

1 Introduction
The 20th century was the era of organic media in synthetic chemistry, which contributed immensely to the flourishing of chemistry science and to the today's human life (welfare in peaceful times and, ill-being and insecurity in war-times). New chemical products and novel synthetic pathways leading to coveted products have been the goal of chemists. Media were always sought to achieve such an objective. Nowadays, it is known that water was already the medium employed in synthesis more than a century ago, but once the organic solvents emerged and were found to exhibit better performance because of the ensued miscibility of the reaction components, that is a homogeneity, water was promptly dismissed. Indeed, a better substitute to water was highly polar organic solvents such as dimethyl formamide (DMF), dimethyl acetamide (DMAc), and dimethyl sulfoxide (DMSO). A common sense was that the course of a homogeneous reaction occurs more neatly than a heterogeneous one. Unequivocally, the success of an organic reaction is partly due to its organic solvent-based liquid phase, obeying to the common belief: corpora non agunt nisi soluta, that is, substances do not interact unless dissolved, and like needs (dissolves) like. A torrent of products coupled with a myriad of chemical reactions was the outcome of using organic solvents. The blossoming of chemistry from its simple synthesis of urea, a simple molecule, to the multi-step synthesis of molecules of architecture-defying structures such as vitamin B12 is partly due to the adequately chosen organic solvent [1,2]. Thanks to the tireless and seemingly endless research endeavor, water was resuscitated and retrieved its unshakable place as a solvent for organic synthesis in 1980 by Professor Ronald Breslow at Columbia University; in their report, Rideout and Breslow [3] described the cycloaddition of cyclopentadiene (CP) with methyl vinyl ketone (MVK) in water and found its rate to be unexpectedly enhanced by a factor of 722 compared with the reaction in isooctane (kwater/kisooctane = 722 at 20 °C). Also, in the same work, the rate constant of the Diels–Alder reaction of cyclopentadiene with acrylonitrile (ACN) in water was 31-fold that in isooctane at 30 °C. This work was indeed a landmark in the history of chemistry. And, accordingly, the popularization of Professor Ronald Breslow and his pioneering work within the chemists’ community resembles, to a great extent, that of Professor Bruce Merrifield and his fascinating work on the solid-phase peptide synthesis (SPPS) using polymeric support; that is, Breslow's name1 is being compulsorily and inevitably invoked in every paper dealing with water-borne organic synthesis.
Quite enticingly, the hydrophobic character of water played an unexpected role in accelerating the Diels–Alder reaction run by Breslow; the hydrophobic effect provides a driving force for association of the molecules of organic reactants. With no delay, this unprecedented finding sparked a great interest to synthetic chemists. Indeed, they rushed on redoing some reputed reactions and others in water and several accounts on water-borne organic syntheses have been reported [4–13]. In nearly all studied cases [9,11], enhancement of the rate was observed. To cite but a few, the studied reactions were the Claisen rearrangement, the aldol condensation, the benzoin condensation, the Wittig reaction, and the [14] Mannich type reaction. This rate of acceleration of organic reactions in aqueous media was ascribed to one or a combination of the following factors and/or phenomena [15]: the high cohesive energy density of water, the high internal pressure within the medium, the hydrogen-bonding ability, the hydrophobic packing of diene and dienophile in cycloaddition reactions, the hydrophobic vs. antihydrophobic effects, the micellar catalysis, the solvophobicity, and the solvent polarity. Polo et al. [16] advanced a theoretical explanation of the role hydrogen-donor solvents in hetero Diels–Alder reactions, namely between acetone and butadiene derivatives. Based on ELF (electron localization function) and NBO (natural bond orbital) analyses, it was elucidated that all solvents form stronger HB interactions (HBhydrogen bond) at the transition state, but only those presenting larger charge-transfer interactions, such as water, methanol, and chloroform, benefit more from the polarization of the carbonyl group than other solvents with less “covalent” character such as dichloromethane and chloromethane.
Recently, it was reported [17] that pyridyl group of RNA bearing 5-(4-pyridylmethyl) carboxamide uridines showed also an accelerating hydrophobic effect on Diels–Alder reaction between anthracene and maleimide derivatives with high turnover, a hydrophobic effect similar to that of water.
Should we not be perplexed about the fact that water is the actual medium of a countless number of in vivo reactions that occur with never any failings, albeit their organic nature? The success of these natural reactions and the synthetic ones infers that water is indubitably acting by its unique properties, that is its high cohesive energy density CED, (550 cal/mL), its high heat capacity (Cp = 4.18 J K−1 cm−1), and its large surface tension, hydrogen bonding ability, and redox stability.
In the present account, effort is expended to survey the water-borne cycloaddition reactions reported over the last two decades. Yet, the results prior to 1990 are equally important. This essay is intended to bring several results, with the emphasis on the latest ones, of the issue closer to the reader in a concise manner. The aqueous systems for cycloaddition reactions herein delineated are in one of the following forms: pure water solutions, on-water media (suspension media), aqueous media (adjunction of an organic solvent), aqueous micellar solution (presence of surfactant), and microemulsion media. One of the features of the on-water reaction is the easy isolation of the product by filtration.
2 Diels–Alder reactions
2.1 General considerations
The Diels–Alder reaction (DA), a [4π + 2π] cycloaddition, is one of the most intensively studied organic reactions and the one affording six-membered products via carboncarbon bond making. Not only from a synthetic standpoint [18,19] is the DA important, but also from a theoretical aspect [20–23]. It was also the first organic reaction run in water by Otto Diels and Kurt Alder themselves in 1931 [24], and, from its retrieval in 1980 up to now, several reports on the issue have tremendously enriched the literature. Variations in rates and yields and variable extents of selectivity were clearly observed in DA reactions when carried out in solvents of different natures. The magnitude of rate and the selectivity of this DA reaction were thought to be ascribed to three factors: solvophobic effects, hydrogen bond donor (HBD) interactions, and dipolarity–polarizability. While the increase in reaction rate observed in aqueous reaction media was explained by the high solvophobic power (Sp) values of these media, it was imputed to HBD ability when the reaction was performed in fluorinated alcohols. In some media, solvent solvophobicity has also been invoked to explain the increase in endo/exo selectivity observed, and HBD power seemed to be the only factor affecting the regioselectivity.
Research has come up with the fact that even a slight amount of water may, in some cases, provoke Diels–Alder reactions to occur. For example, Windmon and Dragojlovic [25] employed a slight amount of water to speed up the solventless Diels–Alder reactions between cyclopentadiene 1 and seven dienophiles 2–8 shown in Scheme 1. It was postulated that, on a multi-gram scale, a minute quantity of water would help controlling the violent reaction in the case of very reactive reactants, particularly the cyclopentadiene; in some instances, some reactions are vigorous and explosive due to their high exothermicity.
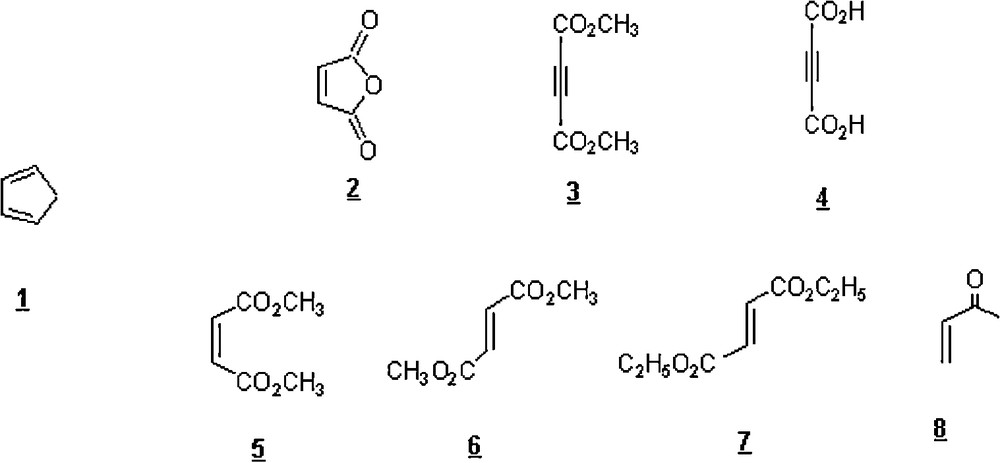
Not only did the addition of a small amount of water result in lower reaction temperature but also in an increased reaction rate and the formation of a higher purity product. The reaction time ranged from 10 to 60 min, depending on the dienophile. The yields of cycloadducts were good to excellent (74–95%). The reaction of CP 1 with maleic anhydride 2 in the presence of a few millilitres of water gave the cis-5-norbornene-2,3-dicarboxylic anhydride in 74% yield with an endo/exo ratio of 98/2.
Other distinguished features of aqueous Diels–Alder reactions were, in some cases, the positive effects of water-tolerant Lewis acids as catalysts and the use of surfactants creating micellar media. Indeed, a myriad of Lewis acids were employed and brought several outcomes of the Diels–Alder reactions and other cycloaddition reactions such as a stereoselectivity prevalence and an enhancement of the yields and kinetics [26]. Effects that surfactants may confer to the Diels–Alder reactions are: substrate solubilisation, concentration effect, cage effect, preorientation effect, microviscosity effect, and polarity and charge effects [27].
The thermodynamic facet of the aqueous Diels–Alder cyloaddition was tackled by Lubineau et al. [28], focusing on the following diene/dienophile pairs: cyclopentadiene (CP)/buten-2-one, 1-β-d-glucopyranosylbuta-1,3-diene (GlcOBD)/ buten-2-one, and 1-methoxybuta-1,3-diene (MeOBD)/ buten-2-one. It was disclosed that the rate enhancement of the aqueous Diels–Alder reaction was exclusively due to a decrease in the activation entropy in going from methanol or water-methanol mixtures to pure water for the three cases studied. The enthalpic parameter ΔH≠ was nearly unaffected by the nature of medium. It was then deduced that the rate enhancement for GIcOBD, which was not as large as that for CP, was more sensitive to the water structure than MeOBD, as reflected by a larger increase in the activation entropy ΔS≠ (9.63 vs. 7.3l kJ mol−1). This sensitivity was thought to be possibly due to the degree of binding of glucose moiety to water structure network, that is, to the degree of water–sugar interaction. The effect of additive was also assessed for the system (GlcOBD)/ buten-2-one and it was realized that methanol decreased the rate constant but glucose and saccharrose increased it. The latter increase was function of the additive concentration, that is, the higher the concentration, the higher the rate constant. For example, glucose and saccharrose with a concentration of 2 M increased the rate constant by 58 and 162%, respectively. These two hydroxyl-containing additives seemed to enhance greatly the hydrophobic interactions.
2.2 Homo Diels–Alder reactions of dienes
2.2.1 With maleimides
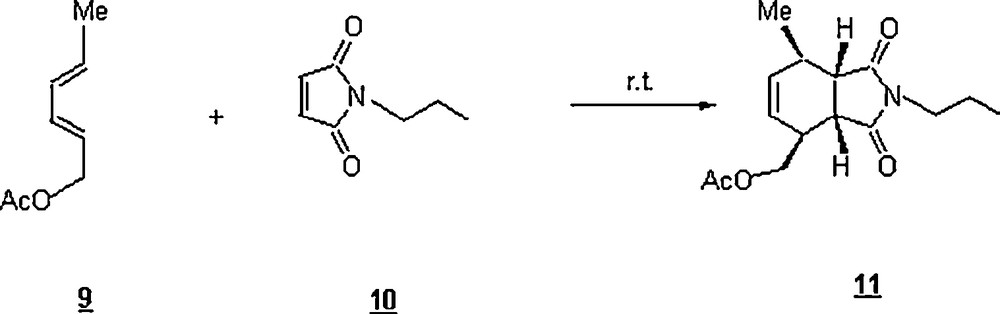
Organic reactions in water have mostly been performed using a co-solvent to engender some solubility. Today's status, the insolubility of organic reactants in water, once considered a drawback, turns out to be advantageously a leading factor for the success of organic reactions in pure water. In 2005, Sharpless coined these heterogeneous reactions as “on-water” reactions [29,30]. The “on water” method consists simply of stirring the reactant(s) with water to generate an aqueous suspension. He carried out some pericyclic reactions in aqueous suspension (on-water systems) and found both kinetics and yields extremely enhanced in most cases, compared with those in organic solvents. One of the many cycloaddition reactions was the reaction between trans, trans-2,4-hexadienyl acetate 9 and N-propylmaleimide 10 to give the cycloadduct 11 (Scheme 2). A protic organic solvent (MeOH) performed better than an aprotic one (toluene, CH3CN, DMF) as shown in Table 1.
Reaction conditions and yields of Scheme 2.
| Solvent | Time (h) | Yield (%) |
| Toluene | 144 (6 days) | 79 |
| CH3CN | >144 | 43 |
| MeOH | 48 | 82 |
| H2O | 8 | 81 |
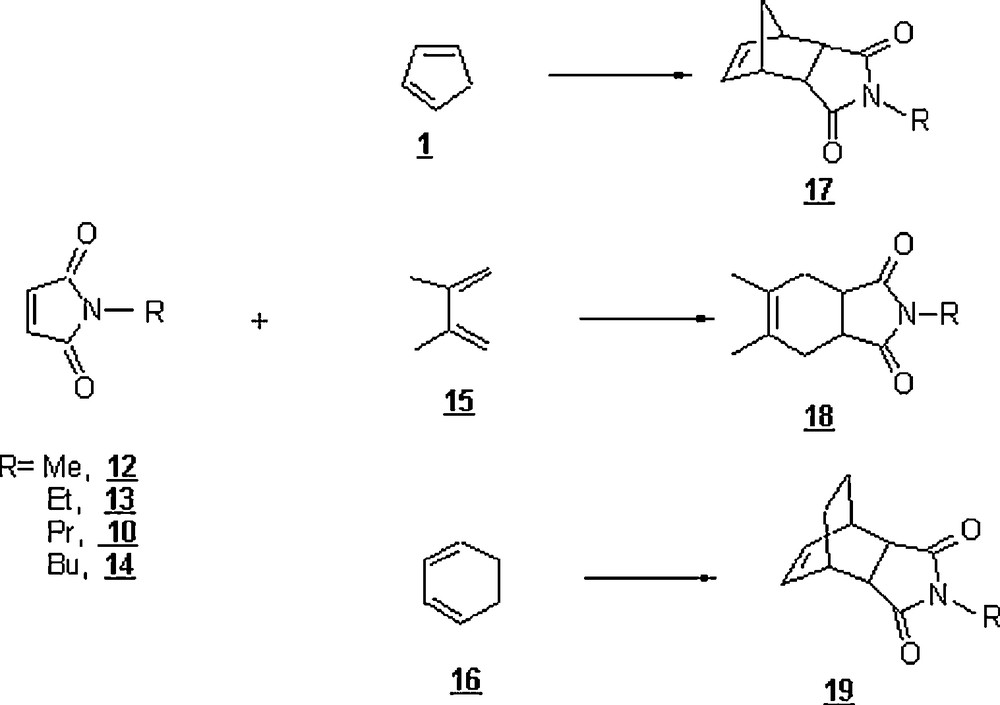
Meijer et al. [31] undertook a study of the kinetics of Diels–Alder reactions between alkyl maleimides as dienophiles and cyclopentadiene 1, 2,3-dimethyl butadiene 15, and cyclohexa-1,3-diene 16 as dienes in different media including water at 25 °C (Scheme 3). The objective was to assess the hydrophobicity of both the diene and dienophile on the course of the DA reaction. The solvents employed were n-hexane, acetonitrile, ethanol, 1-propanol, 1,1,1-trifluoroethanol (TFE), and water. In all cases, a dramatic acceleration of the second-order rate constant was remarked by using water, compared to the remaining solvents. Compared to other organic solvents, TFE enhanced the rate enormously; this finding strongly enlightened the notion of hydrogen-bonding interactions. The reaction rates of alkyl maleimides 10 and 12–14 with 15 were strikingly larger than those with 1 and 16 (Table 2). The rates with the latter maleimides were within the same range. These results indicated the effect of two methyl groups on the 2,3-dimethyl butadiene, 15. Also, the accelerations of reactions of alkyl maleimides with 15 revealed an approximately linear trend with increasing alkyl chain length of R. There was a hydrophobic interaction between the methyl groups of 15 and the N-alkyl group of maleimides during the activation process. In the cases of 1 and 16, which lack methyl substituents, this interaction was not possible, and extending the alkyl chain from ethyl to butyl did not result in an additional acceleration by water. The enhanced hydrophobicity near the reaction center of dienes 15 and 16 compared to 1 resulted in an increased water-acceleration of the Diels–Alder reactions of the former dienes with 12.
Relative rates Kwater/l-propanol of the Diels–Alder reactions of 1, 15, and 16 with 10, 12, 13 and 14.
| 1 | 15 | 16 | |||||||||
| 10 | 12 | 13 | 14 | 10 | 12 | 13 | 14 | 10 | 12 | 13 | 14 |
| 402 | 299 | 408 | 403 | 1684 | 1005 | 1447 | 1881 | 317 | 175 | 326 | 389 |
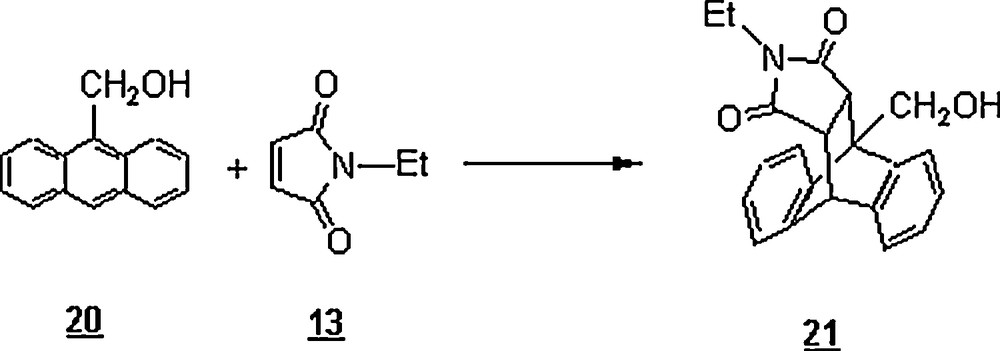
Breslow and Rizzo [32] studied the effects of LiCl, LiClO4, GnCl, and GnClO4 on the rate of the Diels–Alder reaction between anthracene-9-carbinol 20 and N-ethylmaleimide 13 in water at 45 °C as shown in Scheme 4. This study showed a remarkable rate acceleration in water compared to those in methanol (kwater/MeOH ≅ 68) and 2,2,4-trimethylpentane (kwater/TMP ≅ 28.75). The aqueous solution of 4.0 M of LiCl enhanced the rate notably (kwater/LiCl ≅ 2.2), whereas the solution of LiClO4 of same concentration slowed down the reaction compared to that in water ( ≅ 0.70), pointing to the salting-effect of LiClO4 due to the large ClO4− anion. This effect was even more pronounced for GnClO4 compared to the effect of GnCl ( ≅ 0.4; kGnCl/water ≅ 0.56).
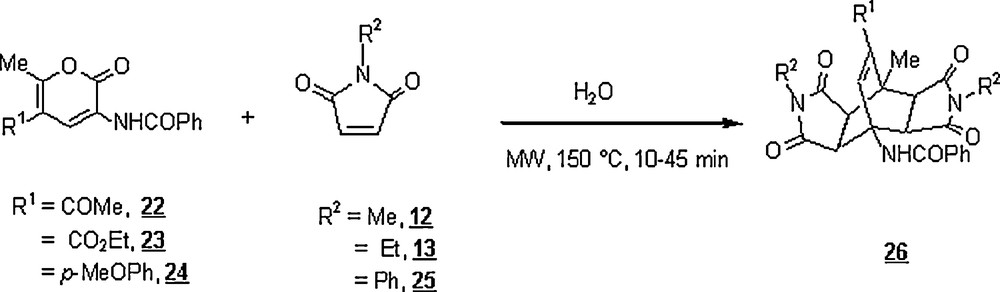
Kranjc et al. [33] developed a green methodology for making sterically constrained didehydroamino acid derivatives 26 containing bicyclo[2,2,2] oct-7-enes and ketone or ester functionality via microwave-assisted Diels–Alder reaction between 2H-pyran-2-ones 22–24 and maleimides 12, 13, 25 in water (Scheme 5). Albeit the water-solubility of diene 22–24 was low, microwave (MW) assistance promoted the reactions to completion at 150 °C for 30–45 min. The yields of the isolated cycloadducts were excellent, 81–94%. It is worth recalling that the conventional methodology required higher temperatures and longer times (boiling decalin, 90–120 min) to afford good yields. In a continuing effort, Kranjc et al. [34] extended the reaction to 2H-pyran-2-ones bearing furfuryl and thienyl groups at the methyl position with R1H. The yields of the corresponding fused-biyclo [2,2,2] oct-7-enes were 99 and 96%, respectively, at the same temperature and for longer reaction times (90 and 150 min).
Otto and Engberts, and Rispens and Engberts [27,35] made an effort to provide insights into the actual sites of the substrates of Diels–Alder reactions (diene and dienophile) within the micellar media. To this end, they worked out the DA reactions of cyclopentadiene 1, sorbyl alcohol 28, and sorbyltrimethylammonium bromide 29 with a series of N-substituted maleimides 12, 14, 27 (Scheme 6) in micellar media made of surfactants CTAB (cetyltetramethylammonium bromide) and SDS (sodium dodecyl sulfate), at 25 °C. The rates of reactions in water kw of 1, 28, and 29 with maleimides seemed to be independent on the substituent in maleimide, and increased in this order 1 > 28 > 29: kw (1) ≅ 2000 kw (28) ≅ (4600–8600) kw (29); kw (28) ≅ (25–43) kw (29).

