

1 Introduction
During work on asymmetric catalytic and heterogeneous hydrogenation of α-ketoesters over Al2O3-supported platinum using a chiral modifier (the Orito reaction [1], known for about 30 years [2]), we found [3] that erythro-piperidyl naphthyl alcohol 1 was, in such reactions, a chiral catalyst almost as efficient as natural erythro-cinchonidine 3. Different routes toward enantiopure aryl piperidyl alcohol 1 were explored [4] but, until now, no efficient and short asymmetric synthesis have been found and resolution of the amino alcohol itself or of the precursor aryl-pyridyl methanol is still necessary.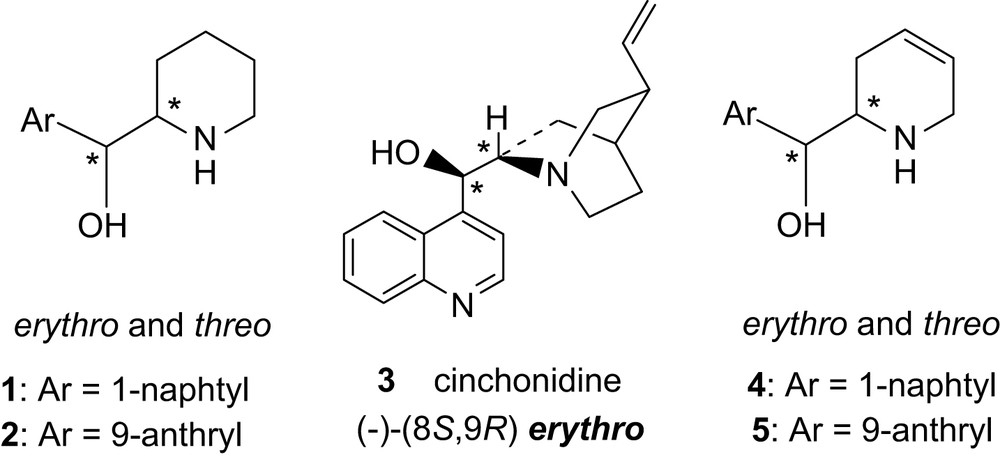
An enantioselective synthesis from d-mannitol of enantio and diastereopure aryl-[1,2,5,6-tetrahydro-pyridinyl] methanols 4 and 5 [5], precursors of 1 and 2 [5], has thus been envisaged and is described here as well as the direct use of these modifiers for asymmetric heterogeneous hydrogenation of ethyl pyruvate.
2 Synthesis
The first five steps of the synthesis are presented in Scheme 1. The starting protected glyceraldehyde 6, Scheme 1, obtained from d-mannitol [6], must be freshly prepared and immediately used to prepare the allyl imine 7 [7]. The addition of allyl Grignard reagent onto 7 proceeds smoothly to give, at −78 °C, compound 8 as an 89/11 mixture of both diastereomers (according to 1H NMR). After Boc protection, the major diastereomer of compounds 9 (9t) was separated by chromatography.
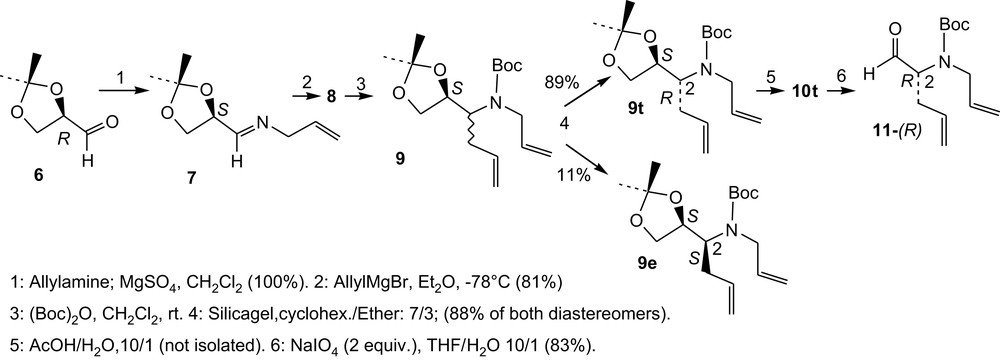
Synthesis of 4H, 5H and 4Me; steps 1–6.
