

1 Introduction
Investigations of the organization of matter at the nano-level are currently under way in many chemical systems with potential applications in nanotechnology. Recently, nanoscale structures were reported for the first time for actinide-containing compounds as well [1–6]. As it is well known, solid-state chemistry of inorganic uranyl oxocompounds is dominated by two-dimensional (2D) layered structures, due to the strong tendency of U6+ cations to form linear actinyl ions, UO26+ [7–9]. The 2D character of polyhedra polymerization makes uranyl oxocompounds attractive from the viewpoint of their potential ability to form nanostructures based upon real-2D and pseudo-2D topologies. For instance, spontaneous formation of nanotubes was observed in a number of systems where exfoliation of lamellar solids into individual sheets can be achieved [10–14].
Organic–inorganic 2D nanocomposites is an emerging group of organic–inorganic materials that contain alternating organic and inorganic substructures [15–20]. One useful concept to analyze structures of organic–inorganic nanocomposites has been the concept of charge-density matching [21,22] at the organic–inorganic interface. The idea is that “…two different materials will self-organize to have similar charge densities at their surfaces and, therefore, achieve local electroneutrality” [22]. This concept has been successfully applied to a large number of organic–inorganic composites, including metal phosphates [23,24], vanadates [22] and mesoporous silica [25]. The unique character of uranyl chemistry, namely, tendency of U6+ ions to form actinyl groups, requires special attention to be paid to the application of the charge-density matching principle to uranyl and, in general, to actinide compounds.
In this paper, we report syntheses and structures of three new organic–inorganic uranyl selenates. The uranyl selenate system is of special attraction for uranyl chemistry since relatively soft synthetic conditions, on the one hand, allow manipulations with many possible compositions, and, on the other hand, result in formation of highly ordered structures accessible via X-ray diffraction single-crystal structure analysis [26–43]. Structure determination makes it possible to analyze structural parameters in more detail which, in turn, allows elaboration of quantitative models of behaviour of matter at the nanoscale. Here we will use the three structures to examine the validity and applicability of the charge-density matching principle for uranyl compounds.
2 Experiment
Single crystals of [C10H26N2][(UO2)(SeO4)2(H2O)] (H2SeO4)0.85(H2O)2 (1), [C10H26N2][(UO2)(SeO4)2] (H2SeO4)0.50(H2O) (2), and [C8H20N]2[(UO2)(SeO4)2(H2O)] (H2O) (3) were prepared by evaporation from aqueous solutions of uranyl nitrate, selenic acid and the respective amines. For the preparation of 1 and 2, the solution contained: 0.124 g of (UO2)(NO3)2·6H2O, 0.040 g of 1,10-diaminododecane, 0.5 ml of 40% solution of H2SeO4 in water, and 2 ml of H2O; for 3: 0.069 g of (UO2)(NO3)2·6H2O, 0.10 ml of octylamine, 0.4 ml of 40% solution of H2SeO4 in water, and 2 ml of H2O. The solutions were left to evaporate in a fume hood. Crystals were formed after approximately 24 h. The compounds were characterized by semi-quantitative electron microprobe analysis. Far-infrared spectra were collected and the presence of uranyl ions and selenate groups was confirmed.
The crystals selected for data collection were examined under an optical microscope and mounted on glass fibers. The crystals of 2 and 3 were extremely air-sensitive and were encased in an epoxy. Data were collected by means of a STOE IPDS II diffractometer using monochromated Mo Kα radiation and framewidths of 2° in ω. The unit-cell dimensions (Table 1) were refined by least-squares techniques. The data were corrected for Lorentz, polarization, and background effects. An analytical absorption correction based on the indexed faces was applied. The structures were solved and refined by means of the programs SIR-92 [44] and SHELXL-97 [45], respectively. Due to the low quality of crystals and their sensitivity to air, only relatively rough structural models could be obtained and R1 values are not of the same order as those obtained from perfect crystals. It is noteworthy that, in most cases, structures of this kind could not be grown as single crystals suitable for conventional X-ray diffraction experiment. For instance, Feng et al. [23] prepared crystals of several composite organic–inorganic Al phosphates but could not localize the positions of diamine molecules separating inorganic sheets. Due to the same reasons, we were unable to localize the positions of H atoms as well. The selected bond lengths are given in Table 2 for all compounds. The supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK (CCDC # 626009, 626010, and 626011 for 1, 2, and 3, respectively).