The rates of reactions of 12 with the three dienes seemed unaffected by addition of SDS into water medium. In contrast, those for the dienophiles 14 and 27 increased with SDS concentration and reached maxima at a concentration slightly above its CMC (critical micelle concentration), and decreased afterwards. The rates with 27 were more importantly enhanced. The trend in micellar rate constants for both 1 and 28 was found in this order: km (27) > km (12) > km (14). On the other hand, the rates of reactions of 12 with 1 and 28 in CTAB micellar medium decreased with increasing CTAB concentration. This rate drop was more pronounced in SDS media, reflecting the fact that organic substrates generally bind stronger to CTAB micelles than to SDS micelles. The reaction in the micellar phase was thought to occur mainly in the region between the core and the Stern layer of micelle.
Engberts et al. [36] tackled the DA reaction of cyclopentadiene 1 with N-ethylmaleimide 13 (Scheme 7) in O/W microemulsions made of isooctane/AOT (sodium bis[2-ethylhexyl] sulfosuccinate)/water at 25 °C. The kinetics of the reaction was studied as a function of W = [H2O]/[AOT] ratio and AOT concentration. The results were that a marked catalytic effect relative to the rate constant in pure isooctane (ko = 2.80 × 10−2 M−1 s−1) was observed. The effect ranged from an increase in rate constant by a factor of 2 in microemulsions with a low water content (W = 2) to one-fifteenth for W = 35. It was found that upon increasing both W and [AOT], the amount of the incorporated reactants rose at the interface. For example, increasing the [AOT] from 0.103 to 0.617 led to an increase of 13 at the interface from 5–10% to 75–95%, depending on W; hence, an increase in the apparent rate constant, kapp. However, the rates in protic and polar solvents were greater than those in microemulsions, as can be noticed from the values of the rate constant (k, M−1s−1): 0.028 (isooctane), 0.0529 (hexane), 0.063–0.44 (interface of the AOT microemulsion), 0.126 (acetonitrile), 0.354 (ethanol), 0.411 (n-propanol), 5.04 (TFE) and 21.6 (water). The explanation advanced for such a discrepancy was that, at the interface of a microemulsion, virtually no hydrogen bonds were present and, therefore, no rate acceleration due to enforced hydrophobic interactions was expected. Moreover, the reactions were slower in organic solvents than in aqueous solutions. The term “enforced” was used to distinguish the hydrophobic bonding of the reactants from other hydrophobic interactions during the activation process [37].
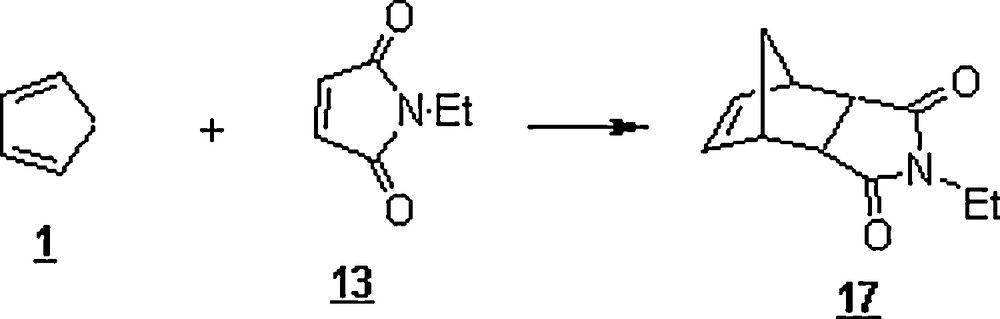
Rudolf et al. [38] studied the Diels–Alder reaction of (η5-cyclopentadienyl)M(CO)x(η1-N-maleimidato) complexes (M = Fe, Mo, W, x = 2 or 3) (32–34) with CP in water–alcohol mixture (3:1 to 9:1) and pure dichloromethane (DCM) (Scheme 8). The yields of isolated cycloadducts were in the range of 50–80% in both solvent systems. However, the reaction in aqueous alcohol was faster than that in DCM; for example, in the case of 32, the reaction in water was complete after 30 min, whereas the same reaction in DCM required 2 h.
The intramolecular DA reactions were also attempted in water by Engberts [39]. Indeed, water was also found to enhance the rate of the intramolecular reaction of N-furfuryl-N-alkylacrylamides 36, 37 (Scheme 9). The rate constants at 25 °C were measured for 36 as kDCM = 2.35 × 10−5 s−1 and kwater = 2.50 × 10−2 s−1, that is, a rate enhancement of 1064. However, for 37, the attempt in EtOH failed to afford the intramolecular adduct even for 3 weeks at 60 °C, but it was successful in water at this temperature with kwater = 2.05 × 10−5 s−1 at the same temperature.

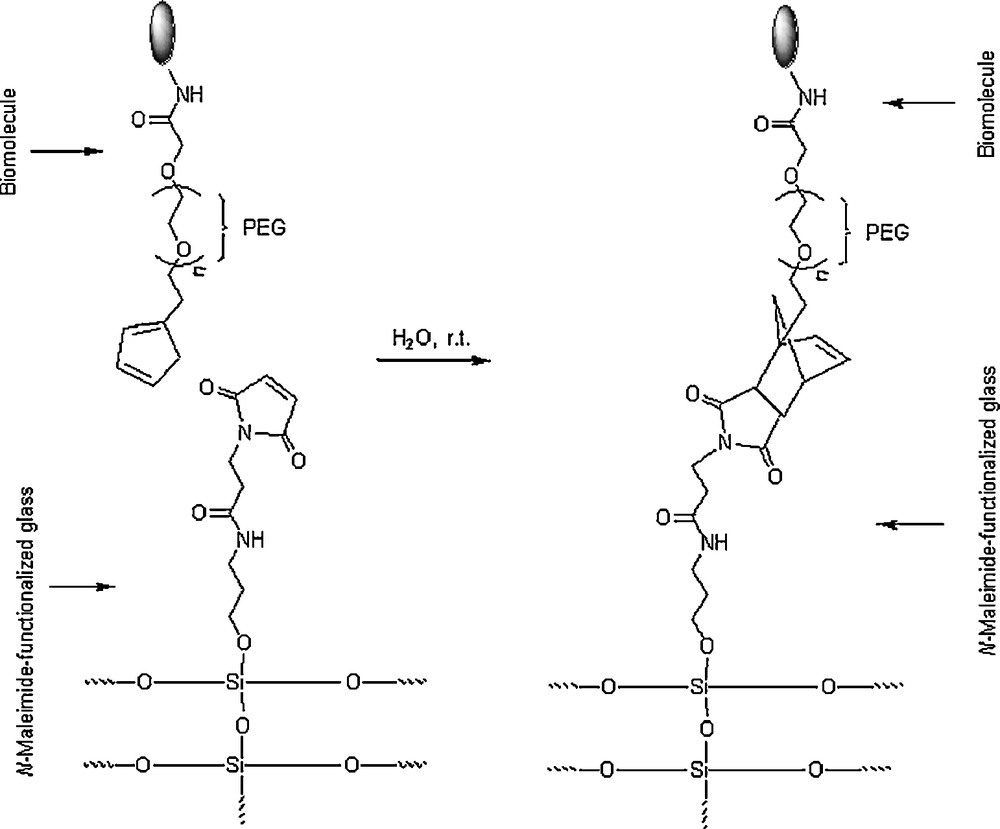
The Diels–Alder reaction using suitably N-substituted maleimides as dienophiles has been applied for the site-specific conjugation of biomolecules, because the reaction is fast, efficient and chemoselective in aqueous media. Sun et al. [40] ingeniously immobilized biomolecules onto a solid surface support via a linkage provided by water-borne Diels–Alder reaction. Surface functionalization of medical devices plays an important role in biological research and applications. The biomolecules (biotin, lactose and protein A) were immobilized via a cyclopentadiene-containing PEG (polyethylene glycol) as a spacer onto the surface of N-maleimide-functionalized glass as shown in Scheme 10. The Diels–Alder reaction was effected with no major difficulty by incubation of N-maleimide-functionalized glass with cyclopentadiene–PEG–biomolecule in water at room temperature for 12 h.
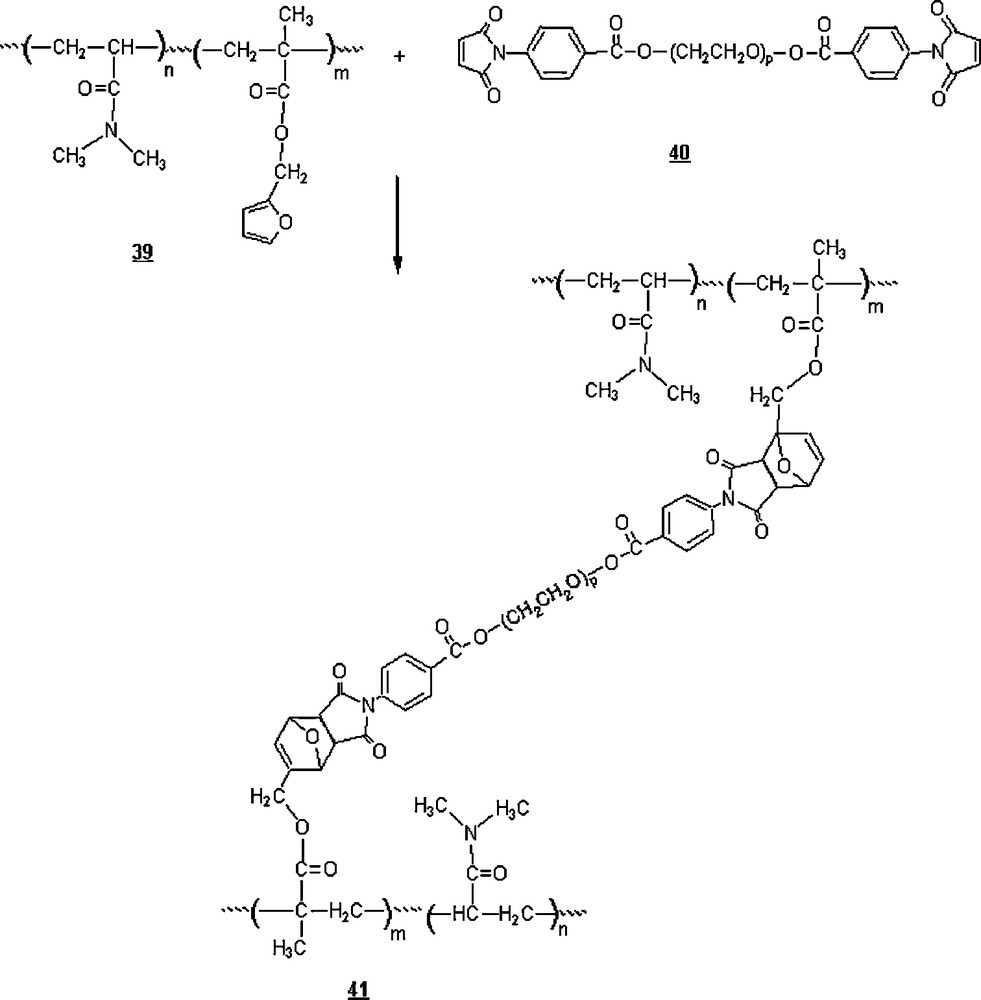
Wei et al. [41] applied the aqueous Diels–Alder cycloaddition approach to devise a polymeric network with hydrogel properties. In this work, a gelation in water of a mixture of the synthesized poly(N,N-dimethylacrylamide-co-furfurylmethacrylate) 39 and N-[4-(formyl polyethylene glycol ester) bis-maleimide 40 induced the formation of a polymeric hydrogel, a result of cross-linking via a Diels–Alder cycloaddition (Scheme 11). The gelation time in water decreased with temperature increase: 160 min (27 °C), 50 min (37 °C), 30 min (47 °C), 20 min (57 °C), 15 min (67 °C), 10 min (77 °C). However, no gel was formed when the experiment was run in DMF even for reaction time as long as four days. It was rationalized that the rate acceleration in homogeneous aqueous solution could be attributed to hydrophobic aggregation, cohesive energy density, ground-state destabilization, and micelle-like aggregation. The retro Diels–Alder reaction of the hydrogel (depolymerisation) in water did not occur even at 100 °C, reflecting the thermosensitive and water-stable property of the hydrogel, but the depolymerization was successful in DMF after 15 min only at this temperature.
Marchán et al. [42] described the application of Diels–Alder cycloaddition in water to the preparation of peptide–oligonucleotide conjugates via the reaction between diene–oligonucleotides and maleimide–peptides (Scheme 12). The reactions were generally completed in 8–10 h. Similar work on biomolecule conjugates was reported earlier by Hill et al. [43].

Grondin et al. [44] reported the Diels–Alder reaction of benzotriazole maleimide 45 (BTM) with seven dienes in aqueous acetonitrile systems (‘on-water systems’) (Scheme 13). The reaction with 9-anthracene carbinol as a diene gave the corresponding cycloadduct, 2-(1H-benzotriazol-5-yl)-4,9-[1′,2′]benzeno-4-(hydroxymethyl)-3a,4,9,9a-tetrahydro-1H-benz[f]isoindole-1,3(2H)-dione 46 in good yield (79%). The yields of 2-(1H-benzotriazol-5-yl)-5,6-dimethyl-3a,4,7,7a-tetrahydroisoindole-1,3-dione 47, 2-(1H-benzotriazol-5-yl)-4-methoxy-3a,4,7,7a tetrahydroisoindole-1,3-dione 48, and 2-(1H-benzotriazol-5-yl)-7-methyl-1,3-dioxo-2,3,3a,4,7,7a-hexahydro-1H-isoindole-4-carboxylic acid 49, the respective cycloadducts of 2,3 dimethylbuta-1,3-diene, 1-methoxybuta-1,3-diene, and hexa-2,4-dienoic acid, were 90, 71, and 43%, respectively. The low yield of cycloadduct 49 was attributed to the electron withdrawing carboxylic acid functionality on the corresponding diene, making the reaction less favored compared to the other two butadienes. The presence of 4.86 M lithium chloride in the acetonitrile–water medium was found to increase the rate of the cycloadditions of butadienes. However, 4-(1H-benzotriazol-5-yl)-10-oxa-4-azatricyclo[5.2.1.0]dec-8-ene-3,5-dione (50) and 4-(1H-benzotriazol-5-yl)-1,7-dimethyl-10-oxa-4-azatricyclo[5.2.1.0]dec-8-ene-3,5-dione (51) were obtained only in the presence of Cu(NO3)2 as a Lewis acid catalyst and in yields of 43 and 74%, respectively. Cycloaddition of BTM with furan-2-carbaldehyde dimethylhydrazone afforded 2-(1H-benzotriazol-5-yl)-1,3-dioxo-2,3-dihydro-1H-isoindole-4-carbaldehyde dimethylhydrazone (52) in 70% yield; the chemical structure of this cycloadduct was formed as a result of spontaneous dehydration, owing to the created highly conjugated system by the electron-donating group on the furan ring.
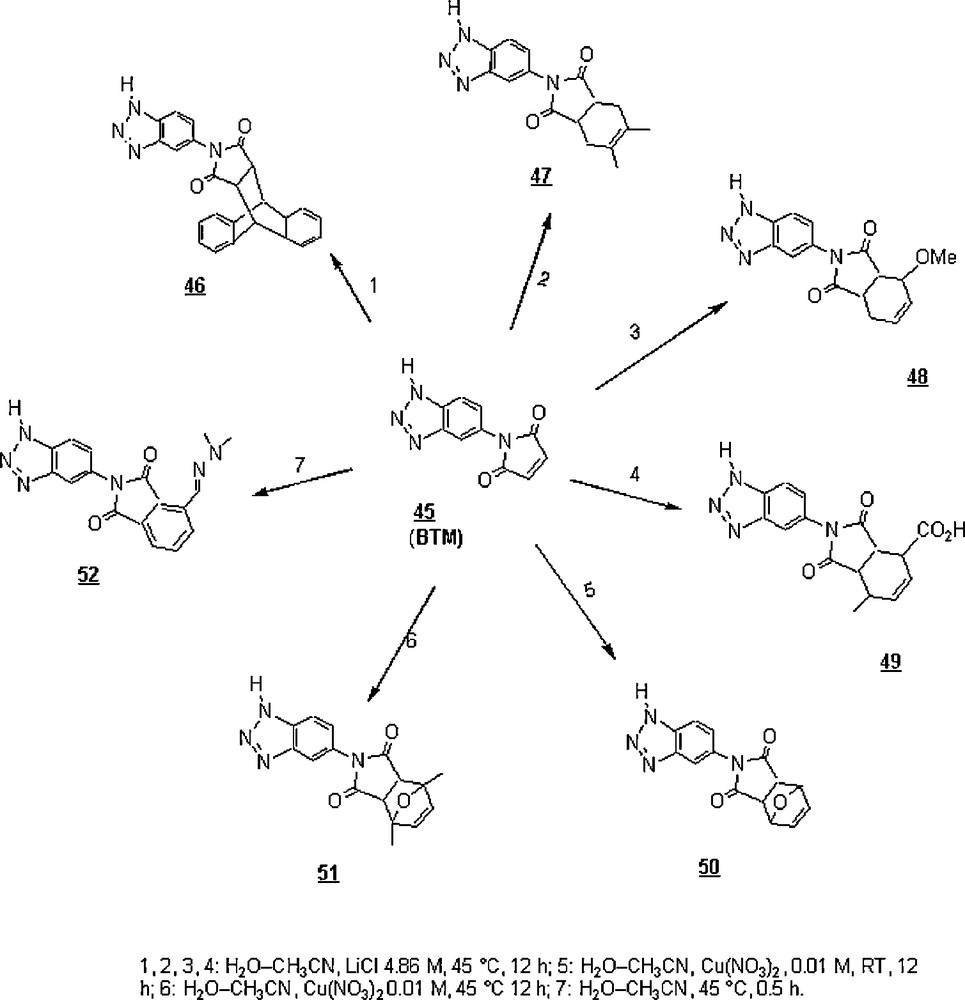
The different synthesized cycloadducts were characterized by surface enhanced Raman scattering (SERS) by adsorbing them onto a metal surface via the triazole group; analysis of the different SERS spectra confirmed the cyclisation occurrence.
Zhang and Bruice [45] undertook a computational work on the ribozyme catalysis of Diels–Alder between maleimide and anthracene in water. It was demonstrated that, during the course of the catalyzed reaction, the maleimide molecule is held in a hydrophobic pocket while the anthracene molecule approaches to the maleimide through the back side of the active site; the latter active site is so narrow that the anthracene molecule has to adopt a tilted position while drawing near maleimide. For this ribozyme Diels–Alder catalysis, theoretical results revealed that the kinetic of the reaction in water is due to the creation of a reactive conformer at the active site.
2.2.2 With α,β-unsaturated ketones
2.2.2.1 p-benzoquinones
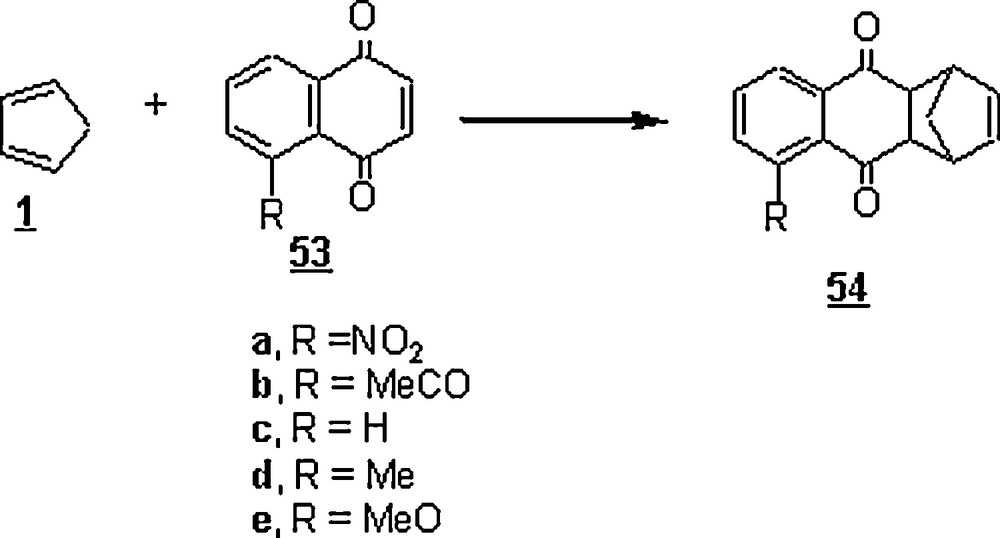
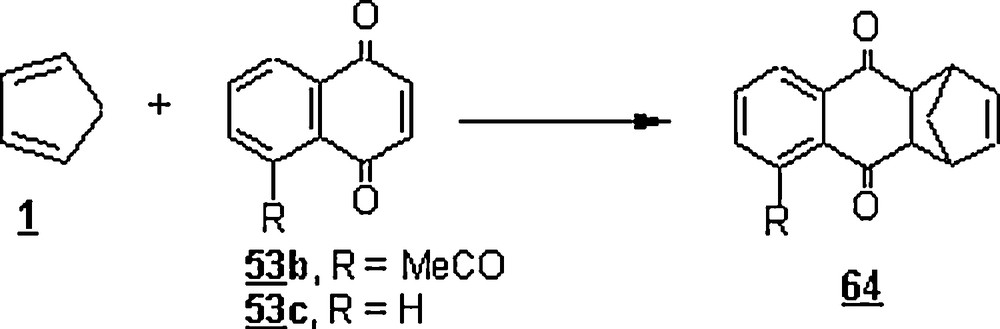
Otto et al. and Engberts [37,39] launched a series of DA reactions in the aim to ascertain the factors affecting the rate increase when the reactions were run in water. The cycloaddition reactions of the cyclopentadiene 1 with naphthaquinone derivatives 53 (Scheme 14) to give the anthracenediones 54 were studied in n-hexane, acetonitrile, ethanol, n-propanol, 1,1,1-trifluoroethanol (TFE), 1,1,1,3,3,3- hexafluoro-2-propanol (HFP), and water. The results were that the reaction rate increased from n-hexane up to water and the rate enhancement was estimated as kwater/kn-hexane at 25 °C and was: 1180 (53a), 1651 (53b), 4583 (53c), 7250 (53d) and 12,780 (53e). Both the nature of substituent on the dienophile, that is, the naphthaquinone derivative, and the hydrogen bond-making capacity of the solvent played major roles in the acceleration process. There was a dependence of the reaction rate on the electronic nature of substituent. Indeed, the methoxy group, being an electron-releasing group, slowed down the reaction in n-hexane. However, in water and HFP, it speeded it up, reflecting a solvent-mediated interaction between the substituent and the nearby carbonyl group. In general, the substituent effects as given in the scheme decreased with increasing rate of the reaction in a specific solvent, indicating that the charge separation in the activated complex in water was not much different from that in the other solvents [37].