Compound 9t-(1S,2R) was then transformed into aldehyde 11-(R) in two steps and 41% overall yield from 6, Scheme 1.
The (2R)-absolute configuration of compound 11 was assigned using 9t. A single crystal of 12t, obtained in two steps from 9t (Scheme 2), was analyzed by X-ray diffraction and, as seen in Fig. 1, shown to be the threo isomer; therefore, the configuration at C1 (C14 or C2 in Fig. 1) being (1S), the configuration at C2 (C15 or C3 on Fig. 1) is (R). Moreover, 12t (Scheme 2) was also transformed into the known (R)-(+)-aldehyde 13.

Chemical correlation for assignment of the R-configuration at C2.
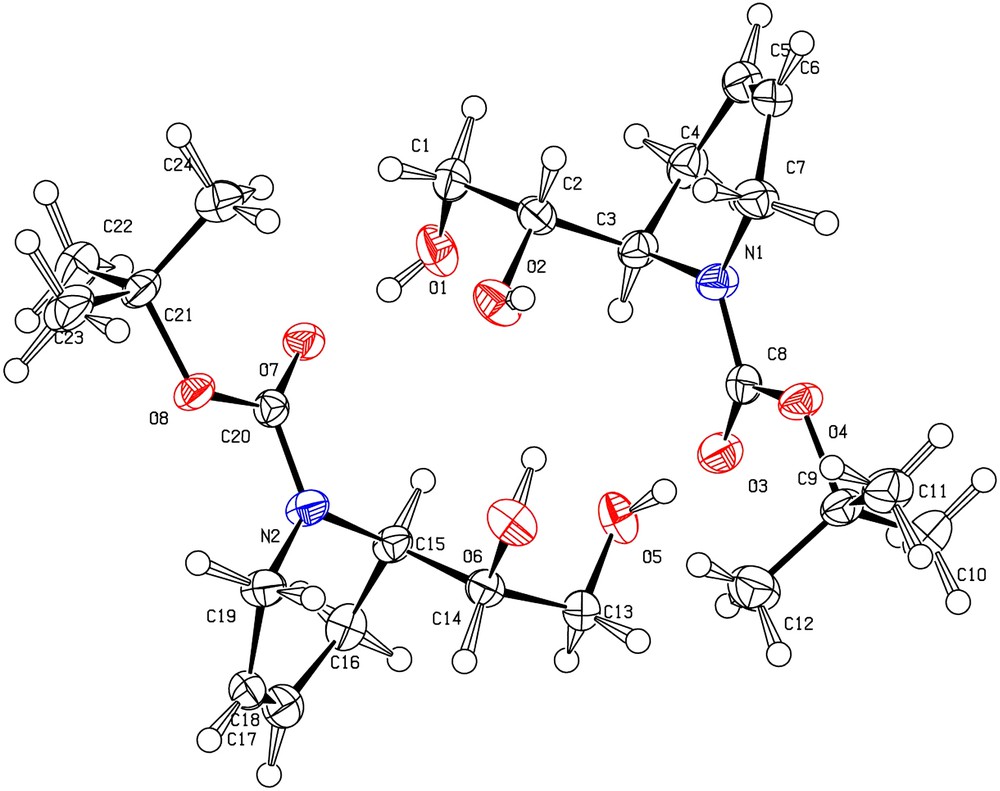
Asymmetric unit of the crystal structure of 12t with labelling scheme. The ellipsoids enclose 50% of the electronic density.
The next three steps are given in Scheme 3. Addition of naphthyl Grignard reagent onto (R)-11 provides higher yield (14a = 83%) than addition of the anthryl Grignard reagent (14b = 55%). Mixture of diastereomers was obtained and separated by chromatography. In both cases, the erythro isomer (II) is major (cf. below for assignment). Formation of the six-membered ring was performed using the Grubbs (1st generation) catalyst [8] and provided satisfying yields in both the cases (94% of 15a-II and 85% of 15b-II).