Crystallographic data and refinement parameters for amine-templated uranyl selenates: [C10H26N2][(UO2)(SeO4)2(H2O)] (H2SeO4)0.85(H2O)2 (1), [C10H26N2][(UO2)(SeO4)2] (H2SeO4)0.50(H2O) (2), and [C8H20N]2[(UO2)(SeO4)2(H2O)] (H2O) (3)
| Compound | |||
| 1 | 2 | 3 | |
| a (Å) | 7.5461(6) | 29.280(2) | 7.498(3) |
| b (Å) | 14.9910(12) | 13.3013(10) | 11.897(4) |
| c (Å) | 22.3789(17) | 11.4513(7) | 32.056(14) |
| α (deg) | 77.678(6) | 90.00 | 89.69(3) |
| β (deg) | 85.463(6) | 93.295(5) | 90.05(4) |
| γ (deg) | 82.717(6) | 90.00 | 88.80(3) |
| V (Å3) | 2449.8(3) | 4452.5(5) | 2859(2) |
| Space group | C2/c | ||
| Formula weight | 833.81 | 811.27 | 852.48 |
| μ (mm−1) | 10.307 | 11.437 | 8.278 |
| Z | 2 | 4 | 2 |
| Dcalc (g/cm3) | 2.26 | 2.42 | 1.98 |
| Crystal size (mm3) | 0.12 × 0.08 × 0.02 | 0.08 × 0.06 × 0.01 | 0.12 × 0.11 × 0.01 |
| Diffractometer | Stoe IPDS II | Stoe IPDS II | Stoe IPDS II |
| Radiation | Mo Kα | Mo Kα | Mo Kα |
| Total Ref. | 16244 | 16248 | 9359 |
| Unique Ref. | 9541 | 4733 | 6644 |
| 2θ range (deg) | 3.01–53.72 | 2.78–53.90 | 3.64–50.30 |
| Unique |Fo| ≥ 4σF | 7910 | 3461 | 3669 |
| Rint | 0.034 | 0.094 | 0.108 |
| Rσ | 0.034 | 0.074 | 0.151 |
| R1 | 0.054 | 0.091 | 0.087 |
| wR2 | 0.135 | 0.161 | 0.231 |
| S | 1.095 | 1.167 | 1.022 |
Selected bond lengths in the structures of 1, 2 and 3
| 1 | |||
| U1–O19 | 1.755(8) | Se1–O6 | 1.609(8) |
| U1–O22 | 1.761(7) | Se1–O11 | 1.622(9) |
| U1–O17 | 2.349(7) | Se1–O3 | 1.638(9) |
| U1–O14 | 2.363(9) | Se1–O17 | 1.647(7) |
| U1–O12 | 2.369(7) | Se2–O10 | 1.617(8) |
| U1–O18 | 2.379(8) | Se2–O8 | 1.621(8) |
| U1–O1 | 2.481(8) | Se2–O18 | 1.635(8) |
| U2–O21 | 1.758(8) | Se2–O12 | 1.655(7) |
| U2–O20 | 1.767(8) | Se3–O9 | 1.607(9) |
| U2–O3 | 2.353(9) | Se3–O4 | 1.623(9) |
| U2–O7 | 2.364(8) | Se3–O14 | 1.633(9) |
| U2–O13 | 2.365(8) | Se3–O13 | 1.648(8) |
| U2–O15 | 2.381(9) | Se4–O16 | 1.615(9) |
| U2–O2 | 2.479(8) | Se4–O15 | 1.625(9) |
| Se4–O5 | 1.628(9) | ||
| Se4–O7 | 1.655(8) | ||
| 2 | |||
| U1–O6 | 1.759(14) | Se1–O5 | 1.621(12) |