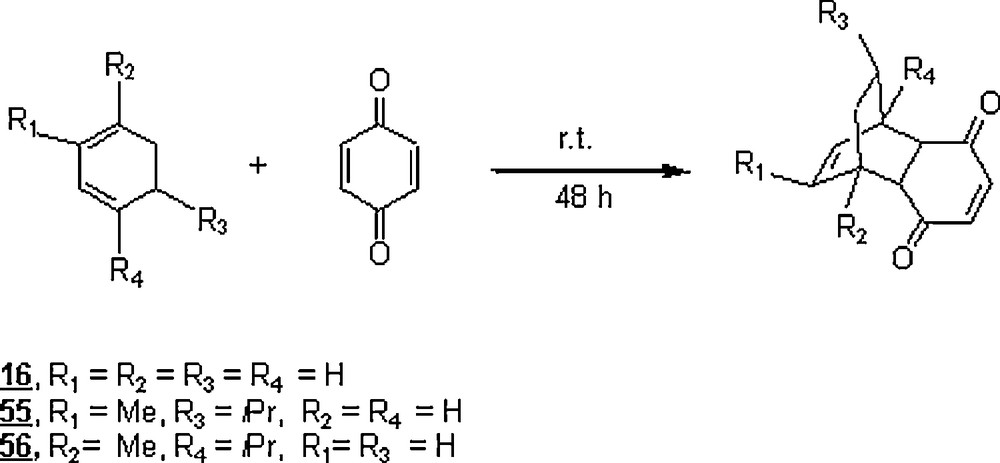
The DA reactions of the cyclohexa-1,3-diene 16 and two of its derivatives, α-phellandrene 55 and α-terpene 56, with p-benzoquinone 57 (Scheme 15), showed hydrophobic and steric hindrance effects on the yields [46]. Still, water promoted better reaction yields than the organic solvent (toluene) did as illustrated in Table 3. In the case of the cyclohexa-1,3-diene 16, the yield and the reaction time were improved to 83% and 20 h, respectively, by using Ti(IV) Lewis acid as a catalyst [47]. The occurrence of a hydrophobic packing of the diene and the dienophile in the transition state would account for the greater yields of water-promoted DA reactions, particularly in the case of 16. However, the relatively lower yields in the case of 55 and 56 were imputed to the steric interactions imposed by the methyl and the isopropyl radicals, disfavoring the packing phenomenon. For 56, examination of the effects of NaCl (a salting-out additive) and GnCl (a salting-in additive) disclosed distinct acceleration and retardation, respectively. However, the yields were substantially enhanced with GnCl additive and depreciated with saturated solution of NaCl.
Yields (%) of the DA reactions of cyclohexadienes with p-benzoquinone 57 in 1:1 molar. Time: 2 days.
| Toluene | H2O | Sat, NaCl (aq.) | GnCl (15 M) | |
| 16 | 15 | 67 | 69 | 83 |
| 55 | 15 | 27 | 25 | 52 |
| 56 | 3 | 28 | 13 | 67 |
Yu et al. [48] embarked on a set of DA reactions in water involving six dienes and six dienophiles (Scheme 16) promoted by [O = P(2-py)3W(CO)(NO)2](BF4)2 as a water-soluble organotungsten Lewis catalyst 59 (Scheme 17).
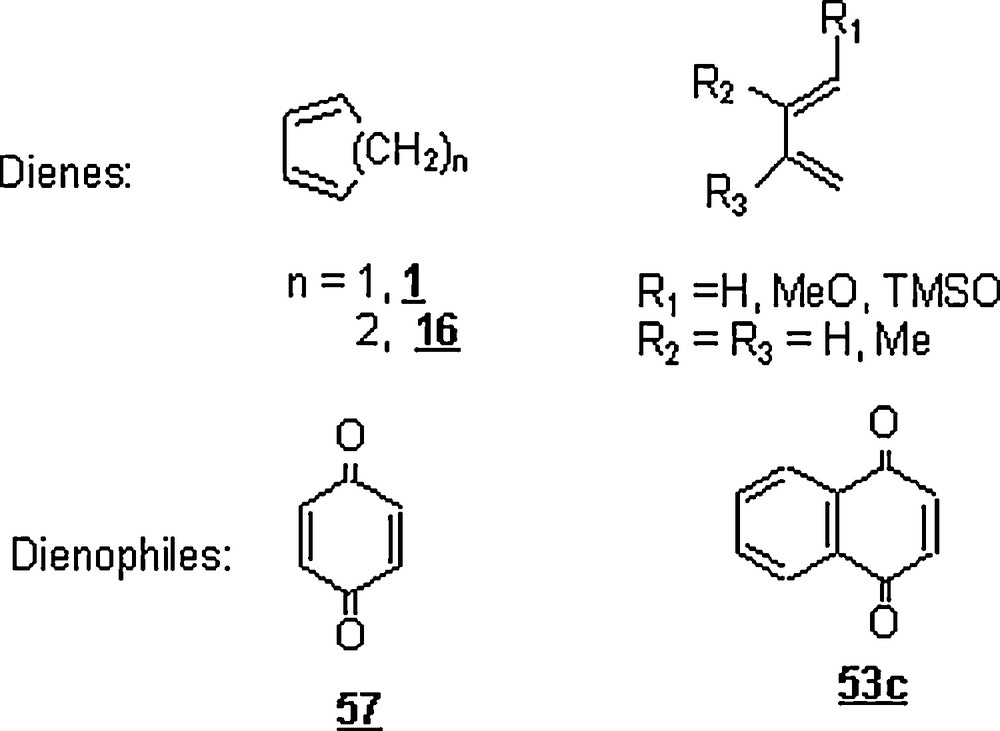
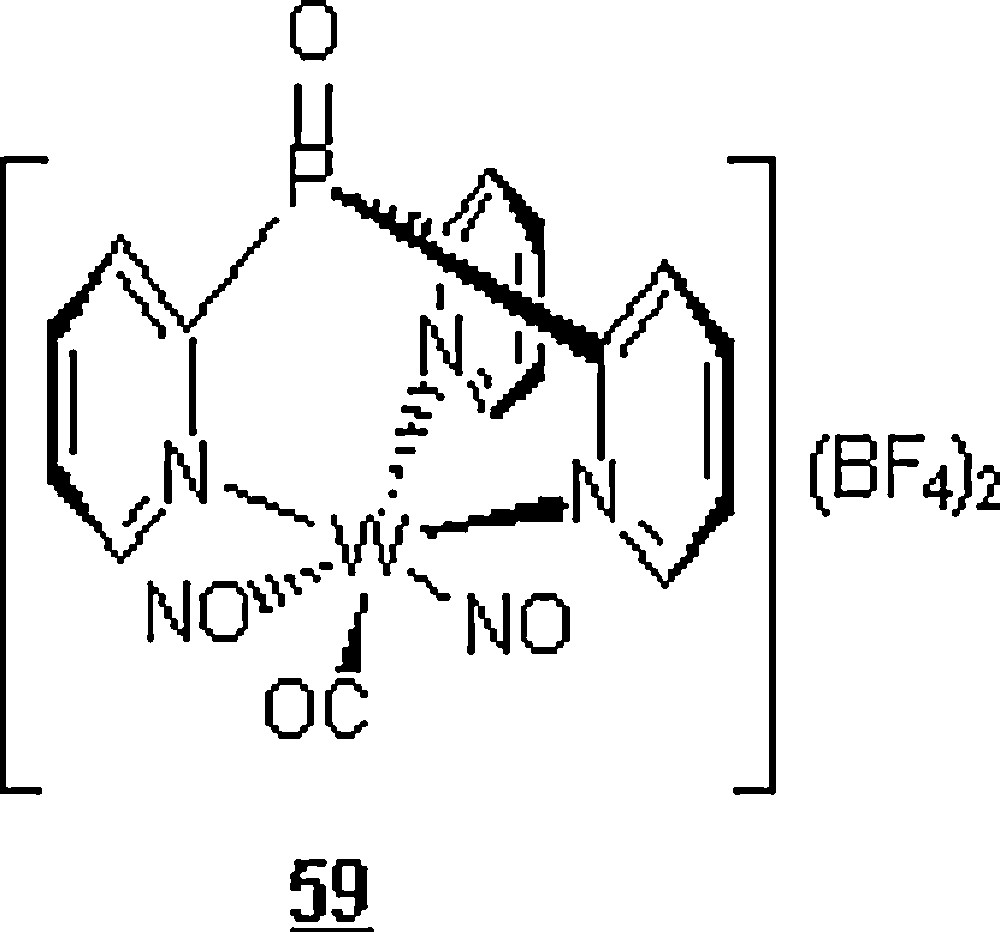
The corresponding cycloadducts of different DA reactions run in water and in the presence of the mentioned catalyst were obtained in good to excellent yields (75–99%) and in higher endo selectivities (95 to 100%), at temperatures of 25 and 50 °C. A competitive polymerization of some dienes such as methyl acrylate 60 and isoprene 61 by the catalyst during the DA reactions was observed. As to the kinetics of reactions, they were reasonable (1–2 h) when compared to the reactions conducted in organic solvents. For instance, the reaction at room temperature of cyclopentadiene 1 with vinyl methyl ketone 8 was completed in 2 h with an endo/exo selectivity of 95:5, while the controlled reaction in nitromethane under similar conditions was over after 5 h with about the same selectivity (97%). Also, a loss of methoxy group in the cycloadduct from the diene trans, trans-1,3-butadienyl methyl ether 62 due to the unstability of the adduct, and the deprotection of trans, trans-1,3-butadienyl trimethylsilyl 63 (the loss of TMS, the trimethylsilyl group) by the BF4− anion of the catalyst occurred during the reactions. Microwave heating induced a significant DA reaction acceleration as compared to the thermal heating. Within a reaction time of 50–60 seconds, the yields of cycloadducts were 83–97%, the conversions of 92–99%, and the endo extents of 100%.
Another aspect of the Engberts’ group research [49] was to assess the effect of cationic surfactants with hydrogen-donating ability on the Diels–Alder reaction between cyclopentadiene and dienophiles with hydrogen-bond-accepting capability, such as substituted naphthoquinones 53, (Scheme 18). An increase in rate was observed for cationic surfactants with hydrogen-donating capacity such as DDAB (dodecyldimethylammonium bromide) and DMAB (dodecylmethylammonium bromide), particularly with 5-acetyl-2,4-naphthoquinone 53b which has three carbonyl groups.
Chiba's group investigated the effect of aqueous sodium dodecylsulfate (SDS) micelles in an electrochemical medium on the Diels–Alder reaction between in situ generated quinones with dienes [50]. While the reaction of an equimolar mixture of quinone (57 or 65) and myrcene 66 (Scheme 19) in an organic solvent (benzene or acetonitrile) gave no product after 40 h standing at 25 °C, very small amounts of cycloadducts were obtained in water. Of the surfactants experimented, SDS promoted significant reaction acceleration, particularly at higher concentrations. The yields of cycloadducts from the reaction of quinone with myrcene were 8 and 35.1% for a concentration of 20 mM of sodium dodecylsulfonate and sodium dodecylsulfate, respectively. However, the yield increased to 99.8% at a concentration of 200 mM of SDS. A very attracting result was the effect of electrochemical medium on the global outcome of the reaction. Indeed, the Diels–Alder reaction between the in situ electrochemically produced benzoquinones of the hydroquinones 67, 68, and 69 with dienes 15, 28, 61, 70–73, in a SDS micellar solution; the nafion-coated anode in the SDS micellar solution provided selective oxidation of hydroquinones to give the desired cycloadducts in quantitative yields (84–100%) (Scheme 20). Albeit hydrophilic p-quinones that possess electron-withdrawing groups were unstable in aqueous media due to the plausible Michael addition of H2O, it was found that hydrophobic p-quinone generated by cerium (IV) oxidation in the SDS micelle gave the desired cycloadducts in excellent yields.
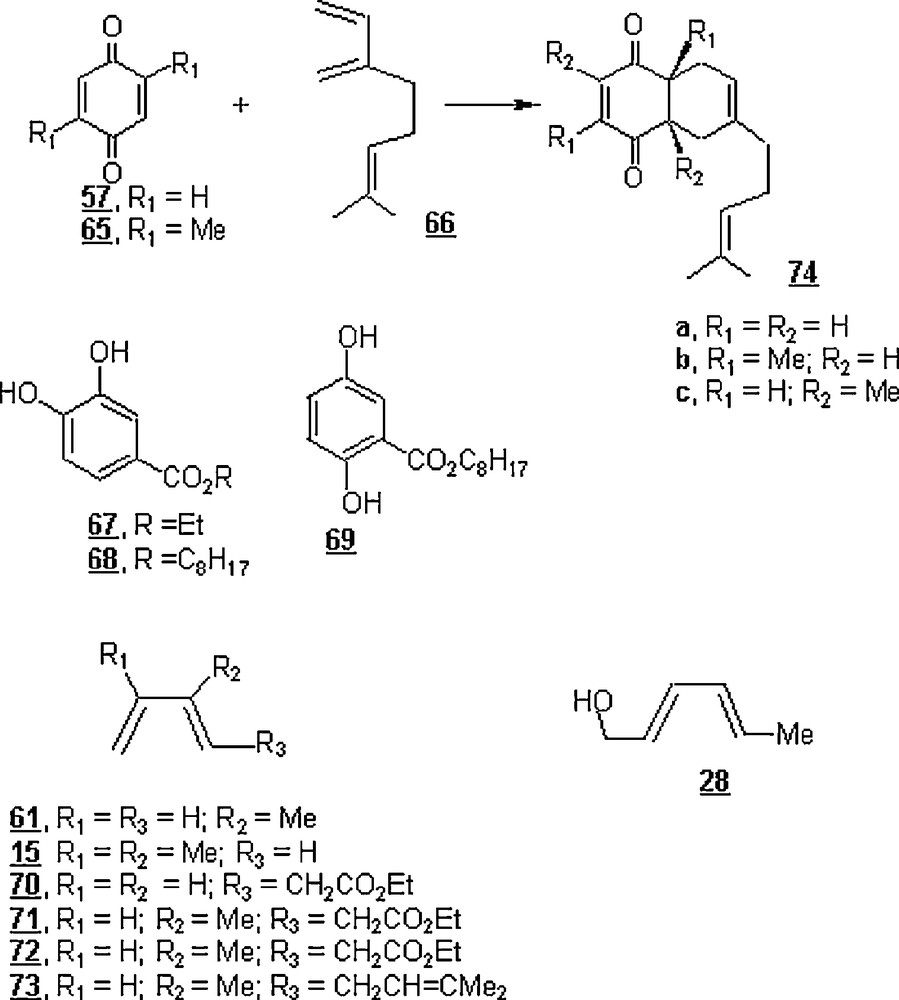
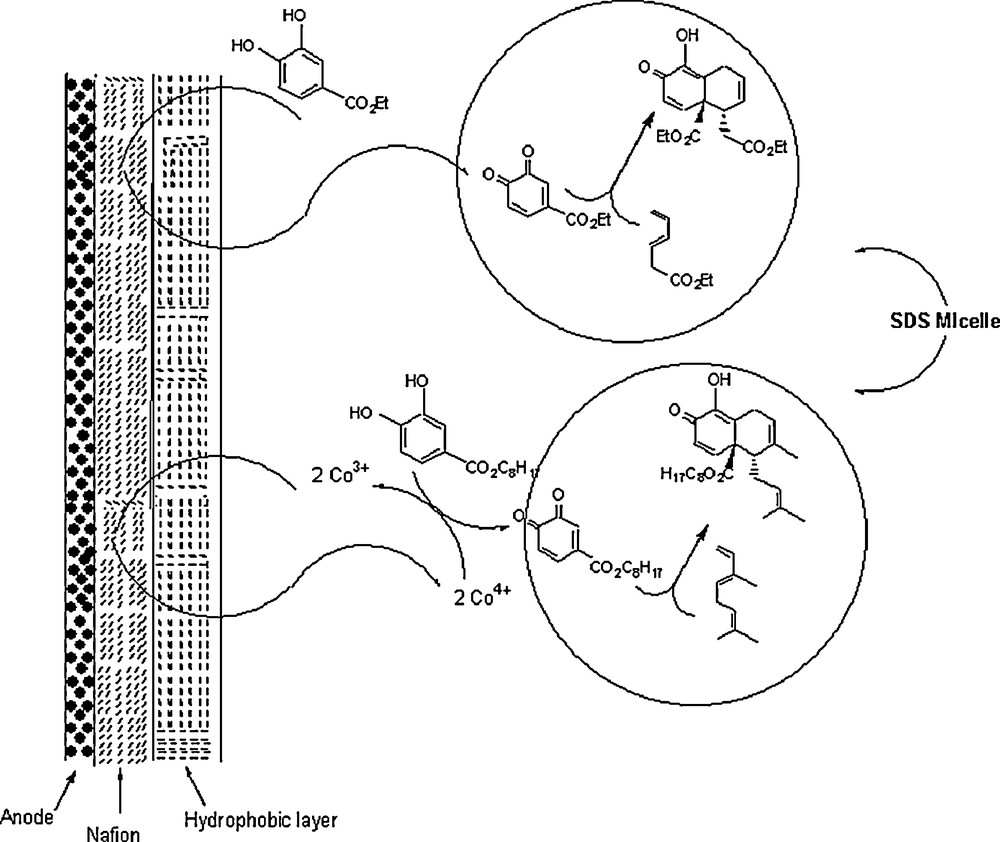
In another work, Chiba and his co-workers [51] presented results of Diels–Alder reaction between 2-methylnaphthoquinone 75 and 2,3-dimethyl-1,3-butadiene 15 in an aqueous micellar system composed of perfluorohexane (PFH) and lithium perfluorooctanesulfonate (LiFOS) (Scheme 21). At 25 °C and for 24 h, the reaction rates in the presence of PFH (100 mM) and LiFOS (100 mM) were nearly 0.2 and 8.0 times that in pure water. However, the rate was tremendously enhanced by working in aqueous fluorous emulsions composed of 500 mM PFH and 500 mM LiFOS; it was about 100-fold greater than that in water. The rate was found to rise with the concentrations of PFH and LiFOS.
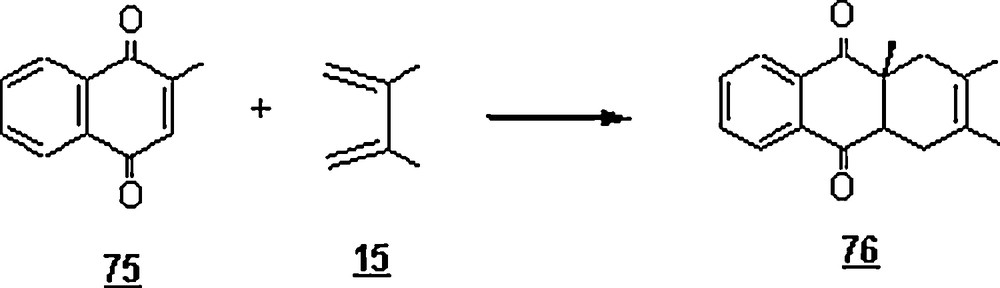
Nematollahi and Ghorbani [52] performed the electrochemical oxidation of 1,4-hydroquinone in the presence of cyclopentadiene 1 in water/ethanol mixture using cyclic voltammetry. The DA reaction between the generated p-benzoquinone 57 and cyclopentadiene gave the endo-anti-endo cyclo-bis-adduct 1,4,4a,5,8,8a,9a,10a-octahydro-(1,4),(5,8)-dimethano-9,10-anthraquinone 77. The results indicated that the observed homogeneous rate constants for the reaction increased with increasing temperature, and the activation energy was estimated to be 34.4 kcal/mol. Also, the rate constant of the reaction was found to be dependent on the water content, that is, it increased proportionally with increase of water percentage. From the established voltammograms, the peak current ratio (cathodic peak current intensity/cathodic peak current intensity) decreased with increasing water in the mixture, resulting from the acceleration of the Diels–Alder reaction in aqueous solutions.