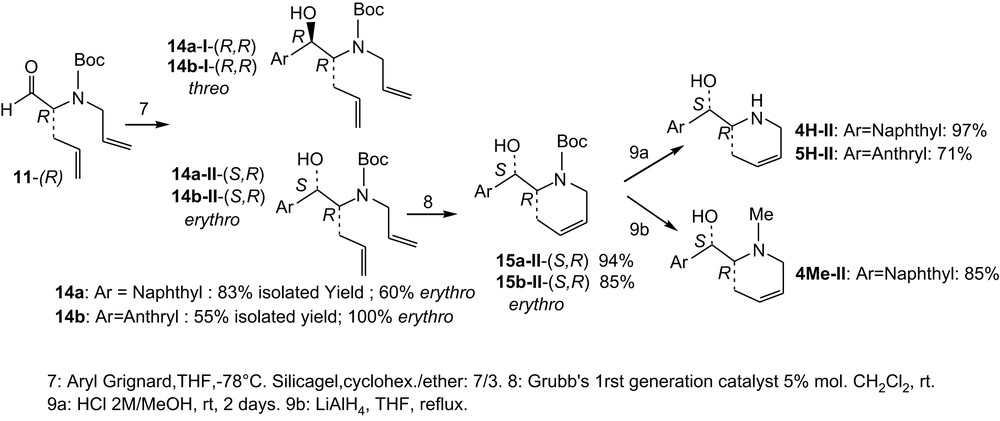
Synthesis of 4H, 5H and 4Me; last three steps.
Finally, deprotection of 15a-II and 15b-II provided the desired diastereo and enantiopure amino alcohols 4H-II and 5H-II in good to satisfying yields (97% and 71%, respectively). LiAlH4 reduction of 15a-II provided diastereo and enantiopure 4Me-II in good yield, but reduction of 15b-II did not proceed under similar conditions and 5Me-II was not obtained.
3 Determination of the erythro structure of 4H-II and 5H-II
The erythro structure of 4H-II and 5H-II has been determined using NMR. In previous studies concerning enantiopure erythro and threo amino alcohols 16, some of us have shown by VCD [4a] that the diastereomer determined as being erythro by NMR (on the basis of the smaller value of the 3J coupling constant: 4 Hz compared to 6 Hz for the threo isomer) was indeed erythro. The known amino alcohols 17 [4a] exhibit also a smaller coupling constant for the erythro isomer than for the threo isomers (5 Hz versus 7.5 Hz) as well as 18 [4b] (7 Hz versus 9.5 Hz). Therefore, compared, respectively, to 16, 17 and 18, 4H-II, having a coupling constant of 3 Hz, and 5H-II, having a coupling constant of 7 Hz, were assigned the erythro structure.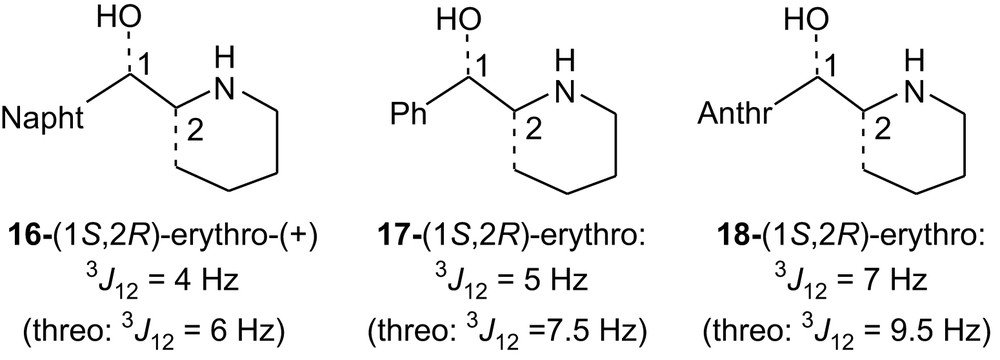
4 Heterogeneous hydrogenation of ethyl pyruvate
To test the amino alcohols 4H-II, 5H-II and 4Me-II, we have chosen a well-defined catalysts from Engelhard: 5% Pt/Al2O3 – 4759 (E4759). The results are summarized in Table 1.