| U1–O4 | 1.771(14) | Se1–O10 | 1.636(15) |
| U1–O2 | 2.356(12) | Se1–O7 | 1.653(12) |
| U1–O9 | 2.372(13) | Se1–O1 | 1.650(13) |
| U1–O1 | 2.387(12) | Se2–O8 | 1.608(15) |
| U1–O3 | 2.408(12) | Se2–O3 | 1.632(12) |
| U1–O7 | 2.414(12) | Se2–O9 | 1.652(13) |
| Se2–O2 | 1.669(13) | ||
| 3 | |||
| U1–O6 | 1.78(3) | Se1–O22 | 1.58(2) |
| U1–O1 | 1.83(3) | Se1–O13 | 1.59(2) |
| U1–O17 | 2.33(3) | Se1–O3 | 1.64(3) |
| U1–O14 | 2.37(2) | Se1–O17 | 1.65(3) |
| U1–O9 | 2.39(2) | Se2–O9 | 1.61(2) |
| U1–O20 | 2.43(2) | Se2–O7 | 1.61(2) |
| U1–O12 | 2.47(2) | Se2–O16 | 1.63(2) |
| U2–O2 | 1.79(3) | Se2–O19 | 1.66(3) |
| U2–O4 | 1.79(3) | Se3–O20 | 1.56(3) |
| U2–O3 | 2.34(2) | Se3–O10 | 1.59(3) |
| U2–O11 | 2.36(3) | Se3–O15 | 1.61(2) |
| U2–O21 | 2.37(2) | Se3–O11 | 1.64(3) |
| U2–O7 | 2.38(2) | Se4–O8 | 1.63(2) |
| U2–O5 | 2.53(2) | Se4–O21 | 1.65(2) |
| Se4–O18 | 1.66(2) | ||
| Se4–O14 | 1.67(3) |
3 Results
As typical for uranyl selenates [26–43], the structures of the compounds 1, 2, and 3 contains U and Se atoms in pentagonal bipyramidal and tetrahedral coordinations, respectively. The U6+ cations form two short U6+–O bonds resulting in linear uranyl ions, [UO2]2+. The uranyl ions are coordinated in the equatorial plane by five anions each. The Se6+ cations are tetrahedrally coordinated by four O atoms each. The UO7 and SeO4 polyhedra polymerize by sharing common O atoms to form chains (compound 1) or sheets (compounds 2 and 3) (Fig. 1). Despite the fact that the uranyl selenate units in the structure of 1 are one-dimensional, the [UO2(SeO4)2(H2O)]2− chains are arranged into pseudo-2D-layers by strong hydrogen bonds between H2O groups and O atoms of the adjacent chains.
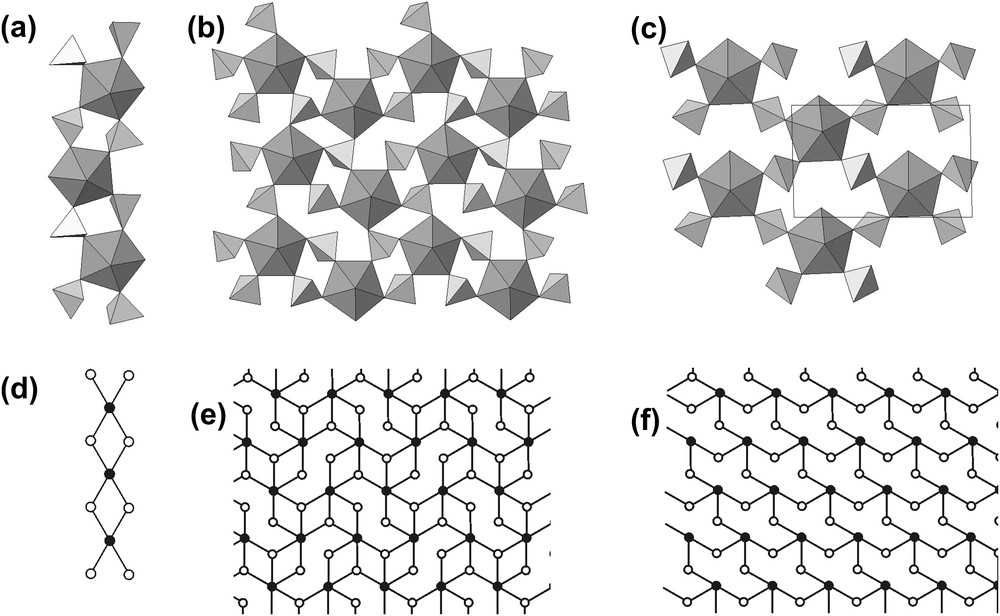
Uranyl selenate units observed in the structures of organic–inorganic uranyl selenates: (a) uranyl selenate chain in 1; (b) and (c) uranyl selenate sheets in 2 and 3, respectively. Below are given black and white graphs describing topology of structural units [d, e and f for a, b, and c, respectively].