2.2.2.2 Vinyl ketones
With dienophiles such as methyl vinyl ketone 8 (MVK) and methyl vinyl sulfone 78 (MVS) (Scheme 22), the rate enhancements were less pronounced as revealed by kwater/korg.solvent ratios and followed the same order with respect to the solvents studied; for example, kwater/kacetonitrile at 25 °C was calculated for 8 and 78 as 285 and 71, respectively [37,39]. This difference in rate is mainly due to the insulating effect of the sulfur atom in MVS, which made its reactivity much less affected by hydrogen bonding than the reactivity of MVK. Compared to pure water, a micellar system decreased the reaction rate as found with SDS and CTAB aqueous solutions; the rate decline was more pronounced with CTAB solutions than with SDS ones.
A common observation was that, in contrast to the reaction in organic solvents, the water-based DA reactions proceeded with higher endo/exo selectivities. For example, the DA reaction of MVK 8 with CP 1 yielded the Diels–Alder adduct with an endo/exo ratio higher in water than in methanol and n-butanol, 20 versus 10 and 7, respectively. Besides, the thermodynamic parameter ΔG≠ (Gibbs energy of activation) was found to be about 10–15 kJ/mol greater in water than in n-propanol.
Cativiela et al. [53] handled a thorough work on the solvent effects on the Diels–Alder reaction of several diene-dienophile systems: cyclopentadiene/methyl vinyl ketone (MVK), cyclopentadiene 1/(1R,2S,5R)-menthyl acrylate 80, isoprene 61/MVK 8, cyclopentadiene 1/isoprene 61, and cyclopentadiene 1/acrylonitrile 81 (ACN). The first three systems were conducted in aqueous fluorinated alcohols, 2,2,2-trifluoroethanol (TFE) and 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP), at different volume ratios. In the case of 1 and 8, the kinetic rate constant increased with water content, particularly, for mixtures with 50% or more in water. This was attributed to an important solvophobicity effect on the reaction rate. However, the rate of the reaction between 1 and 80 did not vary appreciably but tended to increase with water proportion, pointing to a lower solvophobicity effect. The calculated solvophobicity (Sp) of aqueous fluorinated alcohol was found to fall exponentially with the proportion in alcohol, indicating the disruption of the internal structure of water by the alcohol via its hydrogen-bond donating ability. A linear correlation between Sp of the medium and the reaction rate was established, indicating that solvophobicity was the main influencing parameter.
As to the endo/exo selectivity, a little variation was observed with the alcohol content, but it increased to a greater value when the reaction was run in pure water. For the reaction of 1 with 80, this selectivity also remained nearly unchanged upon working at a varying extent of water content, but, surprisingly, it dropped drastically in pure water. This endo/exo selectivity profile confirmed the solvophobicity as the prevailing factor.
The para/meta regioselectivity of the reaction isoprene 61/MVK 8 (Scheme 23) uniformly increased with fluorinated alcohol content in the reaction medium, and was greater for HFIP than for TFE media. Accordingly, this result indicated the HBD ability as the main factor influencing this regioselectivity. Because of its low HDB value (α = 1.17), water could not promote an increase of the regioselectivity, compared to those of TFE (α = 1.51) and HFIP (α = 1.96).

The rate of the Diels–Alder reaction of ACN with CP was higher in water than in TFE and HIFP, as suggested by kwater/TFE = 4.5 and kwater/HFIP = 2. This was the opposite of what has been found for the other reactions, and dipolarity–polarizability and HBD ability were invoked as the main factors, instead of solvophobicity factor. Also, the endo/exo selectivity was also higher in water than in the other two alcohols, 2.34 versus 1.73 and 1.60, respectively. Such a selectivity trend was assigned mostly to the dipolarity–polarizability parameter.
In preparing 11-oxabicyclo[6.2.1]-undec-1,5,9-triene (84), Wang and Roskamp [54] exploited the intramolecular Diels–Alder reaction of 83 in water in the presence of silica-gel as shown in Scheme 24.

2.2.2.3 Phenyl-1-(2-pyridyl)-2-propen-1-one
Lewis acids were found to affect significantly the outcome of DA reactions. Otto et al. [55] examined the influence of Co2+, Ni2+, Cu2+, and Zn2+ on the course of DA reaction between CP and substituted and unsubstituted 3-phenyl-1-(2-pyridyl)-2-propen-1-one 85 (Scheme 25). The latter was purposely chosen because of its bidentate potentiality through the oxygen atom of α,β-unsaturated ketone group and the nitrogen atom of pyridyl group. A general observation was that the rate of Lewis acid-catalyzed DA reactions was greatly enhanced compared to that of uncatalyzed ones. The water rate promotion was substantial as revealed by the rate constants ratio kwater/kacetonitrile = 287 for the reaction with X = NO2 at a temperature of 25 °C. The extent of rate enhancement of DA reactions depended on the Lewis acid; the effect of experimented Lewis acids was as follows: Zn2+< Co2+< Ni2+< Cu2+ for the reaction of X = H with 1. Indeed, the rate constant observed for Cu2+ was 55 times that of Zn2+, 52 times that of Co2+, 28.5 times that of Ni2+ for the following conditions: temperature of 25 °C, ionic strength of 2 M, and [M2+] of 10 mM. Compared to acetonitrile, the rate in 0.010 M aqueous solution of Cu(NO3)2 was about 250 000 faster [56]. Also, Cu2+ catalysis showed better endo/exo selectivity; the latter parameter was estimated in this order: 84/16 (uncatalyzed reaction), 86/14 (Zn2+), 86/14 (Ni2+), 87/13 (Co2+), 93/7 (Cu2+).
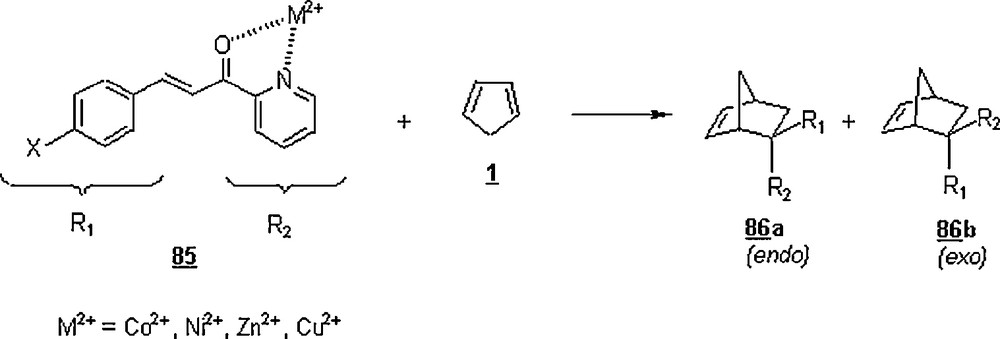
Otto et al. [57] deepened their research on the aqueous Lewis-acid catalyzed Diels–Alder reaction between 3-phenyl-1-(2-pyridyl)-2-propen-1-one 85 and 1, elucidating the “enantioselectivity” aspect of the reaction. The reaction was conducted in water and in the presence of 10% Cu (II) complexes of glycine, l-valine, l-leucine, l-phenylalanine, l-tyrosine, l-tryptophan, and N-α-methyl-l-tryptophan (l-abrine), at 0 °C for 48 h. The cycloadduct was isolated in higher yields, about 90%. Also, an enantioselectivity was observed for the chiral ligands as measured by their enantiomeric excess (ee): 14% (l-phenylalanine), 26% (l-tyrosine), 25% (l-tryptophan), and 74% (l-abrine). Higher enantioselectivities were achieved exclusively for α-amino acids containing aromatic side groups, suggesting the importance of arene–arene interactions in the pathways leading to the enantiomeric Diels–Alder adducts 87; in this scheme, it is shown the attractive interaction between the aromatic ring of the α-amino acid and the pyridine ring of 3-phenyl-1-(2-pyridyl)-2-propen-1-one. The enantioselectvity was more significant in water than in organic solvent as typified by the use of l-abrine as a chiral ligand, ee (enantiomeric excess): 74% (water), 44% (chloroform), 39% (ethanol), 24% (THF) and 17% (acetonitrile).
A work for elucidating the effect of acid type on the DA reaction course in water was undertaken with hydrochloric acid (Bronstëd acid, specific acid) and Cu2+ (Lewis acid) [58]. The results were that copper (II) catalysis was more efficient than HCl catalysis. That is, at equimolar amounts of copper (II) and HCl (0.01 M) catalysts, the reaction rate for Diels–Alder reaction of dienophile X = H with CP was about 40 times faster for copper than for proton catalysis. Another point was that, at 32 °C and 0.01 M HCl, the Diels–Alder reaction of dienophile X = NO2 with 1 was 21 times faster than uncatalyzed reaction, that is in pure water, under the same reaction conditions.
Rispens and Engberts [59] prepared Cu(dDP)2 vesicles from equimolar amounts of the sodium salt of 5,5-di-n-dodecyl-2-hydroxy-1,3,2-dioxaphosphorinan-2-one (NadDP) and CuCl2, with a size of 40–200 nm. These vesicles were employed as Lewis- acid catalysts in the Diels–Alder reaction between 3-phenyl-1-(2-pyridyl)-2-propen-1-one and 3-(4-methoxyphenyl)-1-(2-pyridyl)-2-propen-1-one with cyclopentadiene, in acetonitrile and in water. The catalytic effect was doubly-faceted: Lewis acid/vesicular catalysis. This metallo-vesicular catalysis induced a rate increase relative to the uncatalyzed reaction in acetonitrile as well as in water; the relative increase in acetonitrile was by a factor of 1 × 106. Also, it was noticed that the maximum rate was 1.5–2 times higher for the Cu(DS)2 micelles (DS = di-n-dodecylsulafte) (metallo-micellar catalysis) than for the Cu(dDP)2 vesicles 88 (metallo-vesicular catalysis), under the same reaction conditions. Another good result was that the catalysis of Cu(dDP)2 was observed at much lower concentrations (by a factor of 10–20) for the metallo-vesicles, as compared to the Cu(DS)2 metallo-micelles. This was explained by the much lower critical vesicle concentration (CVC) of Cu(dDP)2, as compared to the CMC of Cu(DS)2, and consequently, hydrophobic microdomains were present at lower concentrations. Thus, cyclopentadiene will bind predominantly into the hydrophobic core of the vesicle bilayer, and dienophile will be bound at the surface of the bilayer, complexed to copper ions.
Otto et al. [60] resumed their work on DA reaction between 3-(para-substituted phenyl)-1-(2-pyridyl)-2-propen-1-ones 85 and 1, aiming at showing the impact of micelles in aqueous systems on the outcome of DA reactions (Scheme 26). The reactions were catalyzed by micelles made of SDS, CTAB, dodecyl heptaoxyethylene ether (C12E7), and copper and zinc didodecyl sulfate (M[DS]2). A feature of this study was that micelles of latter surfactants were of a great interest because they bear catalytically active transition-metal counterions (Zn2+ and Cu2+), exhibiting also Lewis acid catalysis. At the surfactant concentrations of 7.8 mM above the CMCs and a temperature of 25 °C, a decelerating effect of SDS, CTAB and C12E7 compared to the rate of reactions in pure water was noted; this negative effect could be due to the dilution effect upon increasing the surfactant concentration. The retardation was more pronounced for the following systems: dienophile (X = CH2SO3− Na+)/CTAB (cationic surfactant) and dienophile (X = CH2N+(CH3)3 Br−)/SDS (anionic surfactant); this result was rationalized in terms of favorable electrostatic interactions (that is, the binding of micelle to the dienophile) in addition to hydrophobic interactions. No appreciable influence of the 100 mM micellar solutions of SDS, CTAB and C12E7 was detected on the endo/exo selectivities; they were 86/14, 88/12 and 85/15, respectively, and that in water was 84/16. Even the addition of micelles of these surfactants into DA reactions run in the presence of Lewis acid, Cu(NO)3, induced a decline in the rate constant, indicating the inhibitory effect of these surfactants. However, the rate of reactions in micellar solution of Cu(DS)2 was dramatically and tremendously enhanced, about 1.8 × 106 compared to the uncatalyzed reaction in acetonitrile, and about 3 × 103−6 × 103 compared with uncatalyzed reaction in water. The acceleration resulted from an efficient complexation of the dienophile to catalytically active copper ions, both species being concentrated at the micellar surface.
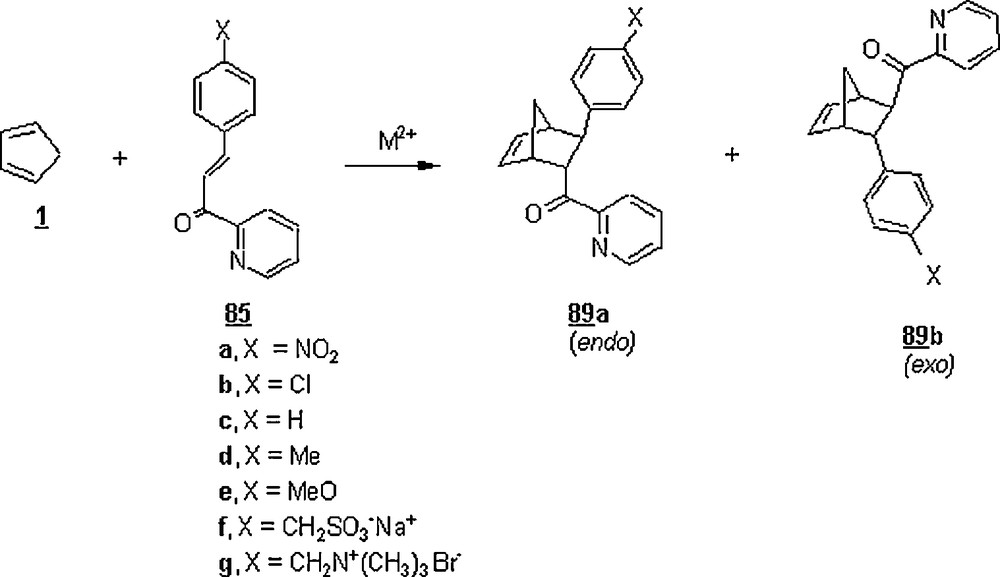
Engberts et al. [49] prolonged their work further on the Diels–Alder reaction between 85a or 85c and 1 (Scheme 26) to the effects of acidified surfactants, SDS, LAS (linear alkylbenzene sulfonic acid), DTAB (dodecyltrimethylammonium bromide), DDAB (dodecyldimethylammonium bromide), and DMAB (dodecylmethylammonium bromide).
At its CMC (8.38 mM at 32 °C), SDS was found to accelerate the reaction of 4-nitrophenyl-1-(2-pyridyl)-2-propen-1-one 85a with 1 at 32 °C, 38.5 times faster than the uncatalyzed reaction rate under similar reaction conditions. This result could be due to the higher concentrations of diene and dienophile in the micellar reaction volume. By lowering the pH of SDS solution to 2, the effect of acidified SDS solutions on the reaction rate was more pronounced: 85 times faster than the uncatalyzed reaction rate and 2.2 times faster than that in the presence of non-acidified SDS. This rate increase upon acidification of the SDS solution would suggest that the combination of hydronium ions and anionic surfactant had led to a more effective specific-acid catalysis for this Diels–Alder reaction. It is worth noting that the use of 10−2 HCl in the absence of surfactant also enhanced the rate to about 21 times faster than the uncatalyzed reaction rate. Best of all was the significant rate improvement when LAS at its CMC was employed in surfactant catalysis; indeed, the rate was 173 times better than the uncatalyzed Diels–Alder reaction. For LAS, its optimal catalytic efficiency was at a relatively low acid strength (pH = 3) and at a concentration of 0.8 mM. The effect of LAS on the rate reaction of 3-phenyl-1-(4-pyridyl)-2-propen-1-one dienophile 90 with 1 (Scheme 27) was about 40 times lower than that of 85a with 1, suggesting the occurrence of hydrogen-bonding interactions in the second case because of the bidentate character of 85a.
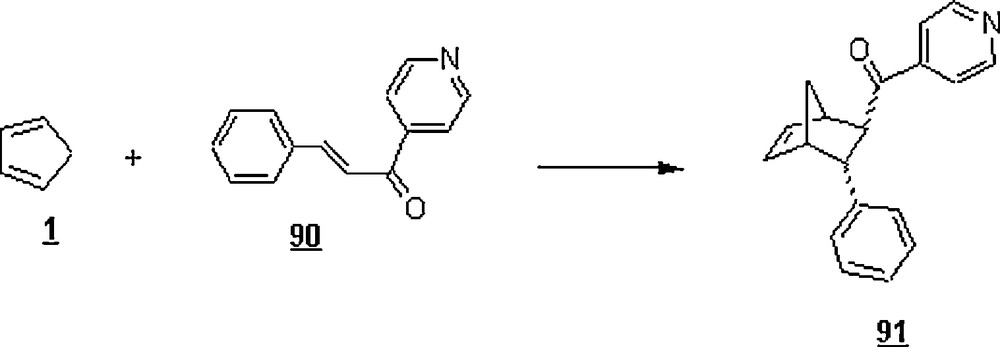
As expected and because cationic surfactants will repel the protons from the Stern region of micelles, acidified DTAB, DDAB, and DMAB were found to inhibit the Diels–Alder reactions in water at pH = 2.
2.2.2.4 Organocatalysis in Diels–Alder reactions of α,β-unsaturated ketones (aldehydes)
Another facet of the development of Diels–Alder reactions is the use of organocatalysts. Ahrendt et al., Northrup and MacMillan, and Wilson et al. [61–63] were the forerunners in developing such a novelty. They made the first highly enantioselective amine-catalyzed Diels–Alder reaction of α,β-unsaturated ketones as dienophiles via iminium formation, proceeded by a LUMO-lowering activation mechanism. Their work involved the reaction in water of a series of α,β-unsaturated ketones 92 with cyclopentadiene 1 using the amine 93 in junction with perchloric acid (Scheme 28). The reactions were conducted at 0 °C. As gathered in Table 4, while the methyl ketone led to a low enantioselectivity (61% ee), the α,β-unsaturated ketones with ethyl, n-butyl, and isoamyl substituents (R2) provided superior enantiocontrol (90–92% ee). However, the isopropyl-substituted ketone afforded racemic cycloadducts in poor yield (0% ee, 24% yield), probably as a result of steric inhibition of iminium formation. The steric contribution of the olefin substituent (R1) did not reduce the enantioselectivity or the reaction efficiency as seen for R1 = Me, n-Pr, and i-Pr (78% yield, 6–25: 1 endo:exo, 90–92% ee).
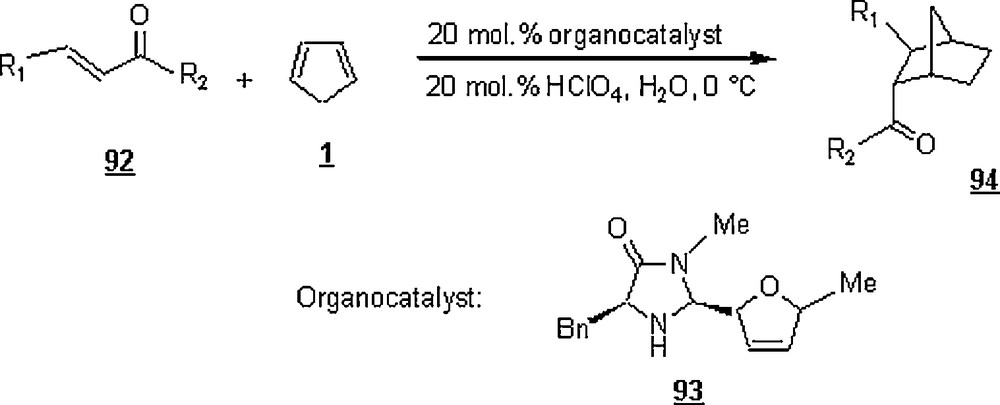
Results of the organocatalyzed Diels–Alder reaction of Scheme 28.
| R1 | R2 | Yields (%) | Endo:exo | ee (%) |
| Me | Me | 85 | 14:1 | 61 |
| Me | Et | 89 | 25:1 | 90 |
| Me | n-Bu | 83 | 22:1 | 92 |
| Me | i-Am | 86 | 20:1 | 92 |
| Me | i-Pr | 24 | 8:1 | 0 |
| n-Pr | Et | 84 | 15:1 | 92 |
| i-Pr | Et | 78 | 6:1 | 92 |
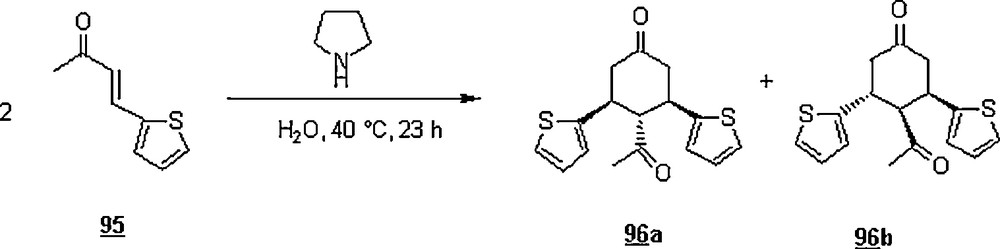
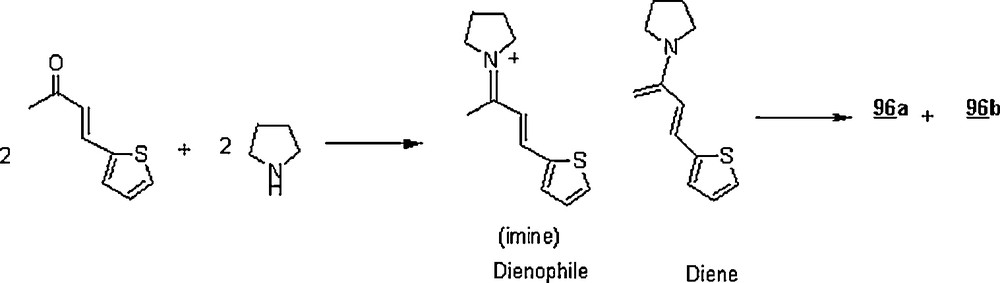
Other Diels–Alder reactions involving this synthetic strategy were reported [64–67]. Ramachary et al. [68] extended the use of organocatalysts to water-borne Diels–Alder reactions as illustrated in Scheme 29. The reaction revealed the amine-catalyzed self Diels–Alder (or double Michael) reactions of α,β-unsaturated ketones, affording pro-chiral acyl-substituted cyclohexanones. Pyrrolidine was tested as an organocatalyst for the Diels–Alder reaction of 95 providing the corresponding cyclohexanones 96a and 96b in 70% yield in 6:1 ratio. When the reaction was carried out in THF for 8.5 °C at 43 °C, the yield and the diastereomeric ratio were 63% and 2:1, respectively. Scheme 30 pictures the in situ formation of the diene and dienophile involved in the cycloaddition.