Hydrogenation of pyruvate in AcOH using E4759 (Engelhard) at room temperature, S/Ca = 100, S/Mb = 2650, of ethyl pyruvate concentration = 3 mol/L, 40 bar, 2 h
| Modifier | Conversion, % | e.e., %) | Lactate (R/S) | o.p.% | Absolute configuration |
| (−)-(9R,8S)-CDc | 100 | 87 | 93.5/6.5 | 87 | R |
| (+)-(1S,2R)-4H | 100 | 75 | 12.5/87.5 | 72 | S |
| (+)-(1S,2R)-5H | 88 | 46 | 27/73 | 45 | S |
| (+)-(1S,2R)-4Me | 80 | 39 | 30.5/69.5 | 40 | S |
| (+)-(1S,2R)-1d | 100 | 72 | 14/86 | – | S |
| (−)-(1R,2S)-1 | 100 | 73 | 86.5/13.5 | 73 | R |
a S/C = substrate/catalyst.
b S/M = substrate/modifier.
c Numbering of atoms are different: the C9 in CD is called C1 in 4H, 5H and 4Me idem: C8 in CD = C2 in 4H, 5H and 4Me.
d cf. Ref. 3c.
The enantiomeric excesses of the ethyl lactate produced were determined by gas chromatography on a Cyclodex-B capillary column (30 m) using a HP 5890 GC-FID instrument; the e.r. values were reproducible within 1% (for the same sample).
The most important feature of these hydrogenations is the high enantioselectivity obtained for the ethyl lactate with erythro-4H (75% e.e.), comparable to that obtained with the saturated homologue erythro-1 (Table 1, compare row 2 with rows 5 and 6). It could be postulated that the double bond of 4H is hydrogenated during the reaction (just as the vinyl group in cinchonidine) but without drastic consequence on the outcome of the reaction.
Therefore, compound 4H can be used directly (no need of a preliminary hydrogenation into erythro-1).
Compound 5H with an anthryl group instead of a naphthyl group (4H) provided lower enantioselectivity and lower yield under identical conditions (Table 1, compare rows 3 and 4).
It appears also that introduction of a methyl onto nitrogen1 (4Me) decreases the enantioselectivity with 39% e.e. for the isolated ethyl lactate using 4Me instead of 75% e.e. using 4H (Table 1, compare rows 2 and 4).
It is worth noting that the enantioselectivities determined by NMR (e.e.%) and from the optical rotation of the lactate (o.p.%) are almost identical (Table 1, compare column 2 and 4).
It must be noted that the reproducibility from one operator to another is about ±0.5 (Table 1, compare rows 5 and 6).
5 Conclusion
The new modifier erythro-4H is as efficient as erythro-1 (compared rows 1 and 5 in Table 1) and is obtained in enantiopure form from inexpensive d-mannitol in 10 easy steps. Moreover, both enantiomers of 4H can been obtained from d-mannitol as it is possible, in running step 2 at room temperature, to obtain a 1/1 mixture and to isolate the erythro 9e in satisfying yield, thus providing the (S)-aldehyde 11. Until now, erythro-4H and erythro-1 are the only two synthetic chiral modifiers available under both enantiomeric forms, which provide enantioselectivities almost as high as those obtained with natural cinchonidine [9], for which only one enantiomer is available.
It must be noted that previous conclusions that the anthryl group should provide more efficient compound have to be reconsidered, as, obviously, in this case the anthryl group is less efficient than the naphthyl: the e.e.% drops from 75% with erythro-4H (naphthyl) to 46% with erythro-5H (anthryl). Moreover, N-methyl substitution (trisubstituted nitrogen as in cinchonidine) leads also to a drop in the e.e.% (from 75% with 4H to 39% with 4Me).
6 Experimental section
1H (400 MHz and 300 MHz) and 13C (75.4 MHz) NMR spectra were recorded on a Bruker AC 400 and AC 300 spectrometers with CDCl3 as solvent. Chemical shifts (δ) are given in ppm downfield from TMS as an internal standard and the coupling constants (J) are given in Hz. Optical rotation were determined on a PerkinElmer 241 MC polarimeter. TLC was performed on Merck's glass plates with silica gel 60 F254. Silica gel Si 60 (40–60 μm) from Merck was used for the chromatographic purifications.