To analyze topological structures of uranyl selenate units considered here, we propose to use a graphical approach that was developed by Krivovichev and Burns [45] and further expanded by Krivovichev [46]. Within this approach, the structure (or a part of it) is represented as a graph with nodes symbolizing coordination polyhedra. Nodes of different colors correspond to geometrically and/or chemically different polyhedra. Two nodes of the graph are linked only if the corresponding polyhedra share at least one common ligand. Black-and-white graphs of uranyl selenate units observed in the structures of 1, 2, and 3 are given in Fig. 1d, e, and f, respectively. Here the black-and-white nodes symbolize the U and Se coordination polyhedra, respectively.
The structure of 1 is shown in Fig. 2a and b. The layers consisting of hydrogen-bonded [UO2(SeO4)2(H2O)]2− chains are separated by mixed organic–inorganic layers comprised of [NH3(CH2)10NH3]2+ molecules, H2O molecules, and disordered electroneutral (H2SeO4) groups. The thickness of the organic–inorganic layers is about 1 nm, which allows us to consider this structure as an organic–inorganic composite. The structure of 2 has a similar architecture but here a purely inorganic layer is represented by a fully connected [UO2(SeO4)2]2− sheet (Fig. 1b).
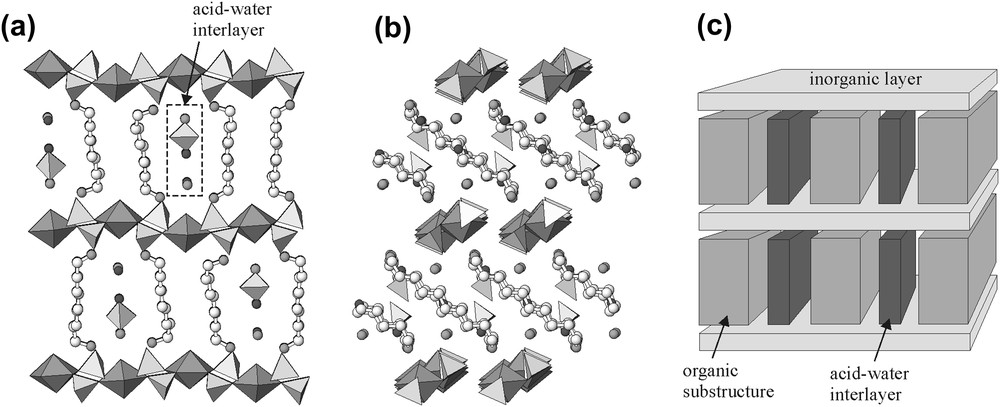
Structure of 1 projected along two mutually perpendicular directions along the uranyl selenate sheet (a, b) and schematic representation of its structural principle (c).
In contrast to 1 and 2, the structure of 3 does not contain disordered (H2SeO4) groups but is based upon alternating [UO2(SeO4)2(H2O)]2− sheets and 1.5-nm-thick organic blocks consisting of positively charged protonated octylamine molecules, [NH3(CH2)7CH3]+ (Fig. 3).
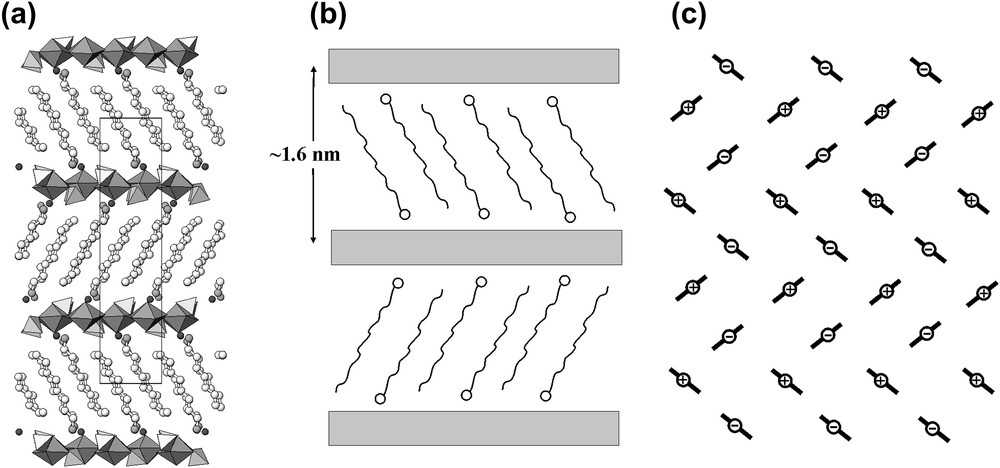
Structure of 3 (a), its schematic representation (b), and orientation of octylamine molecules within the organic nanolayer (c; “+” and “−” signs correspond to the orientations of amine head groups “up” and “down”, respectively).