Recently, Xu et al. [69] published the results of an enamine-activated Diels–Alder reaction of cyclohex-2-enone 97 with β-nitroolefin 98 in water, brine, and seawater (Scheme 31). The organocatalysts 100, 101, and 102 were employed in addition to a carboxylic substance as an additive. The reactions were run at room temperature for a time of 18–24 h. When conducted in water, the catalysts 100 and 102 induced a high enentioselectivity (enantiomeric excess ee = 82–85%) and a high conversion (90–95%), compared with the conversion and the enantiomeric excess of 20 and 75%, repectively, with, the catalyst 101. In seawater and brine (7.5% NaCl aqueous solution), the reaction outcomes were even better with the catalyst 102; that is, a conversion of higher than 98% and an ee of 85–96%. In all cases studied, the exo/endo was greater than 20. A conversion of 20% and ee of 80% were obtained when the reaction was held in THF for four days. The brine and seawater showed a better performance in terms of chemo-, regio- and stereoselectivities.
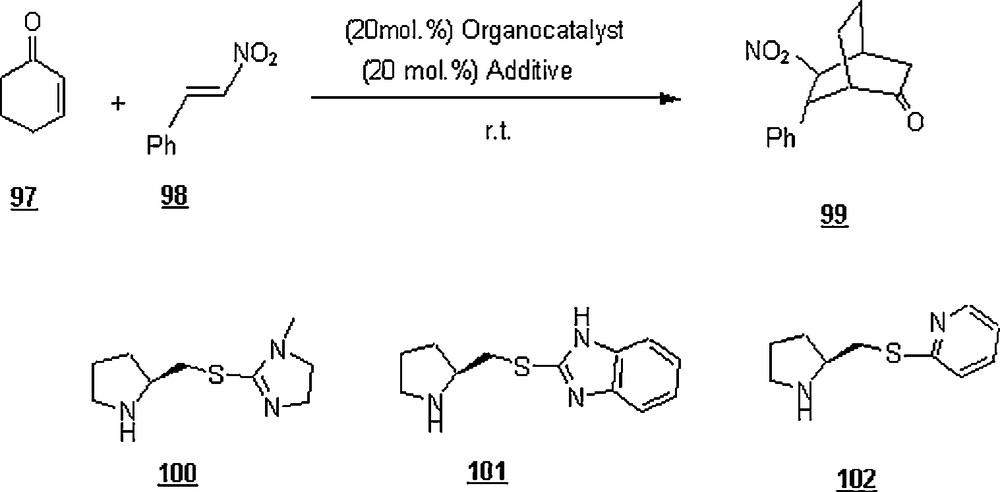
Also, the reactions of cyclohexenone with various substituted-phenyl nitroolefins were investigated in seawater and 7.5% NaCl aqueous solution in the presence of 102 and 4-trifluorobenzoic acid. It was observed that the nitroolefins bearing groups at the para-position of the phenyl ring gave higher yields (92–98%) than those at other positions (55–80%). Moreover, both 3- and 4-methyl-substituted cyclohex-2-enones underwent a Diels–Alder reaction with excellent yields (95 and 96%) and high enantioselectivities (83 and 93% ee).
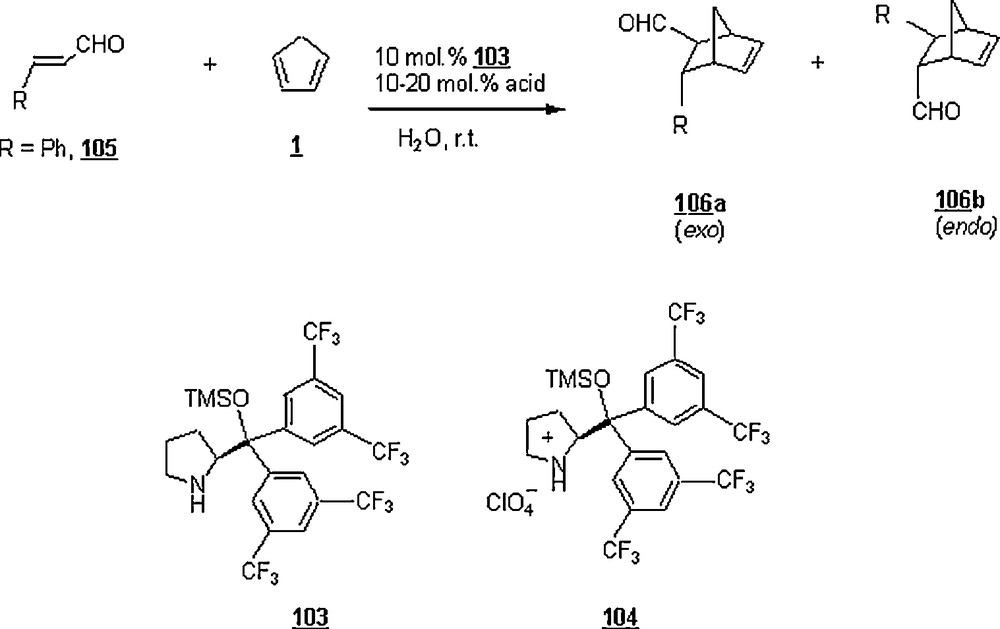
Diarylprolinol silyl ether 103 and its salt 104 were evaluated as organocatalysts in the aqueous cycoaddition reactions of α,β-unsaturated aldehydes with cyclopentadiene in the presence of acid as additive [70] (Scheme 32). Cycloaddition of cinnamaldehyde 105 with 1 in water using 103 and trifluoroacetic acid, trichloroacetic acid, perfluorinated butanesulfonic acid; p-toluenesulfonic acid provided the cycloadducts 106a (exo) and 106b (endo) in 16 to 66% yield with an exo/endo ratio ranging from 41:59 to 82:18. However, after only 4 h, the reaction outcome was enhanced upon using perchloric acid; that is, the yield rose to 100% and the mixture ratio turned out to be 80:20. The enantioselectivities ee of 106a and 106b were 95 and 90%, respectively. A series of α,β-unsaturated aldehydes (R = H, pBrPh, pNO2Ph, 2-Furyl, Me, n-Bu, cyclohexyl) were experimented employing the diarylprolinol silyl ether salt 104 and in the presence of HClO4. The reaction time was 2 to 7 h for all aldehydes with the exception for compounds with R = H and 2-furyl which required 24 and 40 h, respectively. The yields were between 73 to 95% and the exo/endo ratios between 62:38 and 85:15 with enantioselectivity of 92–99% and 86–99% for exo and endo products, respectively. The reaction of 105 with 1 was not successful when attempted in neat conditions and organic solvents (DCM, toluene); in MeOH, however, the yield was limited to 37%.
2.2.3 With α, β-unsaturated esters
Han et al. [71] reported the aqueous Diels–Alder reactions between CP and symmetric diester of fumarate or maleate, to afford cis- or trans- dimethyl and diethyl ester of bicyclo[2,2,1]hept-5-ene-2,3-dicarboxylate. The latter cycloadducts were used as pesticides, and agents in cosmetics and elastomers industries. The reactions were realized in water–acetone (3:2), and at reflux for 12 h. The yields ranged from 74 to 94%. However, the extents of endo/exo selectivity were not estimated.
Pai and Smith [72] made an endeavor to provide some insights into the effects of dienophile concentration, the buffer concentration, the pH buffers, the salts, and the additives at different concentrations on the rate of the Diels–Alder reaction between 2,3-dimethyl-1,3-butadiene 15 and ethyl acrylate 107 in dilute aqueous ethanol solutions (Scheme 33). It was found that, among the salts and additives added, only KH2PO4, NaH2PO4, and LiCl conveyed significant rate enhancements at 7.3 mM concentrations, 46, 38 and 49%, respectively. Both NaHSO3 and tetrabutylammonium bromide showed about a 30% increase in rate at this concentration. The remaining ones (K2HPO4, NaHSO4, Na2SO4, MgSO4, NaCl, ZnCl2, NH4Cl, urea) led to little rate enhancements (10–15%). It must be pointed out in the effects of chloride salts it was not the chloride ion but the lithium ion that was responsible for the rate enhancement (compare the enhancement due to LiCl with those observed with NaCl, NH4Cl, ZnCl2 and MgCl2). Whereas the effect of the dihydrogen phosphates seemed to be due to the anion (H2PO4−) rather than the cation (K+ or Na+). Increasing the ethyl acrylate concentration provoked an increase in rate enhancement up to 0.65 M where a maximum of enhancement of about 30–40% was seen at a buffer concentration of 7 × 10−3 (at ethyl acrylate concentrations higher than 0.7 M, the reactants were visibly insoluble, and therefore the data for these concentrations were unreliable). Unlike the data obtained in toluene, the rate enhancement in this dilute solution decreased at both higher and lower dienophile/diene concentration ratios.

For the 2,3-dimethyl-1,3-butadiene/ethyl acrylate concentration of 0.5 M, results of the effect of buffer concentration (pH 7) showed that the increase in rate diminished from 39% to about 18% as the concentration of buffer was lowered from 1.0 M to approximately 0.1 M. Also, it was estimated that the maximum rate enhancement occurred at neutral pH buffers, suggesting the enhancement was due to the influence of anion or cation of the added salt and not due to base or acid catalysis.
Pawar et al. [73] demonstrated the ionic impact on the stereoselectivity of aqueous Diels–Alder reaction between cyclopentadiene 1 and methyl acrylate 60 in aqueous medium. While the salts LiCl, NaCl, NaBr, and CaCl2 increased the endo/exo ratios, the salts LiClO4 and GnCl (guanidinium chloride) reduced them. In addition, both the increase and decrease in endo/exo ratios were function of the salt concentrations. The first group of salts (salting-out agents) engendered a hydrophibic effect, and the second one (salting-in agents) induced a reverse action. Accordingly, the solubilities of 1 and 60 decreased in aqueous LiCl, NaCl, NaBr, and CaCl2 and, on the other hand, the interactions of ions Gn+ and ClO4− with water increased the solubilities of these reactants in solutions. Thus, the rates were related to the relative changes in the solubilities of diene and dienophile in aqueous salt solutions.
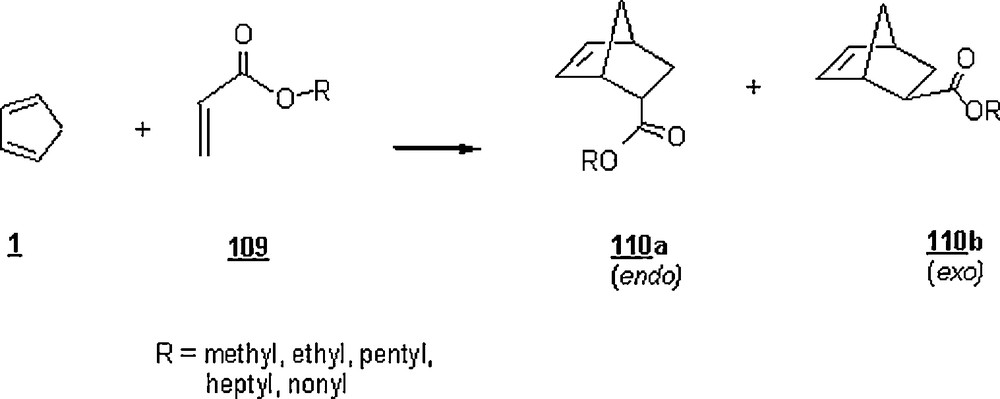
Diego-Castro and Hailes [74] investigated the effect of surfactant on the course of the aqueous Diels–Alder reaction of CP with a range of acrylates 109 (Scheme 34). The study was undertaken with SDS and CTAB at their CMC's (8.3 mM and 0.026 mM) as anionic and cationic surfactants, respectively, and in water and diethyl ether as reaction media. The general trend was that the reactions in the presence of CTAB gave cycloadducts in better yields after 4 h at ambient temperature. The yields were largely improved (70–92%) upon extending the reaction time to 72 h but the endo/exo selectivity ratio was slightly enhanced. Overall, the reactions conducted in diethyl ether failed in most cases or the yields were low; only in the case of 60 and for a reaction time of 72 h, the yield was modest (48%). In the case of nonyl acrylate, CTAB lowered the yield drastically (38%), while SDS enhanced it from 71 to 79%, and the endo/exo selectivity ratio dropped in the presence of these surfactants. The impact of pH of the reaction in water was studied for nonyl acrylate; optimal yield (75%) and endo/exo selectivity ratio (2.3) were estimated at pH = 3. While the effect of SDS was not significant, the presence of surfactant CTAB was important and the highest yields and endo/exo selectivity ratio were at the higher and lower acidity range rather than at neutral pH.
Amantini et al. [75] investigated the cycloadditions of 3-nitrocoumarin (111a), 6-chloro-3-nitrocoumarin (111b), and 6-, 7-, and 8-hydroxy-3-nitrocoumarins (111c) with (E)-piperylene (113), isoprene (61), 2,3-dimethyl-1,3-butadiene (15), 2-methoxy-1,3-butadiene (114), 2,3-dimethoxy-1,3-butadiene (115), and cyclopentadiene (1) in water (heterogeneous system), in organic solvents (homogeneous system), and under neat conditions (Scheme 35). Higher yields were obtained regardless the reaction conditions, 80–95%. At 90 °C, the reactions of 111a with 113, 61, 15 in water were faster than in toluene, 3 h against 24–30 h. With CP, however, the reaction occurred at lower temperature, 30 °C and a shorter time, 3 h. At 20 °C, the times of the reactions with 114 were 1.5 h (water), 218 h (toluene) and 150 h (dichloroethane, DCE). With 115, they were 1 h (water), 24 h (toluene), and 168 h (DCE), but the time was reduced to 6 h for DCE when working at 90 °C.

The cycloaddition of 111a with (E)-piperylene (7) in water and in toluene was regioselective, giving a mixture of exo-adduct (71%), endo-adduct (23%), and a constitutional isomer (6%).
The reactions of 6-chloro-3-nitrocoumarin (111b) with methyl-1,3-butadienes 113, 61, 15 and cyclopentadiene in water afforded nearly the same outcome as with 3-nitrocoumarin (111a).
The cycloadditions of hydroxy-3-nitrocoumarins 111c and 112 with 9 failed in water, toluene and DCE. However, they were successful when worked out in solventless conditions at 150 °C for 2 h, leading to 55–74% yields.
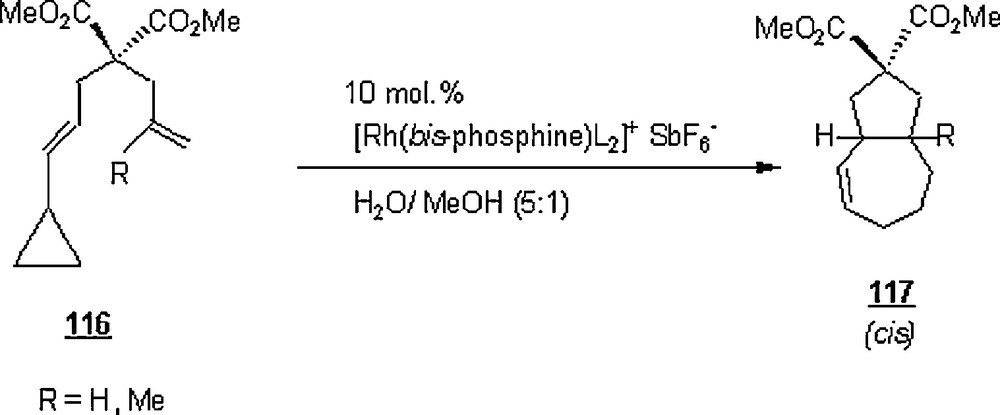
Wender et al. [76] embarked on the use of rhodium complexes to catalyze the [4 + 2], [5 + 2], [4 + 4] and [6 + 2] cycloadditions in view of their applications in the syntheses of molecules of complexed structures such as taxol, phorbol esters, and bryostatin analogs. In Scheme 36, a [5 + 2] cycloaddition of the compound 116 was applied in aqueous methanol in the presence of hexafluoroantimonate rhodium phosphine complex as a catalyst, to give products with high yields, 80–85%.
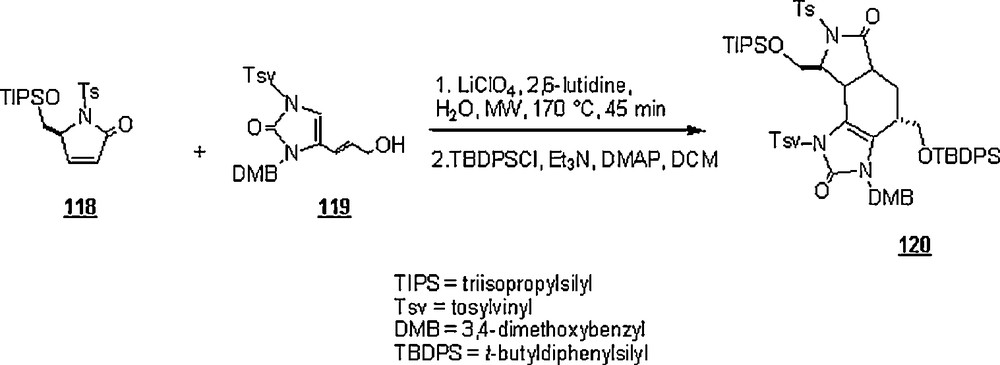
Dransfield et al. [77] took advantage of the Diels–Alder reactions in an aqueous system for making oroidin-derived alkaloids 120 like palau’amine. As shown in Scheme 37, this is a key step for the total synthesis approach for making these alkaloids, involving the DA reaction of 118 with 119 in water in the presence of 2,6-lutidine and LiClO4 as weak Lewis acid, and under microwave-assistance (MW). The reaction at 170 °C and after 45 min, afforded an overall yield of 48%.
2.2.4 With other dienophiles
2.2.4.1 Fumaronitrile
Hunt and Johnson [78] reexamined the Diels–Alder reaction between CP and fumaronitrile 121 in water (Scheme 38). The influence of additives on the aqueous reaction rate was investigated. At 25 °C, lithium, sodium and potassium chlorides, being salting-out additives, were found to increase the reaction rate by nearly 1.18–1.5. Moreover, a linear enhancement of the rate was observed upon increasing the concentration of the salting-out additive. However, triethylammonium chloride, acting as a salting-in additive (the positive charge is buried in the center of molecule), decreased the rate by a factor of 0.77. SDS and CTAB retarded the reaction by a factor of 0.85 and 0.94, respectively. A linear inhibiting effect was found by increasing SDS concentration. In the aim to have an insight into the effect of geometrical structure of the additive on the rate, α-, β-, and γ-cyclodextrins were employed; the latter two forms of cyclodextrins have a cavity size larger (6.0–6.4 Å and 7.5–8.3 Å, respectively) than that of the former form (4.7–5.2 Å). While α-cyclodextrin slowed down the reaction rate by 0.92, β- and γ-cyclodextrins accelerated it by 2 and 1.22. Also, the rate decreased with an increase of α-cyclodextrin concentration and rose with an increase of β- and γ-cyclodextrins’ concentrations.