The selected crystal has been mounted on a Nonius Kappa-CCD area detector diffractometer (Mo Kα, λ = 0.71073 Å). The cell parameters were determined from reflections taken from one set of 10 frames (1.0° steps in Φ angle), each at 20-s exposure. The structures were solved using direct methods (SIR97) and refined against F2 using the SHELXL97 software. The absorption values were not corrected. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were found by Fourier differences.
Crystallographic data (excluding structure factors) have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44) 1223 336 033; e-mail: deposit@ccdc.cam.ac.uk).
6.1 Crystal data and structure refinement details
Colourless crystal; crystal dimension: 0.1 × 0.15 × 0.20 mm3; C12H21NO4; M = 243.30 g mol−1; monoclinic; space group P21; a = 6.012(1) Å; b = 22.091(5) Å; c = 9.798(2) Å; β = 90.66(5)°; Z = 4; Dc = 1.242 g cm−3; μ(Mo Kα) = 0.092 mm−1; a total of 6573 reflections; 2.27° < θ < 30.04°, 3894 independent reflections with 3122 having I > 2σ(I); 475 parameters; final results: R1 = 0.0404; wR2 = 0.0860, Goof = 1.055, maximum residual electronic density = 0.159 eÅ−3.
6.2 General procedure for the synthesis of 9t and 9e (steps 1–4)
To a solution of aldehyde 6 (7.310 g, 54.8 mmol) in anhydrous CH2Cl2 (250 mL) were added anhydrous MgSO4 (25 g) and then, dropwise, allyl amine (10.3 mL, 137 mmol). After stirring for 1.5 h at room temperature, MgSO4 was filtered out. The solvent was evaporated and imine 7 was obtained (9.32 g, yield = 99%). Imine 7: [α]D = +52.0 (c = 0.95, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 1.37 (s, 3H, CH3), 1.42 (s, 3H, CH3), 4.02 (dd, 1H, 2J = 9, 3J = 6), 4.05 (m, 2H), 4.17 (dd, 1H, 2J = 9, 3J = 5.5), 4.56 (ddd, 1H, OCH, 3J = 6, 3J = 5.5, 3J = 5), 5.18 (m, 2H, CHCH2), 5.93 (m, 1H, CHCH2), 7.65 (d, 1H, CHN, 3J = 5). Anal. for C9H15NO2. Found: C, 63.51; H, 9.11. Calcd: C, 63.88; H, 8.93.
To a solution of imine 7 (6.8 g, 40 mmol) in anhydrous Et2O (100 mL) was added dropwise and at −78 °C a solution of allyl MgBr (80 mL, 80 mmol) in anhydrous Et2O (40 mL). The mixture was stirred for 1 h at −78 °C (a precipitate formed rapidly). After addition of a saturated solution of NH4Cl (50 mL), the organic and aqueous phases were separated and the aqueous phase extracted with Et2O (3 × 20 mL). The organic phases were joined, dried over Na2SO4, filtered and the solvent evaporated. Compound 9 was obtained as a diastereomeric mixture (6.92 g, yield = 82%, 8I/8II = 89/11). Compound 8I + 8II: yellow oil. 1H NMR (CDCl3, 400 MHz) signals of I and II overlapped δ: 2.13 (m, 1H), 2.28 (m, 1H), 3.31 (m, 1H), 3.35 (m, 1H), 3.73 (m, 1H), 4.02 (m, 1H), 4.09 (m, 1H), 5.08–5.22 (m, 4H), 5.85 (m, 2H); signals not overlapped δ: 1.36 (s, 3H, CH3, II), 1.35 (s, 3H, CH3, I), 1.40 (s, 3H, CH3, I), 1.41 (s, 3H, CH3, II), 2.72 (q, 1H, I, 3J = 3J = 3J = 6), 2.78 (q, 1H, II, 3J = 3J = 3J = 6).
To a solution of 8(I+II) (4.9 g, 23.5 mmol) in CH2Cl2 (90 mL) was added a solution of (Boc)2O (6.15 g, 28 mmol) in CH2Cl2 (10 mL), stirring was maintained for 12 h at room temperature, then CH2Cl2 was evaporated and the crude product was purified by silica gel chromatography (pentane/ether = 8/2). The major diastereomer 9t was isolated in 88% yield.