It is important to note that the inorganic and organic blocks in the structures under consideration ‘communicate’ to each other via hydrogen bonds only. In fact, ability to form hydrogen bonds does play an essential role in the structural organization. The organic (di)amine molecules are composed of a hydrophilic (amine head groups) and a hydrophobic (carbohydrate) parts. In aqueous media, these molecules tend to self-assemble into specific moieties (spheres, cylinders or lammela) with hydrophilic parts on the surface and hydrophobic parts inside. This is the situation that is observed in the structures of 1, 2 and 3. Thus, in first approximation, we may consider these structures as composed of anionic inorganic sheets (2D blocks) and cationic organic blocks self-organized according to competing hydrophilic–hydrophobic interactions. The analysis of interface interactions between anionic and cationic blocks is of primary importance for understanding structural stability of the compounds under consideration. This can be done using the above-mentioned charge-density matching principle.
4 Charge-density matching between organic and inorganic blocks in uranyl selenates
4.1 General considerations
The charge density of the interface is defined as a formal charge per surface area unit. If inorganic substructure consists of polymerized cation–oxygen coordination polyhedra (as is the case for amine-templated inorganic oxysalts), the larger the size of the polyhedra, the lower the charge density of the surface. In the case of uranyl (and, in general, actinyl) oxocompounds, uranyl ions are coordinated in their equatorial planes by four, five, or six anions Oeq, thus forming tetragonal, pentagonal, or hexagonal bipyramids, respectively. In layered structures, uranyl ions are oriented perpendicular to the plane of the sheets so that equatorial planes of bipyramids are approximately parallel to the sheets. The surface area of bipyramid (SPB) within the plane of the sheet is controlled by the deq = U–Oeq bond length (Fig. 4). For a pentagonal bipyramid, the surface area can be calculated as a surface area of a pentagonal bipyramid with the deq bond length being a distance from its center to its corner. The surface area SPB can be calculated as 2.378 × deq2 [Å2]. For uranyl pentagonal bipyramids, we have deq = 2.36 Å, so SPB = 13.24 Å2.
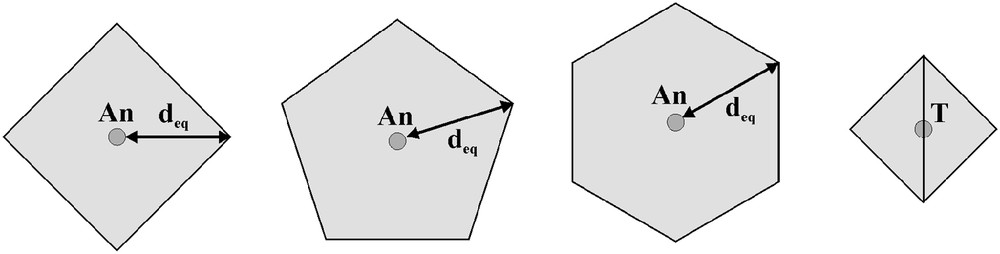
Maximal surface area of uranyl bipyramids (An) in comparison to the maximal surface area of PO4 tetrahedra in metal phosphates (T).
For comparison, let us calculate the surface area (ST) of a tetrahedral unit TO4, which is a common constituent of organically templated lamellar metal phosphates [24]. The surface area is controlled by the average <T–O> bond length, which can vary from 1.5 to 1.7 Å. Since the TO4 tetrahedron may take any position relative to the plane of the sheet, it is convenient to restrict its volume by a sphere with its center in T and its radius being the average <T–O> bond length. The surface area of the tetrahedron will be within the central section of the sphere, i.e. a circle with the radius of <T–O>. By means of some simple calculations, we obtain that the surface area of a metal tetrahedron should be within the range of 7.1–9.1 Å2. These values are significantly smaller than the calculated surface area of the uranyl pentagonal bipyramid. Therefore, charge density of 2D uranyl substructures should be significantly smaller than the charge density of layered metal phosphate units. Let us consider some examples.