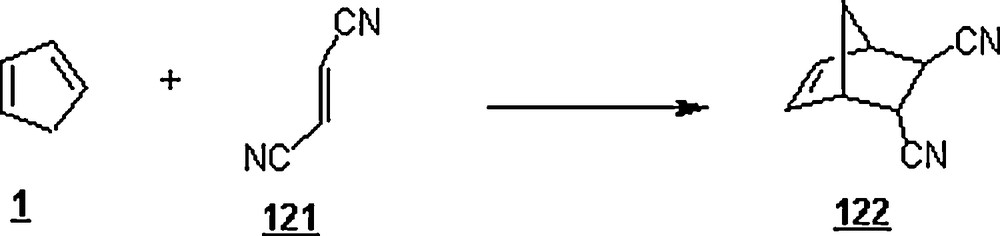
2.2.4.2 Anthracene and tetracene
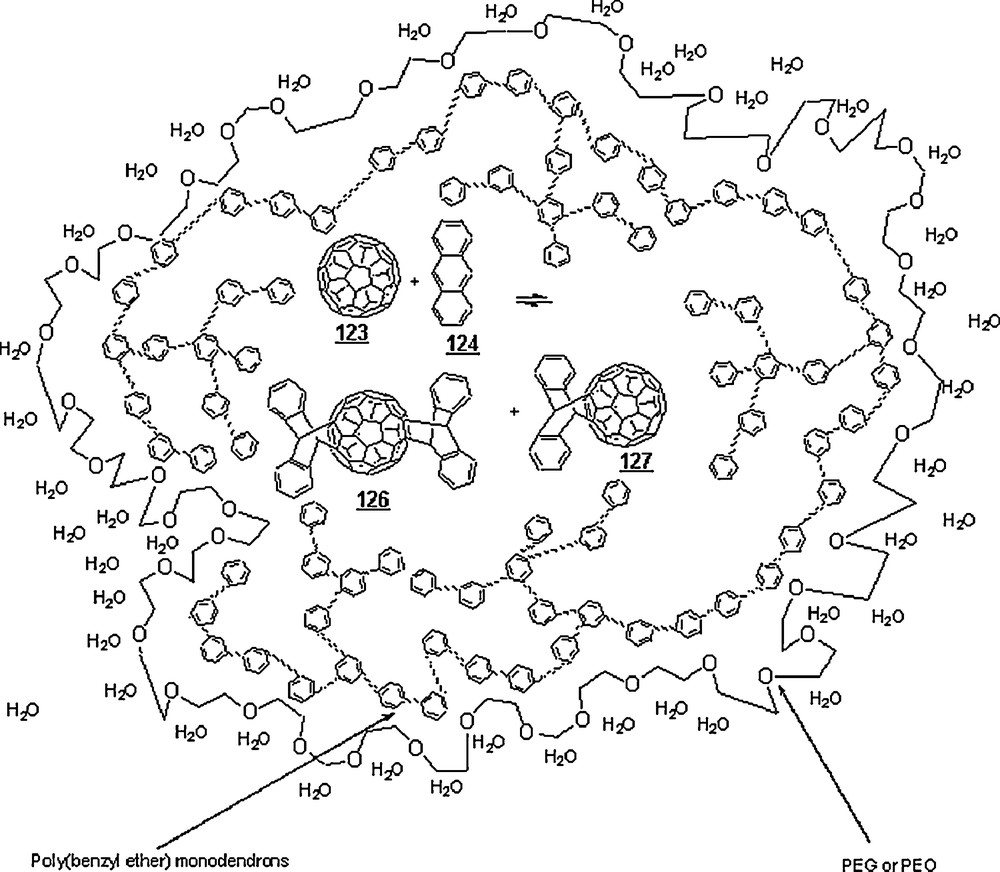
In nanoreactors made of linear-dendritic amphiphilic copolymers with poly(ethylene glycol) [PEG or poly(ethylene oxide) (PEO)] as the hydrophilic blocks and poly(benzyl ether) monodendrons as the hydrophobic segments, Simonyan and Gitsov [79] were able to realize the aqueous Diels–Alder of fullerene C60 123 and anthracene 124 (and tetracene 125), both being highly hydrophilic diene and dienophile, respectively, (Scheme 39). The rate constant for the DA reaction of C60 and tetracene within the G3PEO13k nanoreactor in aqueous medium at 22 °C was markedly higher than in toluene, 208 vs. 1.82 M−1 min−1. The conversions attained 49% for the DA reaction between C60 and anthracene, and 55% for C60 and tetracene. The rate constant in PSt-PEO container (PSt = polystyrene) was 144.9 M−1 min−1. In G3PEO13k and PSt-PEO containers, the reaction proceeded 114 and 80 times, respectively, faster than in organic solvent. Also, it was found that these DA reactions in pure water did not occur. These findings could be imputed to the higher stability and binding capability of the micelles formed by the linear-dendritic copolymers and the relatively higher degree of freedom experienced by the encapsulated reagents in their cores.
2.3 Hetero Diels–Alder reactions
In the same vein, Wijnen et al. [80] reported a thorough study on the hetero Diels–Alder reaction between di(2-pyridyl)-1,2,4,5-tetrazine 128 with a series of substituted styrenes 129 in alcohol–water mixtures and in organic solvents (Scheme 40).
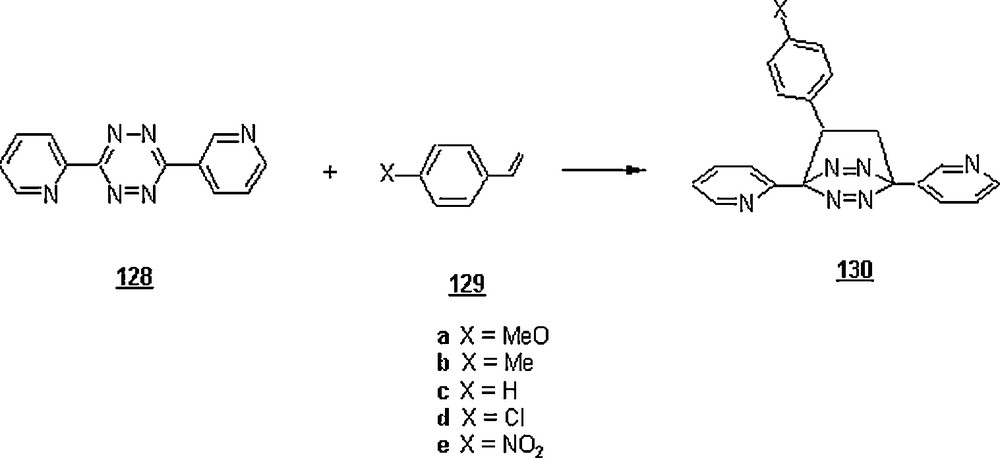
It was found that, beyond a certain water–alcohol ratio, the cycloaddition was dramatically accelerated in water-rich mixtures. The sudden acceleration was observed at different mole fractions of alcohol in the following order: MeOH > EtOH > n-PrOH, reflecting the different hydrophobicities of alcohols. The kinetics of reaction were hydrophobically accelerated; compared to 1,4-dioxane, they were 13 times faster in ethylene glycol, but 131 times faster in water-t-butanol mixture (Xw = 0.95, mole fraction of water). A ρ-value of the Hammett equation was measured as −1.32 of the reaction in the latter water system, suggesting its higher capacity of stabilizing of the activated complex which occurred in the transition state, compared to some media; the ρ-values in the different media were: −0.51 (toluene), −0.56 (THF), −0.58 (CH3CN), −0.99 (n−PrOH), −1.64 (TFE).
Another hetero Diels–Alder reaction in water involving a tetrazine was reported by Devaraj et al. [81], as depicted in Scheme 41. The purpose of carrying out this reaction was to produce a norbornane derivative 133 which was employed to modify a monoclonal antibody destined for targeting human breast cancer cells. The tetrazine–norbornene reaction was viewed as a tool for biological labelling. The reactions of 3-(4-benzylamino)-1,2,4,5-tetrazine 131 with (1S,2S,4S)-bicyclo[2.2.1]hept-5-en-2-yl acetic acid 132 in water and foetal bovine serum (FBS) were smooth, rapid, and clean with an overall conversion of higher than 93% to the corresponding dihydropyridazine products (four isomers; only one isomer is shown in the scheme); the reactions occurred with a concomitant deazotization. In aqueous buffer and FBS, their second-order rate constants were measured as 1.9 and 1.6 M−1 s−1, respectively.

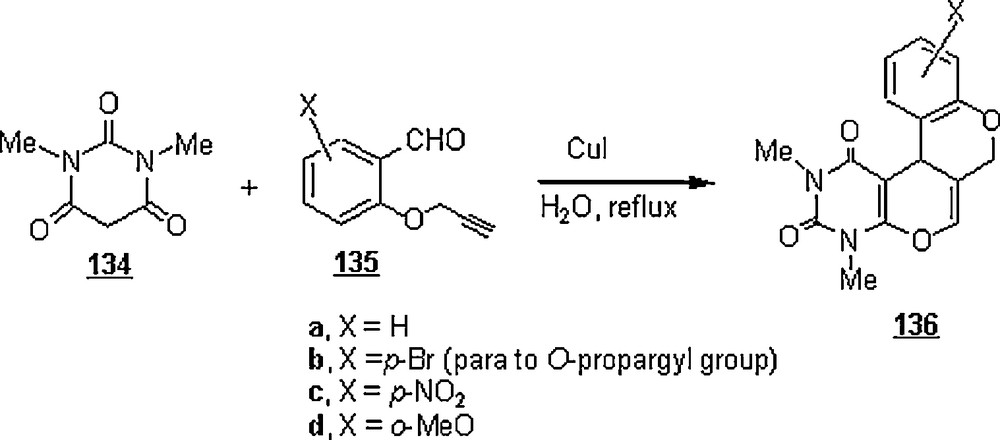
Khoshkholgh et al. [82,83] prepared four tetracyclic uracil derivatives by means of domino Knoevenagel hetero Diels–Alder reactions of 1,3-dimethylbarbituric 134 acid with O-propargylated salicylaldehyde derivatives 135 in water as solvent and in the presence of CuI as a catalyst (Scheme 42). The yields of cycloadducts 136 ranged from 73 to 85% after refluxing for 6–25 h. The advanced mechanism involved was the formation of a copper acetylide, which reduced the electron density of the alkyne and provided the necessary conditions for the hetero Diels–Alder reaction. The yield of reaction with 135a was dependent on the concentration of copper catalyst: 50, 67, and 75% for 20, 30, and 40% of CuI. The reaction in acetonitrile did not take place even in the presence of the following Lewis acids: AgOTf, AgOAc, AuCl3, and CuOTf (OTf, triflate group; OAc, acetate group). However, in the presence of CuI, the reaction occurred but only in 10% yield.
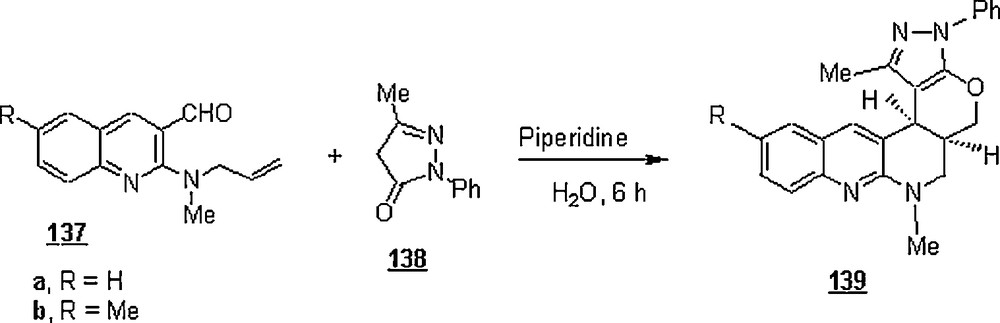
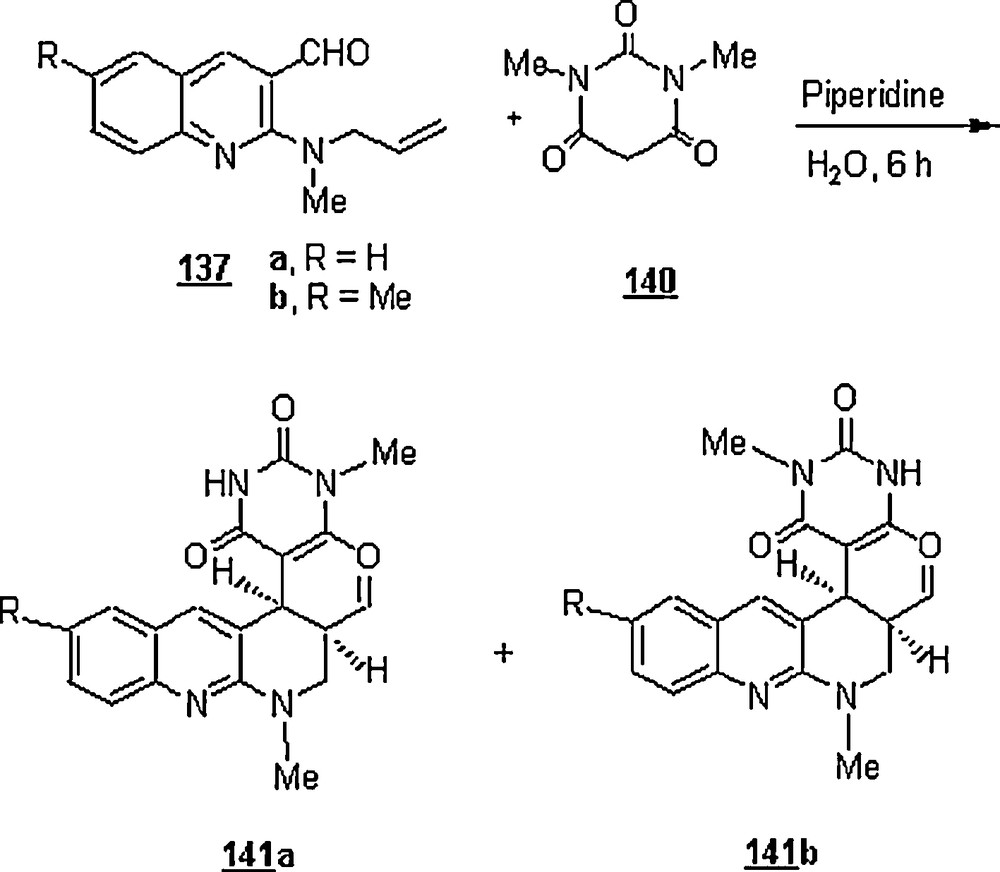
Pyrano[2,3-b]- and pyrido[2,3-b]quinolines were synthesized via intramolecular domino hetero Diels–Alder reactions of 137 with pyrazolone 138 and N-methyl barbituric acid 140 using water as solvent (Schemes 43 and 44) [84]. It is worth recalling that pyrano- and pyridoquinolines are two important classes of many compounds that constitute the basic molecular frameworks of a number of alkaloids of biological significance, such as geibalasine, ribalinine and flindersine. The yields of the desired products were low to good, 26 to 55%, at room temperature and after a reaction time of 5–6 h.
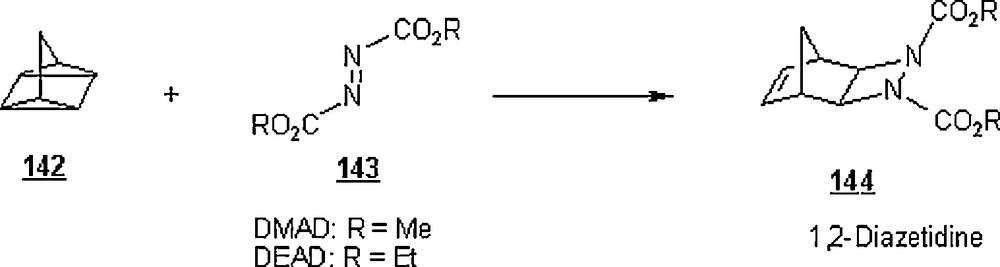
Narayan et al. [29] extended their on-water protocol to the [2σ + 2σ + 2π] cycloaddition. The reaction of quadricyclane 142 with azodicarboxylates 143, DMAD (dimethyl azodicarboxylate) and DEAD (diethyl azodicarboxylate) to form 1,2-diazetidines 144 was studied in organic solvents and in on-water systems (Scheme 45). For the reaction with DMAD, the on-water reaction gave the corresponding 1,2-diazetidine in 82% yield after only 10 min, whereas 74% yield was obtained in toluene after 24 h and no product was detected in neat reaction after 2 h. In general, the polar protic solvents or systems of solvents accelerate the cycloaddition reaction, particularly with water and MeOH/water mixture. This rate acceleration trend can be rationalized in terms of hydrogen bonding, charge stabilization, and dipolar effects. The solvents tested in this reaction were toluene, ethylacetate, acetonitrile, dichloromethane, dimethylsulfoxide, methanol, deuterated water, perfluorohexane. A significant solvent isotope effect was observed: the reaction was slowed down noticeably when D2O (time: 45 min) was used in place of water (time: 10 min). Also, the reaction of quadricyclane with DEAD proceeded faster in “toluene-on-water” than in pure toluene; the yields were 72% and 23%, respectively, when the reaction was carried out at 23 °C. Recently, Domingo et al. [85] reported theoretical results of the [2σ + 2σ + 2π] cycloaddition reaction of quadricyclane with dimethyl azodicarboxylate (DMAD) in water using density functional theory (DFT) methods. In this strategy, solvent effects of water have been modeled by means of a discrete-continuum model. In the gas phase, formation of specific hydrogen bonds between two water molecules and DMAD decreased the activation barrier from 23.2 to 15.1 kcal/mol.
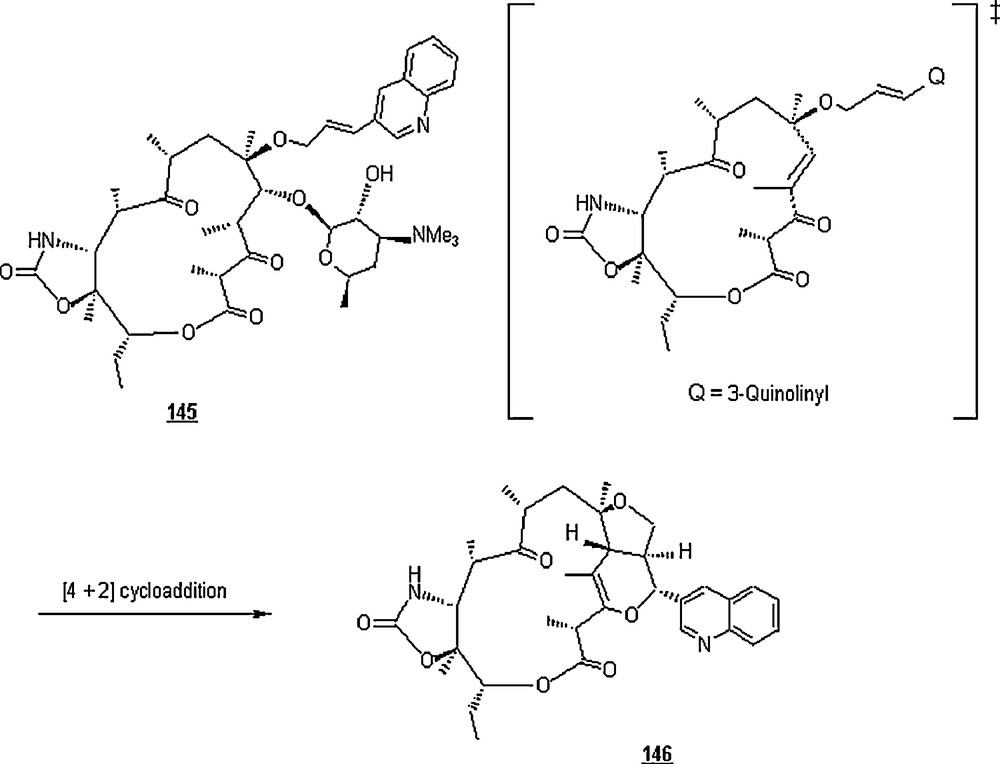
Stoner et al. [86] observed the occurrence of an unusual intramolecular hetero Diels–Alder cycloaddition when ABT-773 (145), an Abbott's ketolide, was heated in aqueous alcohol to afford the cycloadduct 146, a macrolide with an erythromycin–like skeleton (Scheme 46). The aqueous alcohol mixtures (20 to 80% in water) were based on methanol, ethanol, isopropanol or t-butanol. The macrolide was isolated in a limited yield (20 to 50%) at 70–80 °C and after 5 to 7 days. The transformation was temperature sensitive, that is, while the reaction was slow at temperatures of lower than 60 °C, both starting materials and products decomposed at temperatures above 85 °C.