[α]D = +36.0 (c = 0.69, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 1.33 (s, 3H, CH3), 1.40 (s, 3H, CH3), 1.48 (s, 9H, t-Bu), 2.28 (m, 1H), 2.45 (m, 1H), 2.70 (m, 1H), 3.60 (m, 1H), 3.83 (m, 1H), 3.99 (m, 1H), 4.20 (m, 2H), 5.06 (m, 4H, CHCH2), 5.74 (m, 2H, CHCH2). Anal. for C17H29NO4. Found: C, 65.32; H, 9.52. Calcd: C, 65.56; H, 9.38.
6.3 General procedure for the synthesis of 11-(R) (steps 5 and 6)
A solution of 9t (2 g, 6.43 mmol, 1 equiv) in AcOH (30 mL) and H2O (3 mL) was stirred for two days at room temperature. After extraction with CH2Cl2 (5 × 20 mL), the organic phases were joined, washed with a 15% NaOH solution (15 mL) and then with a saturated solution of NaCl (3 × 10 mL). The resulting organic phase was dried over Na2SO4, and, after filtration, the solvent was evaporated. After purification on a silica gel column (hexane/ether = 1/4), 1.62 g of 10t was obtained (yield = 93%). [α]D = +10.0 (c = 1.01, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 1.46 (s, 9H, t-Bu), 2.35 (m, 2H), 3.52 (m, 2H), 3.76 (m, 3H, OCH2 + NCH), 4.13 (m, 1H, OCH), 5.13 (m, 4H, CH2), 5.77 (m, 2H, CH).
To a solution of 10t (1.33 g, 4.90 mmol) in a mixture THF (10 mL)/H2O (1 mL) were added NaIO4 (2 equiv) by portions for over 20 min. After stirring for 1 h at room temperature, the crude mixture was filtered over Celite and the solvent evaporated to give 0.96 g of 11-(R) (yield 82%), which was used rapidly for the next step. 1H NMR (CDCl3, 300 MHz) of a sample purified over silica gel (hexane/ether = 7/3) δ: 1.43 (s, 9H, t-Bu), 2.50 (m, 1H), 2.78 (m, 1H), 3.57 (m, 2H), 4.15 (m, 1H), 5.10 (m, 4H), 5.80 (m, 2H), 9.55 (d, 1H, 3J = 9).
6.4 General procedure for the synthesis of 15a-II and 15b-II (steps 7 and 8)
To a suspension of Mg (5.4 mmol) in anhydrous THF (4 mL) were added dibromoethane (0.1 mL) and then, dropwise, a solution of the desired bromide (1-naphthyl or 9-anthryl) in anhydrous THF (5.4 mmol in 10 mL); after addition the mixture was stirred under reflux until Mg has totally disappeared. Then the mixture was cooled to room temperature (all under argon) and a solution of 11-(R) in anhydrous THF (2.7 equiv in 6 mL) was added dropwise under stirring. The reaction was monitored by CCM and work-up was done when no aldehyde was detected. After addition of NH4Cl (saturated solution), the THF was evaporated and the crude product extracted with Et2O (5 × 15 mL). The combined organic phases were dried over Na2SO4, filtered out, the solvent evaporated and the crude products purified by chromatography over silica gel to give 14a-II and 14b-II.
Compound 14a-II: hexane/ether = 1/1. 1H NMR (CDCl3, 300 MHz): δ 1.49 (s, 9H, t-Bu), 2.72 (m, 1H), 2.95 (m, 1H), 3.56 (m, 3H), 5.05 (m, 4H), 5.60 (m, 2H), 5.63 (m, 1H), 7.48 (m, 3H), 7.75 (m, 1H), 7.87 (m, 2H), 8.03 (m, 1H). Anal. for C23H29NO3. Found: C, 75.19; H, 8.09. Calcd: C, 75.17; H, 7.95.
Compound 14b-II: hexane/ether = 1/1. 1H NMR (CDCl3, 300 MHz): δ 1.40 (s, 9H, t-Bu), 2.58–3.56 (m, 3H), 4.47 (m, 1H), 4.63 (m, 1H), 5.08 (m, 4H), 5.78 (m, 2H), 6.68 (m, 1H), 7.47 (m, 4H), 7.98 (m, 2H), 8.40 (s, 1H), 8.75 (bm, 2H). Anal. for C27H31NO3. Found: C, 77.50; H, 7.55. Calcd: C, 77.66; H, 7.48.