The structure of 2 consists of [(UO2)(SeO4)2(H2O)]2− sheets shown in Fig. 1b. The surface area per one [(UO2)(SeO4)2(H2O)] formula unit is 89.204 Å2, which provides charge density of the sheet as CD = −2/89.204 = −0.0224 e Å−2.
Feng et al. [23] reported a series of lamellar aluminophosphates with general formula [Al13(PO4)18H][NH3(CH2)nNH3]7(H2O)8 (n = 9–12). The surface area of the [Al13(PO4)18H]14− sheet is 237.2 Å2, which results in CD = −14/237.2 = −0.0590 e Å−2.
According to Maggard and Boyle [21], charge densities of layered vanadate units [VxOy]m− vary from −0.0362 to −0.0493 e Å−2. Charge density of the gallophosphate layer in the structure of MIL-35, [NH3(CH2)12NH3][Ga4(PO4)4F4] [48] equals −0.0756 e Å−2.
These examples demonstrate clearly that charge densities of uranyl-based sheets are in general smaller than charge densities of metal phosphate and vanadate units in lamellar compounds. However, the very existence of nanocomposite vanadates and metal phosphates means that the charge-density matching principle in these compounds is observed. Then how does it work for uranyl nanocomposites? There are several possible solutions and two of them are realized in the uranyl selenates under consideration.
4.2 Structures with tail interdigitation (long-chain monoamine structures)
The structure of 3 is shown in Fig. 3. It is based upon the [(UO2)(SeO4)2(H2O)]2− sheets consisting of corner-sharing uranyl pentagonal bipyramids and selenate tetrahedra (Fig. 1c). The sheets are alternating with the arrays of protonated octylamine molecules, [CH3(CH2)7NH3]+, stacked in such a way that amine head groups are oriented towards either overlying or underlying uranyl selenate sheet (see scheme in Fig. 3b). The tails of the octylamine cations interdigitate, so that the charge density at the surface of the organic substructure is two times lower than in the structures without interdigitation. The increase of two times is exactly what is needed to compensate the charge density of the inorganic layer. Thus, the charge-density matching principle is observed. The inclination angle of monoamine chain relative to the plane of inorganic sheet, θ, is about 61°.
4.3 Diamine-templated structures
In contrast to monoamines, long-chain diamines have amine head groups on both sides of the chains. If the molecules are packed in the same way as in the structure of 3, the charge density of the organic surface should be exactly two times higher than that in 3, i.e. should be equal to −0.04 to 0.06 e Å2. However, it seems that charge density at the organic side of the interface can be adjusted by means of the packing mode and the inclination angle θ. Thus, in the structure of MIL-35, CD = −0.0756 e Å−2, whereas, in lamellar aluminophosphates, it is around −0.06 e Å−2. In any case, it is at least two times higher than the charge density of typical uranyl selenate sheets.
The charge-density matching principle is observed by incorporation of acid–water interlayers into the organic substructure. Fig. 2 shows the structure of 1, which contains inorganic uranyl selenate sheets of hydrogen-bonded [(UO2)(SeO4)2(H2O)]2− chains. The charge density of the uranyl selenate sheet is −0.0217 e Å−2. The inorganic sheets are separated by organic nanolayers. The nanolayers are modified by incorporation of additional disordered [H2SeO4] groups and H2O molecules that form slices parallel to the orientations of protonated dodecanediamine molecules, [NH3(CH2)12NH3]2+. We prefer to identify these slices as acid–water interlayers. The structural role of the acid–water interlayers is to modify the organic substructure in order to satisfy the charge-density matching principle. The organic substructure is expanded, which leads to the decrease of its charge density in order to stabilize its interactions with the inorganic layer. The inclination angle of the diamine chain relative to the plane of inorganic sheet, θ, is about 40°. This raises an interesting question: does inclination of the amine chains play any role in the charge-density determination? A simple scheme shown in Fig. 5 allows us to give a positive answer to this question. Inclination of chains results in a larger surface area in comparison with chains oriented orthogonally relative to the surface of an inorganic substructure.