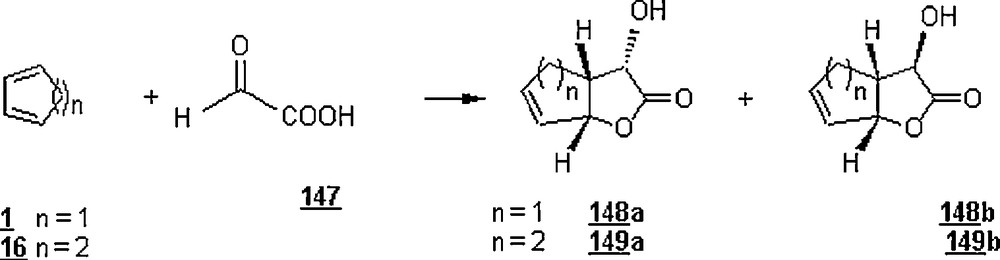
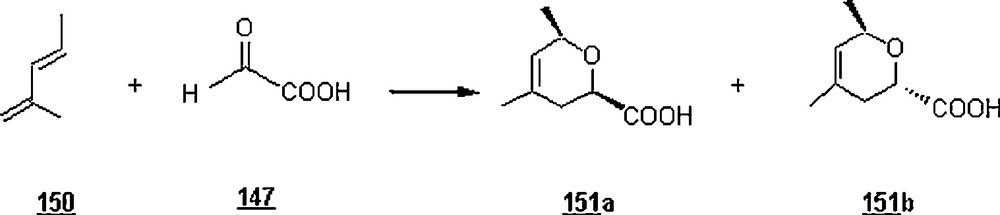
Lubineau et al. [87,88] reported the aqueous hetero Diels–Alder reactions between glyoxylic acid 147 and cyclopentadiene (and cyclohexadiene 16) to give good yields of the corresponding α-hydroxy-γ-lactones 148 and 149 (Scheme 47) as a mixture of epimers. With the first diene and at pH = 0.9 (2.25 M glyoxylic acid in water), the reaction was completed in 90 min at 40 oC, with 83% overall yield of α-hydroxylactones 148a and 148b in a 73:27 ratio. In the case of cyclohexadiene, the reaction in aqueous solution afforded α-hydroxy-γ-lactones 149a and 149b in a 85% yield in a 60:40 ratio. The reaction outcome with cyclopentadiene was pH-dependent: in glyoxylic acid solution (2.25 M) and at a pH = 2.5, the reaction gave α-hydroxy-γ-lactones 148a and 148b (72%; 60:40) after 7.5 h at 60 oC, whereas at pH = 6 the lactones were obtained in the same ratio but in only 16% yield after 24 h at 60 °C. The authors examined the heterocycloaddition reaction of glyoxylic acid aqueous solution with 2-methyl pentadiene 150, a less reactive diene, (Scheme 48). The results were that the reaction yield was quantitative after 1.5 h at 100 °C with a 64:36 epimer ratio 151a/151b. The use of water-tolerant Lewis acids such as the triflates enhanced the yields of reaction; the reaction was quantitative after 12 hours at 60 °C in the presence of 0.1 equivalent of Yb(OTf)3 or Nd(OTf)3, whereas the yield was only 55% under the same conditions and in the absence of a catalyst. On the other hand, the reaction of glyoxylic acid with isoprene required more drastic conditions (100 °C, 18 hours), giving two vinyl lactones in 33% yield and a cycloadduct in 28%.
Pyruvaldehyde 152 and glyoxal 154 were evaluated as heterodienophiles for 2-methylpentadiene (Schemes 49 and 50). The reaction with pyruvaldehyde led to cycloadducts 153a and 153b in 96% and in a 47:53 ratio after 48 h at 100 °C. With glyoxal, the yield of cycloadducts 155 was low after 60 h at 100 °C.

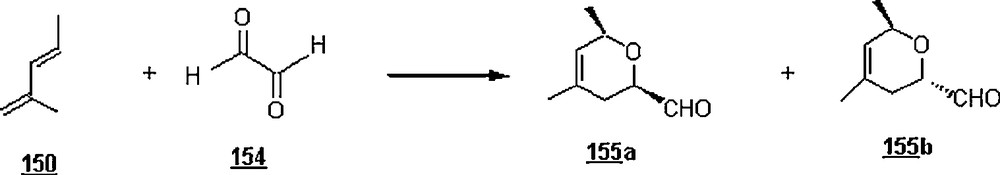
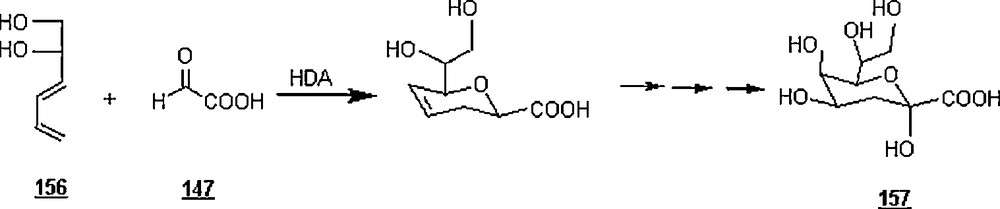
The same research group [89] synthesized 3-deoxy-d-manno-2-octulosonic acid (KDO) 157 with a modest facial stereoselectivity Si/Re of 74/26 via a hetero Diels–Alder reaction (Scheme 51). One step of this sequential synthesis involved the aqueous hetero Diels–Alder reaction (Scheme 52) of water-soluble diene 156 with sodium glyoxylate 158 at pH = 6 went to completion at 110 oC within 18 h, giving after esterification an inseparable mixture of four cycloadducts in 54% yield and in the proportions 42/32/19/7. The features of this heterocycloaddition were the use of sodium glyoxylate, a water-soluble diene, instead of alkyl glyoxylate whose preparation is tedious, and a relatively lower temperature, 110 °C versus 130 °C. To recall, KDO is an essential component of the outer membrane lipopolysaccharide of all Gram-negative bacteria and is found in the primary cell wall of higher plants.
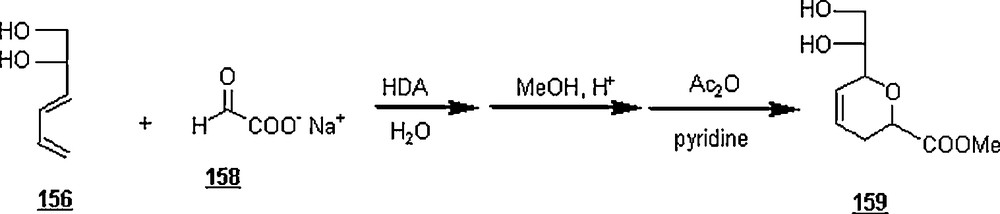
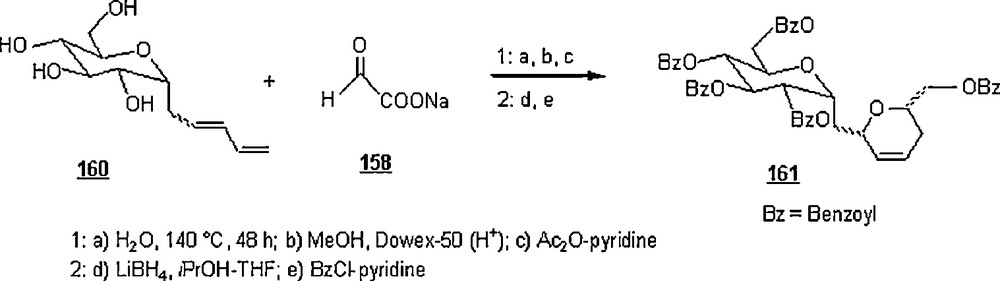
In an extended work on carbohydrate chemistry, Lubineau et al. [90] prepared C-disaccharide analogs of α,α-trehalose through a sequential synthesis involving an aqueous Diels–Alder cycloaddition. The α-d-C-dienylglucoside 160, made from (2,3,4,6-tetra-O-acetyl-α-d-glucopyranosyl)-ethanal, reacted with sodium glyoxylate 158 in water to lead to the C-disaccharide 161 after work-up (Scheme 53); a mixture of four diastereoisomers in the proportions 41:24:21:14 was isolated in a 74% yield.
Akiyama et al. [91] worked out the Brønstëd acid-promoted aza-Diels–Alder reactions of aldimines 162 with Danishefsky's diene 163 in water to give dihydro-4-pyridones 164 (Scheme 54). As shown in Table 5, the yields of latter products of two-component cycloaddition reactions were substantial. Also, a number of three-component aza-Diels–Alder reactions (Scheme 55) were conceived and were successful in aqueous medium, affording the same cycloadducts in good to high yields (Table 5).
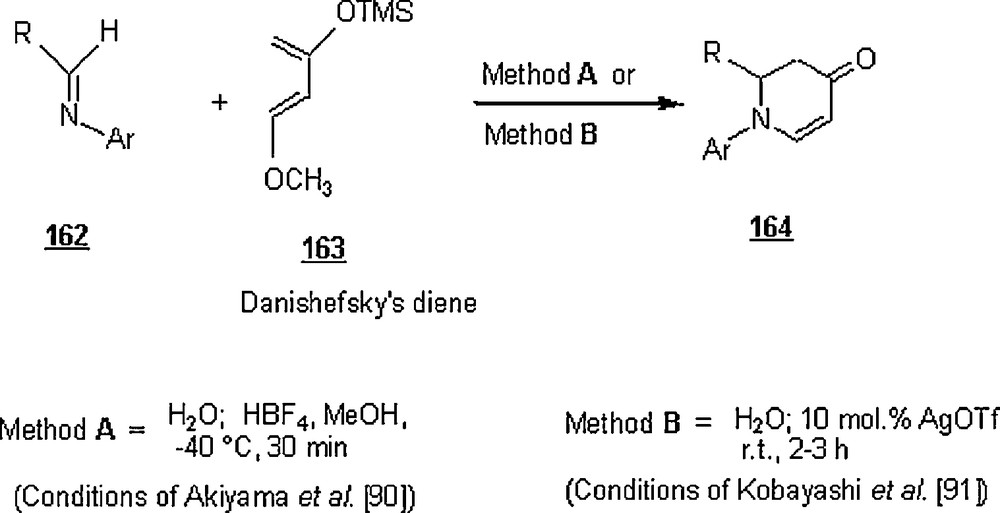
Yields (%) of the aza-Diels–Alder reactions.
| Two-component reaction | Three component reaction | ||||
| R | Ar | Method A | Method B | Method A | Method B |
| Ph | Ph | 98 | 83 | 95 | 63 |
| Ph | p-MeOC6H4 | 90 | – | 88 | 56a |
| p-CH3C6H4 | Ph | 95 | – | 79 | – |
| p-NO2C6H4 | Ph | 87 | 69 | 65 | – |
| c-C3H11 | Ph | – | – | 80 | 70 |
| PhCH2CH2 | Ph | – | – | 75 | 53 |
| (CH3)2CH | Ph | – | – | 77 | – |
| 2-Furyl | Ph | – | – | 58 | – |
| PhCHCH | Ph | 89 | – | – | – |
| p-MeOC6H4 | Ph | – | 77 | – | – |
| p-BrC6H4 | Ph | – | 75 | – | – |
| Ph | p-BrC6H4 | – | 83 | – | 90 |
| PhCHCHCH2 | Ph | – | 92 | – | – |
| (CH3)2CHCH2 | Ph | – | – | – | 72 |
a With addition of 10 mol.% Triton × 100.
Later in 2003, Loncaric et al. [92] undertook a similar investigation on aza-Diels–Alder reactions as Akiyama et al. [91], using silver-triflate catalysis in both two-component and three-component reactions. The different AgOTf-catalyzed aza-Diels–Alder reactions of Danishefsky's diene with imines (two-component system) in water (Schemes 54) afforded the corresponding dihydro-4-pyridones in high yields (up to 92%). Among the silver salts (AgOTf, AgClO4, AgOAc, AgSbF6, AgBF4, AgNO3, Ag2CO3), AgOTf gave the best results in terms of yield; the reactions were unsuccessful either in the presence of AgOAc and Ag2CO3 or in the absence of the remaining silver salts. It was advanced that the high efficiency of the reactions in water was probably a result of the slower hydrolysis of Danishefsky's diene under heterogeneous reaction conditions, preventing the formation of side products.
Also, the cycloadducts from the three-component reactions (Scheme 55) were isolated in 50–90%. In some cases, addition of a non-ionic surfactant and increasing concentration of Danishefsky's diene raised the reaction outcome to higher values. The role of the surfactant was believed to help the formation of the imine in water, since its addition to the two-component reaction did not improve the yield.
It is worth noting that the aza-Diels–Alder reactions with HBF4, a Brønstëd acid occurred in conditions milder than with AgOTf, −40 °C and 30 min versus room temperature and 2–3 h.
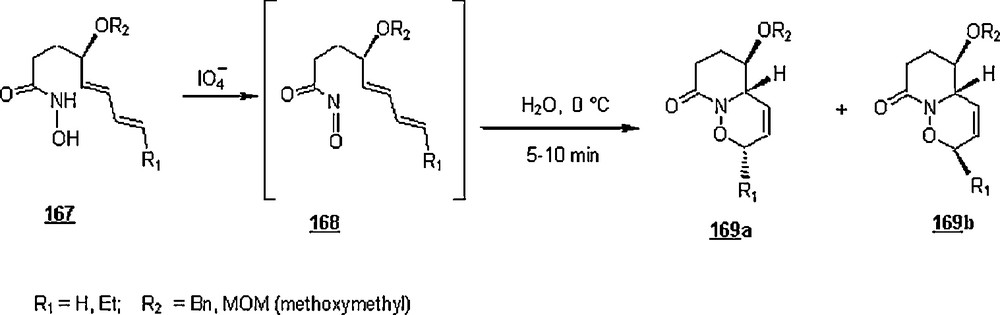
Owing to its low LUMO energy and weak π bond, the acylnitroso group may have a powerful dienophilic affinity, which allows the nitroso to undergo a rapid cycloaddition with diene in water. In this line, Naruse et al. [93] took adavantage of this excellent property for improving the trans stereoselectivity of intramolecular acylnitroso Diels–Alder cycloaddition. In the reactions shown in Scheme 56, the acylnitroso compounds 168 were produced in situ by oxidation of hydroxamic acids 167 with periodate. The whole reactions occurred at 0 °C and were complete within one minute to provide the cycloadducts 169a and 169b. In general, the yields in pure water and aqueous systems (87–97%) were higher than those in chloroform (75–86%). Also, the trans:cis was significantly enhanced in pure water, 4.4:1–5.0:1 against 1.3:1–1.9:1 in chloroform.
The first inverse-electron-demand Diels–Alder reaction in water, opposing to the normal-electron-demand ones (the common Diels–Alder reactions), was reported by Attanasi et al. [94]. It dealt with the reaction of electron-deficient (E)-3-diazenylbut-2-enes 170 (heterodienes) with a variety of electron-rich dienophiles (1, 172–175) in different solvents, including water (Scheme 57). At 15 °C, the reactions in aqueous medium (heterogeneous conditions) were faster than in organic solutions (dichloromethane, ethylacetate, toluene, THF), with higher yield conversions. In most cases, the ‘on water reactions’ were accomplished with high degrees of stereochemical and regiochemical control.
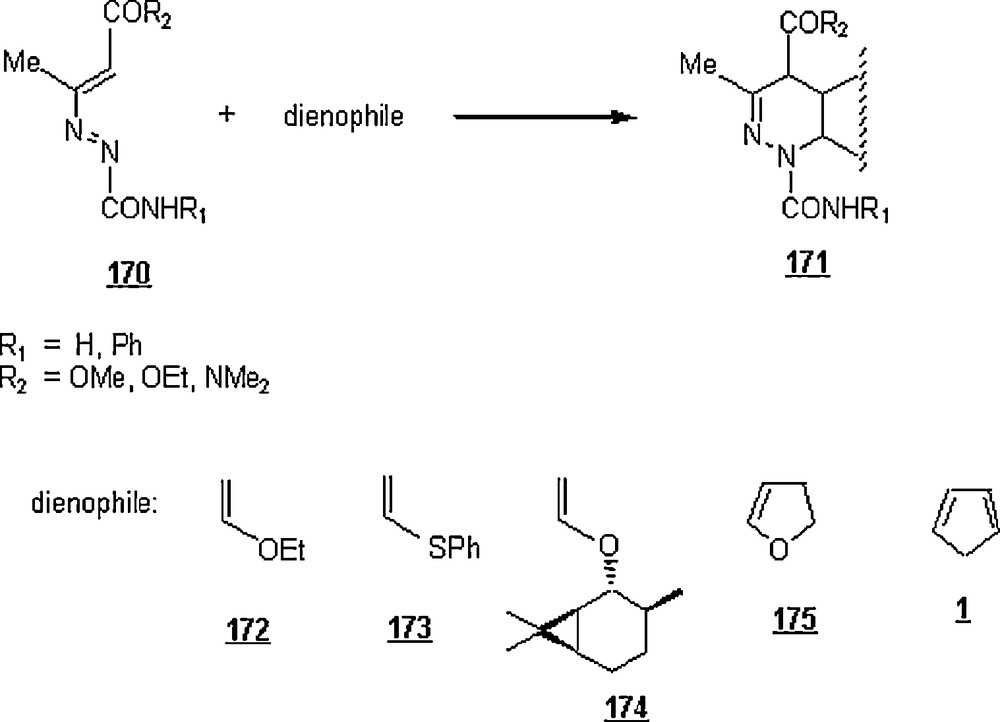
In this study, the first example of an asymmetric ‘inverse electron-demand’ Diels–Alder reaction in pure water was also disclosed, involving the cycloaddition of (E)-3-diazenylbut-2-ene (with R1 = H; R2 = OEt) with (+)-2-(ethenyloxy)-3,7,7-trimethylbicyclo[4.1.0]heptane, 174.
3 1, 3-dipolar cycloaddition reactions
3.1 Formation of [1,2,3]-triazoles
The 1,3-dipolar cycloaddition, also known as the Huisgen cycloaddition, is an organic2 reaction3 that belongs to the class of cycloaddition4 to make five-membered rings. The 1,3-dipolar cycloadditions of organic azides with alkynes are thermodynamically favored by 30–35 Kcal/mol to lead to 1,2,3-triazoles. Chemistry and synthetic approaches of these triazoles in conventional solvents were already surveyed [95]. The importance of 1,2,3-triazoles as agrochemicals, anticorrosive agents, dyes, optical brighteners, pharmaceuticals, photostabilizers, antimicrobial activity, synthons for the preparation of antitumor dehydropyrrolizidine alkaloids and for the modification of nucleosides as antiviral agents including anti-HIV activity. The cycloaddition reactions between azides with alkynes, well-known as Huisgen cycloadditions5.
Li et al. [98] evaluated the Huisgen cycloaddition reaction of azides with electron-deficient alkynes in water to make 1,2,3-triazoles. The authors launched the 1,3-dipolar cycloaddition of 5-azidovalerate 176 with a series of electron-deficient alkynes 177 (Scheme 58). The 1,2,3-triazoles 178 were obtained in good to excellent yields, 67–94%. The use of Cu(I) catalyst was found to enhance both yield and reaction rate for cycloaddition with terminal alkynes; for example, the reaction involving alkyne with R1H and R2CH2OSO2Ph was completed in one hour only with a yield of 85%, compared to 67% for uncatalyzed reaction.
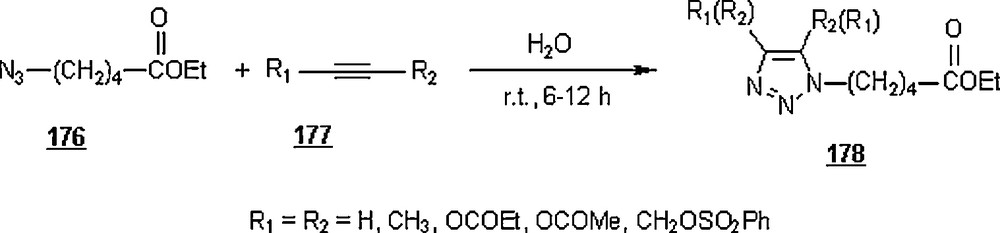
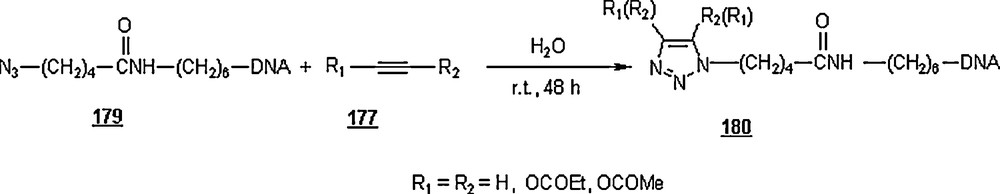
Under identical conditions, the Sharpless’ click chemistry was applied to the cycloaddition of an azido-DNA 179 with electron-deficient alkynes as pictured in Scheme 59. Yields of 180 were generally moderate, 45–67%. The use of Cu(I) for the reaction using alkyne with R1H and R2OCOMe did not improve the yield, but lowered it from 67 to 60% after 24 h.
Lia and Wang [99] claimed the success of the preparation of 1,4-disubstituted 1,2,3-triazoles starting with terminal alkynes, organohalides and sodium azide, in water and in the absence of a catalyst, namely Cu(I). The work-up seemed to be very simple: the mentioned reactants in 1:1:1.1 molar ratios were suspended in distilled water with vigorously stirring at 100 °C for 24 h. Terminal arylalkyne, such as phenylacetylene, p-methylphenylacetylene, p-bromophenylacetylene, or p-chlorophenylacetylene was employed to react with benzyl halides including benzyl bromide, benzyl chloride, p-methylbenzyl chloride, and p-nitrobenzyl chloride. Most of the reactions gave higher yields (>80%), with higher regioselectivity (1,4 isomers as major products). However, when the reactions were conducted in DMF and EtOH, the reaction rates were slower, affording a mixture of regioisomers in 88 and 15% yields, respectively. It was noticed that the extent of production of 1,4-disubstituted regioisomer rose with the increase of alkyl chain in primary aliphatic bromides.