To a solution of first-generation Grubbs catalyst (0.079 g, 0.097 mmol) in anhydrous CH2Cl2 (5 mL) was added dropwise a solution of 14a-II (or 14b-II) in anhydrous CH2Cl2 (1.93 mmol in 40 mL). After stirring for 2 h at room temperature, the solvent was evaporated and the residue purified by chromatography (silica gel, hexane/ether = 1/1) to give 15a-II (94% yield) or 15b-II (85% yield) as colourless oils.
Compound 15a-II: 1H NMR (CDCl3, 300 MHz): δ 1.43 (s, 9H, t-Bu), 2.32 (m, 2H), 2.66 (m, 2H), 4.13 (m, 1H), 4.80 (m, 1H), 5.78 (m, 1H), 5.89 (m, 1H), 7.50 (m, 4H), 7.83 (m, 1H), 7.86 (m, 1H), 8.39 (d, 1H, 3J = 8). Anal. for C21H25NO3. Found: C, 74.14; H, 7.75. Calcd: C, 74.31; H, 7.42.
Compound 15b-II: 1H NMR (CDCl3, 300 MHz): δ 1.44 (s, 9H, t-Bu), 2.46 (m, 2H), 3.03 (m, 1H), 3.78 (m, 1H), 5.33 (m, 1H), 5.73 (m, 1H), 5.99 (m, 1H), 6.23 (m, 1H), 7.50 (m, 4H), 7.99 (m, 2H), 8.41 (s, 1H), 8.70 (m, 2H). Anal. for C25H27NO3. Found: C, 76.82; H, 7.05. Calcd: C, 77.09; H, 6.98.
6.5 Preparation of 4H-II, 5H-II and 4Me-II (steps 9a and 9b)
A solution of 15a-II or 15b-II (0.6 mmol) in MeOH (20 mL) and 2 M HCl (20 mL) was stirred overnight at room temperature. After cooling at 0 °C, a 2 M NaOH solution was added until pH 8, then CH2Cl2 (50 mL) was added and the organic phase recovered. The aqueous phase was then further extracted with CH2Cl2 (10 × 15 mL), the combined organic phases were dried over Na2SO4, filtered and the solvent evaporated providing 4H-II (97% yield) or 5H-II (71% yield).
Compound 4H-II: [α]D = +29.4 (c = 0.36, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 1.55 (m, 1H), 2.34 (m, 1H), 3.38 (dt, 1H, 2J = 11, 3J = 3.5 and 3.5), 3.54 (b, 2H), 5.00 (bs, 2H, NH + OH), 5.63 (m, 1H), 5.67 (m 1H), 5.90 (d, 1H, 3J = 3.5), 7.47 (m, 3H), 7.78 (m, 2H), 7.85 (m, 1H), 8.09 (d, 1H, 3J = 7). 13C NMR (CDCl3, 75 MHz): δ 24.0, 44.5, 56.5, 71.0, 123.0, 123.9, 125.3, 125.6, 126.2, 127.9, 128.8, 130.2, 133.6, 136.5. Anal. for C16H17NO. Found: C, 80.03; H, 7.31. Calcd: C, 80.30; H, 7.16.
Compound 5H-II: [α]D = not determined because of too much light absorption. 1H NMR (CDCl3, 300 MHz): δ 1.30 (m, 1H), 2.54 (m, 3H), 3.21 (b, 2H, NH + OH), 3.78 (m, 1H), 5.66 (m, 1H), 5.84 (m, 1H), 6.22 (d, 1H, 3J = 7), 7.47 (m, 4H), 8.01 (m, 2H), 8.45 (s, 1H), 8.77 (b, 2H). 13C NMR (CDCl3, 75 MHz): δ 23.5, 43.0, 55.6, 70.0, 124.2, 125.7, 128.3, 129.2, 130.2, 131.6, 132.5. Anal. for C20H19NO. Found: C, 82.81; H, 6.87. Calcd: C, 83.01; H, 6.62.
