an alkylperoxo-iron(III) complex. After that, the two possible candidates for the H-abstracting species are the high valent (oxo)ironcomplex [Fe(III)O] or the alkoxyl radical derived from the homolytic cleavage of the O–O bond, respectively. We believe that, in this reaction, both radical species are involved in the H-abstraction. The so-formed [2-nitrito-2-propanolato-iron(III)] species decomposes into acetone and [Fe(III)NO2]. The nitrito group may then be displaced by the oxidant TBHP to give [Fe(III)OOtBu] and HNO2 to close the catalytic cycle.
4 Kinetics and mechanism of the oxygenation of potassium nitronates [66]
The potassium salts of some alkylnitronates were prepared in THF by reacting metallic potassium with the appropriate nitroalkane. The salts were characterized by IR spectra. The IR spectra of the potassium salts show intense sharp bands around 1600 [ν(CN)], 1150 and 1120 [νas(NO2)], and 940 [νs(NO2)] cm−1 corresponding to the deprotonated aci form of the nitroalkane ligands. The oxygenation of potassium salts of alkylnitronates in DMSO leads to potassium nitrite, nitrate and the corresponding aldehydes or ketones.
| (14) |
Kinetic measurements for potassium-1-propylnitronate (K-1-PN) resulted in an overall second-order rate equation (−d[K-1-PN]/dt = k2[K-1-PN][O2], k/M−1 s−1 = (1.43 ± 0.04)). Values of the activation parameters (ΔH≠/kJ mol−1 = 57 ± 10, ΔS≠/J mol−1 K−1 = −60 ± 32) indicate that the oxygenation of potassium nitronates is a bimolecular reaction involving an associative transition state of nitronates and molecular oxygen. We could not detect the superoxide radical by EPR, even at low-temperature measurements; however, the reduction of nitroblue tetrazolium (NBT) to blue formazan was used as an indicator of O2− (the blue formazan has a characteristic absorption around 550 nm, and this test indicated the presence of free O2− anions [60]).
On the basis of chemical, spectroscopic and kinetic data, a suggestion can be made for the possible mechanism of the oxygenation reaction (Fig. 4).
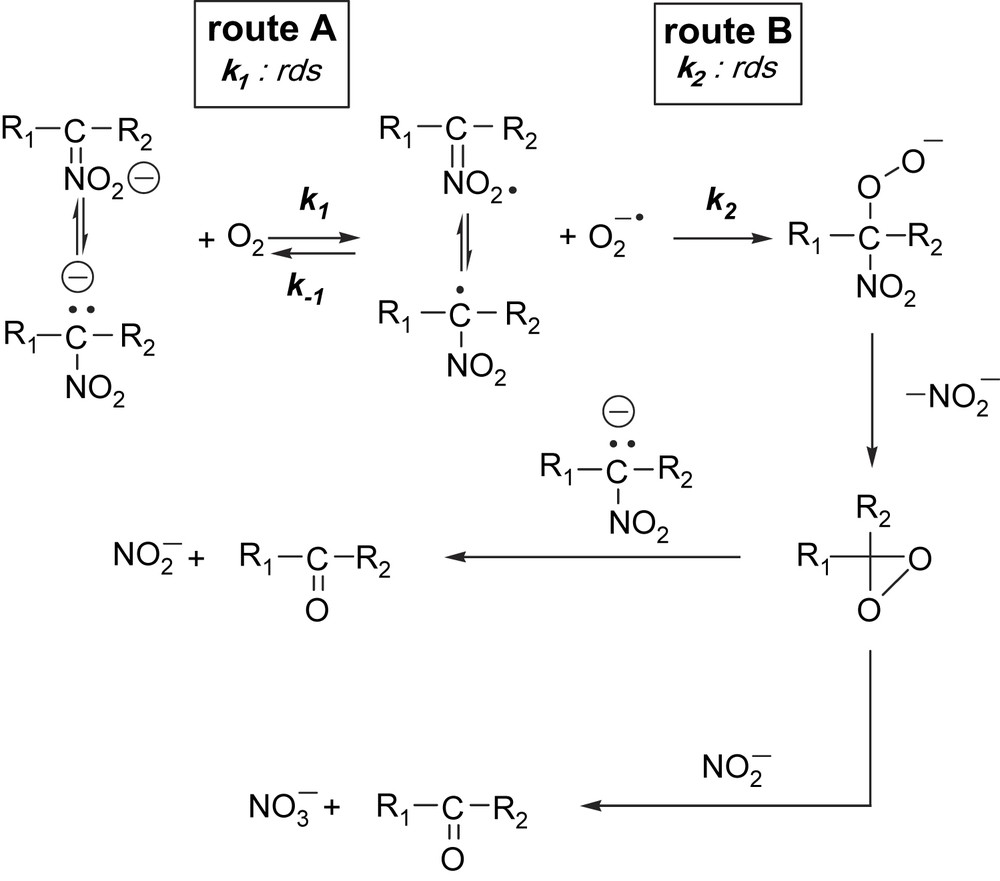
Proposed mechanism of the oxygenation of potassium nitronates.