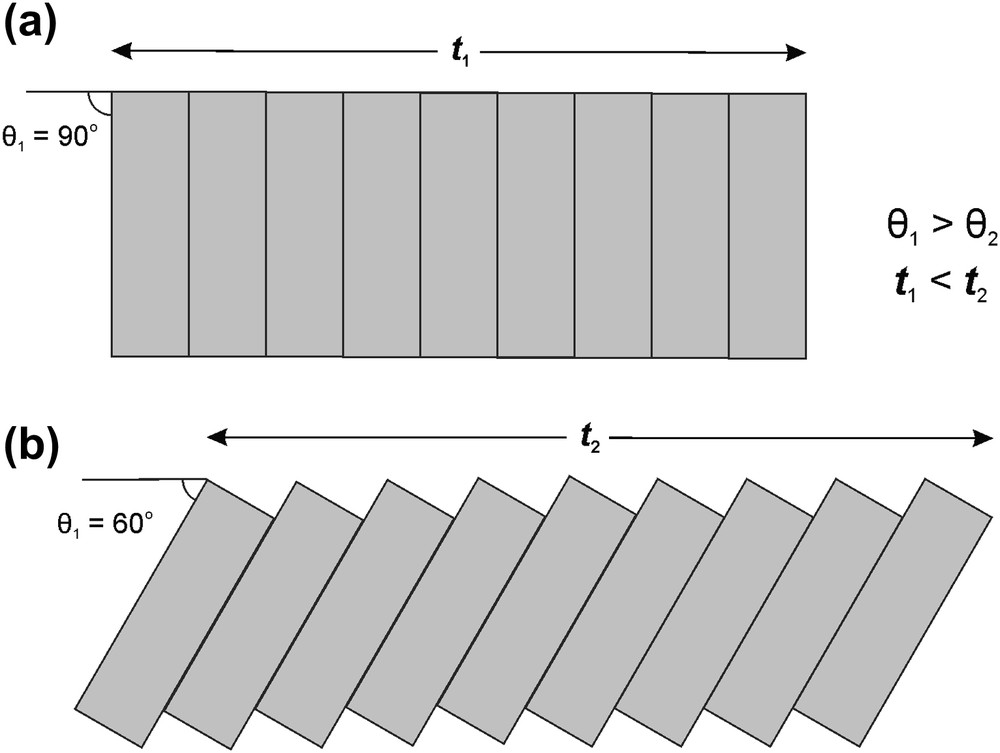
Scheme illustrating the influence of the inclination angle θ upon the surface area of stacking of long-chain molecules (shown as rectangles). The smaller the angle (a versus b), the longer the distance t, the larger, the surface area.
5 Conclusions
Using three uranyl selenate structures templated by long-chain amine and diamine molecules as an example, we have demonstrated that the charge-density matching principle is observed in uranyl compounds. However, in order to satisfy some basic peculiarities of uranyl (in general, actinyl) chemistry, it requires specific additional mechanisms that are employed by nature:
- (a) in long-chain-amine-templated compounds, protonated amine molecules interdigitate;
- (b) in long-chain-diamine-templated compounds, incorporation of acid–water interlayers into an organic substructure is necessary;
- (c) inclination angle of the amine chains may vary in order to modify surface area of an organic substructure.
It is important to note that the mechanisms described here are valid for lamellar uranyl selenates with alternating 2D inorganic and organic layers. Cylindrical self-assembly of long-chain protonated amine molecules had also been observed and was believed to be responsible for the formation of uranyl selenate nanotubules [3]. In general, hydrophilic–hydrophobic interactions are of great importance in uranyl selenate chemistry. For instance, we have recently demonstrated that branched structure of short-chain amine molecules is important in controlling the topology of 2D uranyl selenate units with the U:Se ratio of 2:3 [48].
Acknowledgements
S.V.K. thanks the Russian Ministry of Science Education for financial support through the Grant RNP 2.1.1.3077 and MD 4886.2007.5, and contract # 02.442.11.7301, and the Russian Foundation for Basic Research (Grants 06-03-32096, 06-03-97000, and 06-03-42725). This work has been performed in the framework of the program ‘Innovative Education Environment in a Classic University’ of the St. Petersburg State University (National project ‘Education’).