To a solution of 15a-II (0.66 mmol) in anhydrous THF (10 mL) was added by portion LiAlH4 powder (∼0.1 g, 1.2 equiv), and the mixture was stirred under reflux for 24 h. The work-up was done through addition of H2O (0.1 mL), NaOH 15% (0.1 mL) and H2O (0.3 mL); after stirring until the precipitate formed becomes powdered and white (∼2 h), the solution was filtered out and the precipitate carefully rinsed with Et2O. The organic phases were joined and the solvent evaporated to provide 4Me-II (85% yield).
Compound 4Me-II: [α]D = +33.3 (c = 0.60, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 2.35 (m, 2H), 2.76 (s, 3H, Me), 2.87 (m, 1H), 3.07 (m, 1H), 3.65 (b, 1H, OH), 3.78 (m, 1H), 5.59 (m, 2H), 6.09 (d, 1H, 3J = 3), 7.50 (m, 3H), 7.81 (m, 3H), 7.99 (m, 1H). 13C NMR (CDCl3, 75 MHz): δ 24.0, 41.8, 61.7, 65.4, 67.6, 122.5, 123.2, 124.1, 125.0, 125.2, 125.4, 125.8, 127.6, 129.0, 133.6, 136.0. Anal. for C17H19NO. Found: C, 80.38; H, 7.93. Calcd: C, 80.59; H, 7.56.
6.6 Synthesis of 12t and 12e (Scheme 2)
Deprotection of 9t and cyclisation have been done using the procedures described above.
Compound 12t: [α]D = −38.0 (c = 0.6, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 1.47 (s, 9H, t-Bu), 2.07 (m, 1H), 2.49 (m, 1H), 3.53–3.73 (m, 4H), 4.19 (m, 1H), 4.45 (m, 1H), 5.72 (m, 2H). Anal. for C12H21NO4. Found: C, 59.03; H, 8.96. Calcd: C, 59.24; H, 8.70.
Compound 12e: [α]D = +29.0 (c = 0.67, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 1.47 (s, 9H, t-Bu), 2.17 (m, 1H), 2.35 (m, 1H), 3.50–3.70 (m, 4H), 4.22 (m, 1H), 4.30 (m, 1H), 5.65 (m, 2H).
6.7 Synthesis of 13-(R) (Scheme 2)
Hydrogenation of the double bond was done on 12t using H2/Pd/C: in a 100-mL autoclave were introduced 12t (1.11 g, 4.57 mmol), AcOEt (25 mL) and the catalyst Pd/C (50 mg). After having purged three times (successive vacuum and H2), the H2 pressure was adjusted to 20 bar and the mixture was maintained under this H2 pressure under stirring for 2 h. After filtration, the catalyst was rinsed with AcOEt, the solvents were joined and then evaporated to provide the desired hydrogenated compound (1.007 g, 90% yield), which was directly transformed into the desired known aldehyde 13 (90% yield), using the same conditions as for preparation of 11-(R).
Compound 13-(R) (Ref. [10]): [α]D = +49.0 (c = 0.6, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 1.27 (m, 2H), 1.47 (s, 9H, t-Bu), 1.62 (m, 3H), 2.11 (m, 1H), 2.89 (bm, 1H), 3.93 (m, 1H), 4.56 (m, 1H), 9.57 (s, 1H). Anal. for C11H17NO3. Found: C, 62.28; H, 8.32. Calcd: C, 62.54; H, 8.11.
6.8 Heterogeneous hydrogenation of ethyl pyruvate
All heterogeneous hydrogenations have been conducted with a well-defined catalyst from Engelhard: 5% Pt/Al2O3 – 4759 (E4759). AcOH was used as solvent, the reactions were run at room temperature and under 40 bar H2, stirring was maintained at ∼500 rpm and the pyruvate, purchased from Aldrich, was distilled before use. Commercial cinchonidine (CD) was used as a reference.
Acknowledgments
We are grateful to Ministère de l'enseignement supérieur et de la recherche for a grant (MNERT) to KA.
1 A methyl group was introduced to get closer to cinchonidine (CD) where the nitrogen atom is trisubstituted.