From the kinetic point of view two reaction routes seem to satisfy the kinetic findings. Route A involves a slow rate-determining single electron transfer from the mesomeric carbanion to dioxygen, resulting in the nitroalkyl radical and superoxide ion. This is then followed by a radical coupling reaction between the two radicals formed, ending up in the peroxide intermediate. In route B, the mesomeric carbanion undergoes a fast and reversible electron transfer with dioxygen, yielding the nitroalkyl radical and superoxide ion. This is followed then by the rate-determining step between the two radicals. The presence of superoxide ion was proved by NBT test. Considering the two reaction pathways, it is known that carbanions with electron affinities = 20 kcal react with 3O2, but those of higher do not. On the other hand, O2− is a good reducing agent probably able to reduce the nitroalkyl radical. These facts seem to suggest reaction route B as the most probable event under these conditions. However, the SET reaction as rate determining cannot be excluded. All the consecutive reactions of the nitroalkyl radical and superoxide ion are shown in Fig. 4.
In a radical–radical coupling reaction, the nitroalkyl radical reacts with the superoxide ion, leading to peroxide intermediate. This then decomposes to potassium nitrite and 1,1-dialkyldioxirane. The 1,1-dialkyldioxirane then can oxygenate both the alkylnitronate and the nitrite ion, resulting in nitrate and aldehyde, or ketone. Previous studies have shown that dimethyldioxirane oxidation of nitronate anions affords the corresponding carbonyl product [61].
5 Conclusion
2-Nitropropane dioxygenase is an iron-containing flavin-dependent enzyme, which catalyzes the oxidation of nitroalkanes to the corresponding carbonyl compounds and nitrite. The generation of an anionic flavin semiquinone and superoxide anion as intermediates in the enzyme reaction was found.
Since the 2-nitropropane dioxygenase is an iron-containing enzyme and the role of the iron ion in the active site of enzyme is still obscure, we initiated studies on copper- and iron-mediated oxygenations of nitroalkanes. In order to gain more insight into the mechanism of these reactions, kinetic studies on the oxygenation of potassium alkylnitronates in aprotic solvent were also carried out.
The following conclusions can be drawn from the model studies carried out:
- • The copper-assisted intermolecular oxygenation of the CN double bond of the coordinated aci-2-nitropropanato ligand with triplet dioxygen may indicate that the presence of copper(I) is essential in the activation of triplet dioxygen, and reduced oxygen species is ready for oxidative splitting of the CN double bond. We believe that there is a valence tautomerism between [CuII(NP)] and [CuI(NP)] due to intramolecular electron transfer from the ligand to the metal ion. In this sense of the activation process, the ligand is transformed into a radical and copper(II) to copper(I). The latter then reacts with dioxygen to form a peroxidic species, which is transformed to the (nitrito)copper(II) complex and acetone. The catalytic reaction may be regarded as a functional model for 2-nitropropane dioxygenase and provides an alternative preparative method for the acetylation of alcohols. Oxygenation of 2-nitropropane was also catalyzed by iron(III) nitronate complexes, which represent the first examples of iron-catalyzed oxygenation of 2-nitropropane resembling enzyme action, and indicate the possible role of iron ion as a cofactor. In the oxygenation of nitroalkanes with TBHP catalyzed by iron(II), complex radical species (tBuO and Fe(III)O) are involved in the H-abstraction. The catalytic process is rather similar to the enzyme catalyzed reaction, where the anionic flavin semiquinone species reacts with dioxygen, resulting in the oxidation of flavin and formation of superoxide, which as a secondary oxidant reacts with the nitroalkane substrates.
- • Kinetic results of the oxygenation of potassium nitronates provide further support to the key role of the nitroalkyl and superoxide ion radical in these reactions, which may be regarded as functional models for 2-nitropropane dioxygenase.
Acknowledgements
Financial support by the Hungarian National Research Fund (OTKA No. T04314 and T042875) is gratefully acknowledged.