Wang and Qin [100] examined 20 1,3-dipolar cycloaddition reactions of a series of dipoles 181 with a number of dipolarophiles 182 in water (Scheme 60). The reaction temperatures were either 85 or 120 °C. For terminal arylalkynes such as phenylacetylene or p-methylphenylacetylene, only 1,4-disubstituted 1,2,3-triazoles were obtained in 81–97% yields after 24 h of reaction. Prolonging the reaction time or raising the reaction temperature promoted higher yields. Indeed, the yield of reaction of p-methylazidobenzene with phenylacetylene was 38% at 85 °C for 24 h, 72% at 120 °C for 4 h and 85% at 120 °C for 24 h. The rate-accelerating effect of electron-withdrawing group on the phenyl ring of azide was observed for the reactions with m-nitroazidobenzene. However, reaction of aliphatic alkynes such as HC≡CC(O)OEt or HC≡CCH2OC(O)Me with most aryl azides at 120 °C usually gave a mixture of regioisomers with the ratio of 1,4- to 1,5-isomers ranging from 3:1 to 28.6:1.
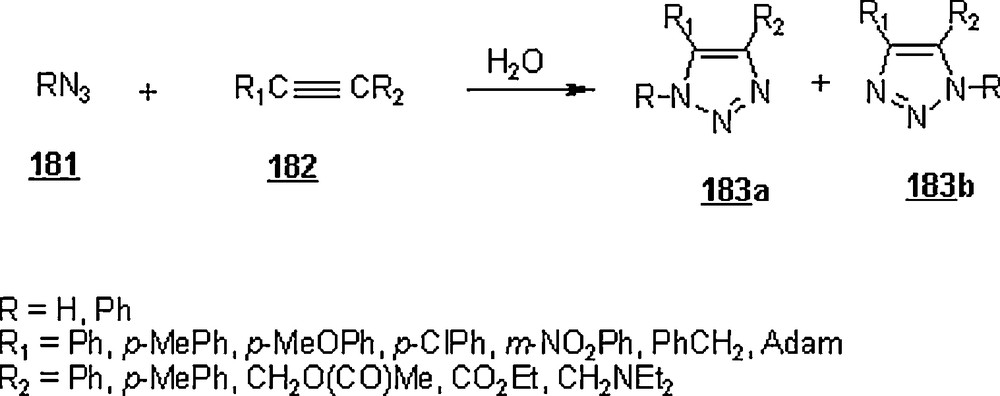
As to the disubstituted acetylenes, they afforded 1,4,5-trisubstituted 1,2,3-triazoles in hot water in low to excellent yields (36–92%). The results showed that the electronic effects of substituents on the phenyl ring of azides affected the reactions; the electron-withdrawing groups promoted the reaction, whereas the electron-donating groups impeded it.
Molteni and Ponti [101] carried out the 1,3-dipolar cycloadditons in aqueous medium between phenylazide and p-substituted derivatives 184 with methyl propiolate 185 in the presence of tetrahexylammonium chloride (THAC) as a phase transfer catalyst and at a temperature of 90 °C (Scheme 61). The reaction times were chosen as a function of substituent: 60 min (R = H), 40 min (R = Me), 30 min (R = MeO), 120 min (R = F), 300 min (R = NO2). These reaction times were shorter, compared to the results for uncatalyzed reactions obtained by Wang and Qin [100]. The overall yields of 186 were excellent (>92%) and were independent on the substituent. However, the reactions were regioselective as the cycloadducts 186a were the major regioisomers; the regioselectivity extent was substituent-dependent, but did not follow the electronic trend. The regioselectivities were explained in the light of the HSAB principle (hard-soft acid-base) developed within the framework of density functional theory (DFT).

Sulfoximidoyl-substituted 1,2,3-triazoles 188 were realized via Huisgen 1,3-dipolar cycloaddition in water, toluene and water–dichloromethane mixtures [102]. The reactions involved organoazides and sulfoximidoyl alkynes 187 (Scheme 62) and were run at reflux and for a time of 3 h. The triazole 188 was isolated in low yields (7–31%) when benzyl azide was used as the azide compound and toluene, water, and water–dichloromethane mixtures were employed as reaction media. However, an increase in the yield to 59% was observed when the reaction was run in pure water with benzyl azide formed in situ from benzyl bromide and sodium azide. When the same conditions were applied to the reaction conducted in water–dichloromethane (3:1), the yields went up to 68%. A feature of this work was that the work-up did not require copper (I) catalysis.

The optimal reaction conditions were exploited for reactions of various sulfoximines in a Huisgen 1,3-dipolar cycloaddition with in situ-generated benzylazide derivatives (Scheme 63). The results in terms of yield suggested that the reactivity be dependent on the substituent affixed on sulfoximine 189, whereas (N-tosyl)-1-hexynylmethylsulfoximine (189a) and (N-tosyl)-3-methoxypropynyl- phenylsulfoximine (189b) gave the corresponding triazoles 148 in 73% and 51% yield, respectively. The fact that (N-tosyl)-2-tert-butylethynyl-phenylsulfoximine (189c) and (N-tosyl)-2-phenylethynyl-phenylsulfoximine (189d) were unreactive was explained on the basis of steric hindrance and electronic character.
Wijnen et al. [103] reported kinetic data on the 1,3 dipolar cycloaddition of phenyl azide 191 to norbornene 192 in organic solvents and water-rich water–alcohol mixtures at 40.3 °C, to give a dihydrotriazole derivative 193 (Scheme 64). The second-order rate constant was within a range of 4.7 × 10−5 to 17.5 × 10−5 M−1s−1 for organic solvents (from apolar aprotic to polar protic solvent), whereas that for water–alcohol mixtures was in the range of 35 × 10−5 −83 × 10−5 M−1s−1. The addition of 1 mol % of l-cyclohexyl-2-pyrrolidinone (NCP) induced a high rate (250 × 10−5 M−1s−1), that is, about 53 times faster than in n-hexane.
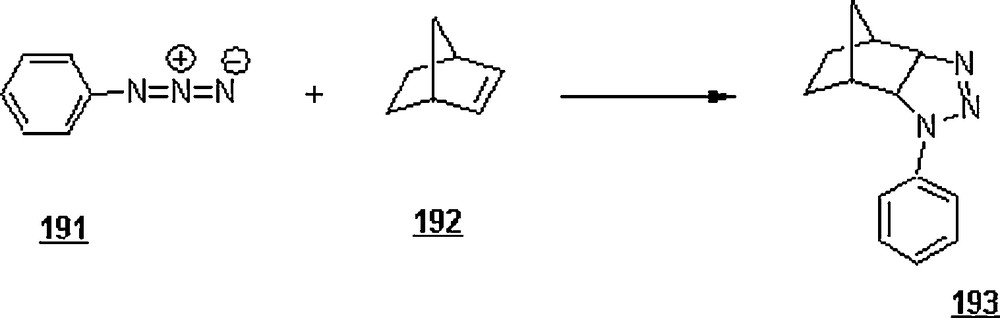
At low water concentrations of water–alcohol mixtures, a moderate rate enhancement was observed, whereas a substantial rate increase was gained at higher concentrations. The specific mole fraction of water in water–alcohol systems at which an optimal increase in rate was measured, increased in the order MeOH < EtOH < 2-PrOH < t-BuOH, reflecting the increasing hydrophobicity of the alcohols.
Recently, Aït-Youcef et al. [104] exploited the positive facet of aqueous micellar catalysis in the synthesis of ribavirin analogues (ribosyl-1,2,4-triazole) 196 by applying Huisgen cycloaddition to the reaction of C-alkynyl ribosides 194 with benzylazide 195. The micellar catalysis was first demonstrated on the 1,3-dipolar cycloaddition between carbohydrates 194 and 195 and benzylazide to yield two isomeric adducts, 1,4- and 1,5-adducts, as illustrated in Scheme 65.
The reaction did not succeed when run in pure water, but the addition of a surfactant caused the reaction to take place. The yields of triazole adducts were 56, 88, 73, and 82% for 1.5 mM solutions of SDS, DTAC (dodecyltrimethylammonium chloride), CTAB, and OGH (octoylglucosylhydrazide), respectively, after a time of 24 h and at a temperature of 70 °C. The reaction under aqueous micellar catalysis always favored the 1,4-adduct. Under these conditions, the 196c/196d ratio was: 58/42 (SDS), 83/17 (DTAC), 59/41 (CTAB), and 87/13 (OGH). The micellar catalysis was then applied to the 1,3-dipolar cycloaddition between ribosylpropiolate 197 and benzylazide leading to the 1,4,5-trisubstituted triazoles 198a and 198b as traced in Scheme 66.
The results were that the yields of adducts 198 were in the range of 46–58% and did not vary systematically as SDS concentration was increased under the following conditions: 70 °C and 24 h. In contrast to the above reaction, the 198b adduct was the major product, about 74%. However, the yield was enhanced upon prolonging the reaction time to 3 days without affecting the regioselectivity.
Alkynes with electron-deficient groups are highly reactive with azides in click chemistry. The 2-chloroacrylonitrile 200, a cyanoacetylene equivalent, reacted with a number of azido compounds 199 [105] and 202 [106] in water to give the corresponding 1-substituted 4-cyano-1,2,3-triazole cycloadducts 201 and 203, respectively (Schemes 67 and 68). The compound 201 was the only triazole regioisomer obtained in the former scheme. The reaction shown in the latter scheme was also carried out in n-heptane, toluene, ethanol, DMF, and the yields (46–78%) were lower than that in water (83–98%).
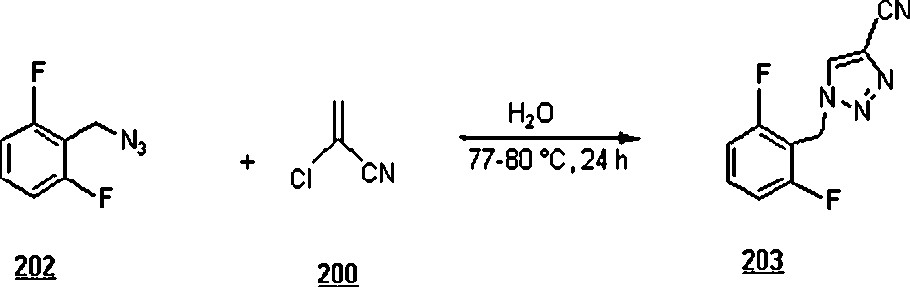
Azido alcohols 204 and 205 which were obtained from their corresponding isomeric diepoxides were subjected to Huisgen cycloaddition with diethyl acetylene dicarboxylate 206 on water [106] (Scheme 69). The crystalline solids bistriazoles 207a and 207b were isolated in quantitative yield (>95%). The dipolarophile 206, being electron-deficient alkynes, reacted readily and cleanly, even in the absence of a catalyst, with the azides on water.
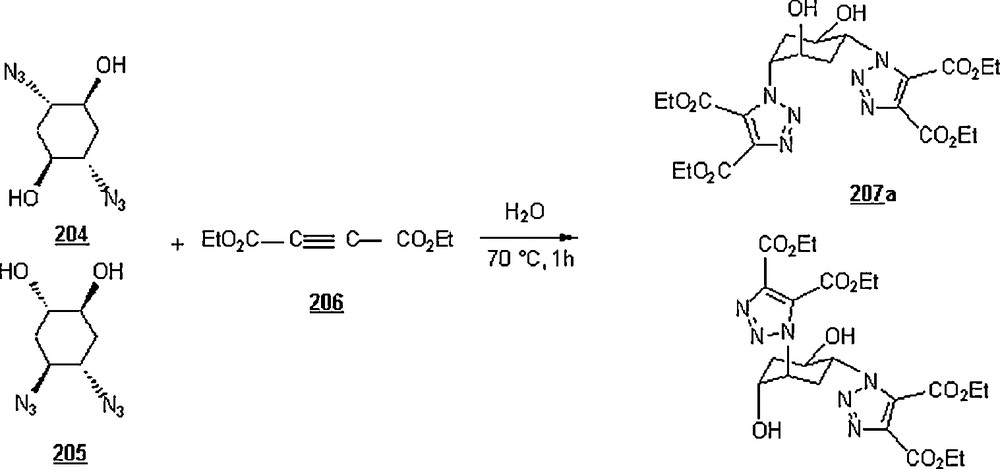
3.2 Formation of tetrazoles
Demko and Sharpless [107] made 5-aromatic tetrazoles 209 by reacting aromatic nitrile 208 with sodium azide in water and in the presence of zinc chloride as a catalyst (Scheme 70). The yields were generally good to excellent, 67 to 96%; in most cases, the reactions were carried out under reflux and for a varying reaction time, 2 to 48 h. It was noted that the more electron-poor the nitrile, the faster it reacted. Also, the reactions with electron-poor aromatic and heteroaromatic nitriles, such as 2-cyanopyridine and cyanopyrazine, were complete within a few hours, but some electron-rich aromatic nitriles required higher temperatures. One of the features of this proceedure was that the formation hydrazoic acid was minimized even at a temperature of 100 °C. Another remark was the exclusive catalytic role of zinc bromide; among other zinc salts and other metallic salts, zinc bromide was found to give better results in terms of reactivity.
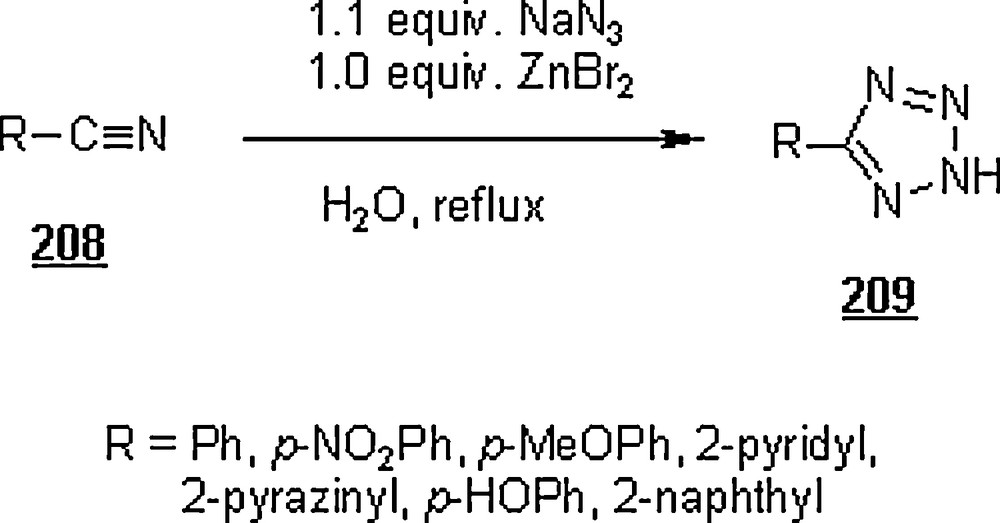
Myznikov et al. [108] applied the above Sharpless’ strategy for making substituted tetrazoles from a series of nitriles and sodium azide. Their reaction conditions were: water as the reaction medium, ZnCl2 as a catalyst, a temperature of 92–95 °C, and under microwave-assisted conditions (Scheme 71). Indeed, the yields of substituted tetrazoles were moderate to good, 60–83%, after a reaction time between 2 and 14 h.
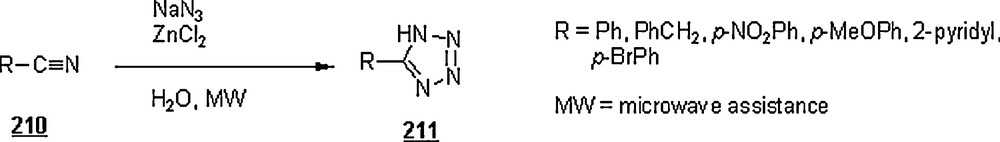
3.3 Formation of pyrazoles and pyrazolines
An account on syntheses and chemistry of pyrazoles in appropriate media was reported [109]. Jiang and Li [110] developed a water-borne 1,3-dipolar cycloaddition for making pyrazoles which are well known for their insecticide and herbicide potency and anti-tumor, anti-inflammatory, anti-microbial and anti-psychotic activity. Their first attempt was the reaction of ethyl diazoacetate 212 with ethyl propiolate 213 (Scheme 72) via an intermolecular 1,3-dipolar cycloaddition. The results were that pyrazole 214 was obtained in 87% yield when the reaction was conducted in water and in the presence of 20% InCl3 at room temperature for 24 h. The use of InCl3 as a reaction catalyst was to lower the LUMO of alkyne dipolarophile, inducing a decrease of HOMO–LUMO energy gap between the reactants of cycloaddition. A featuring aspect of this work was the reuse of catalyst while in the aqueous phase without loss of its catalytic activity (89% and 90% yields of pyrazole 214 for second and third run, respectively). The postulated mechanism for the formation of pyrazole 214 was an initial 1,3-dipolar cycloaddition to form 3H-pyrazole, which underwent a spontaneous 1,3-hydrogen migration. It is found that the use of organic solvent, namely benzene and dichloromethane, afforded traces of the targeted cycloadducts.
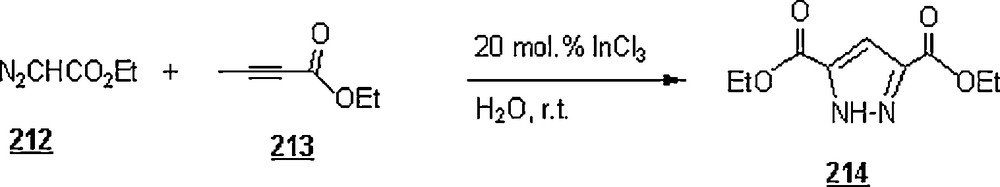
The protocol was extended to the reaction of methyl α-diazophenylacetates 215 and methyl propiolate 216 (Scheme 73). The reactions were more regioselective, leading to the product 217a predominantly with yields of 77–90%, and 217b with yields of 4–12%. The results indicated a slight electronic effect of substituent on the phenyl group.
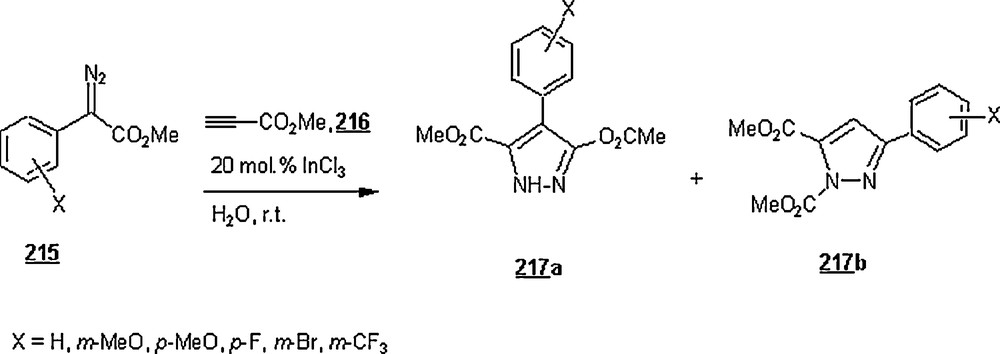
Since the reaction of phenylacetylene with α-diazophenylacetates failed to give the desired product, suggesting that InCl3 actually activated the alkynes by coordinating its neighboring carbonyl group and thus lowers the LUMO of alkyne group. The success of InCl3-catalyzed intermolecular cycloaddition of α-diazophenylacetates with alkynes with adjacent carbonyl groups was confirmed by reaction of a set of α-diazocarbonyl-bearing substances, which gave the corresponding products in moderate to good yields (37 to 87%).
Molteni et al. [111] tackled the synthesis of a number of 1-aryl-5-substituted-4,5-dihydropyrazoles (pyrazoline) 221 via a 1,3-dipolar cycloaddition between an array of substituted nitrilimines 219, produced in situ from hydrazonyl chlorides 218, with the appropriate vinyl dipolarophiles 220 in aqueous media and in the presence of a surfactant or a phase transfer catalyst and at room temperature (Scheme 74). The reaction outcome depended on the electronic factors of both nitrilimines 219 and alkenyl dipolarophiles 220: the yields fluctuated from a very low yield (3%) to a quantitative one (95%) and the reaction time was in the range of 10–360 min. The cycloaddition between nitrilimines 219a–c (with electron-donating groups) and ethyl acrylate 220a afforded the corresponding 4,5-dihydropyrazoles 221aa, 221ba and 221ca in 93–95% yields after 10 min only. On the other hand, the reactions of 219a–c with 1-hexene 220c and butyl vinyl ether 220d afforded 221ac, 221bc, and 221cc in poor to modest yields (17, 31, 52%) at a reaction time of 90–120 min, and 221ad, 221bd, and 221cd in poor yields (0, 24, 27%) after reaction times of 120–150 min; these results would infer the governance of dipole HOMO (nitrilimine) or/and of the dipolarophile LUMO (alkenyl subtrate). To assess the catalytic effect of surfactant of the phase transfer catalyst on the extent of cycloaddition reactions, the reaction of 218b with acrylonitrile (220b) was also performed in the presence of such additives. It was found that no cycloadduct was formed in the absence of these additives. The yields were surfactant concentration-dependent. Indeed, increasing the concentration of SDS from 2 to 14 mM, the yields jumped from 75 to 88% for a reaction time of 420 min. Also, varying THAC (tetrahexylammonium chloride) concentration from 1 to 17 mM induced an almost linear increasing in yield from 62 to 94% for a reaction time of 120 min.
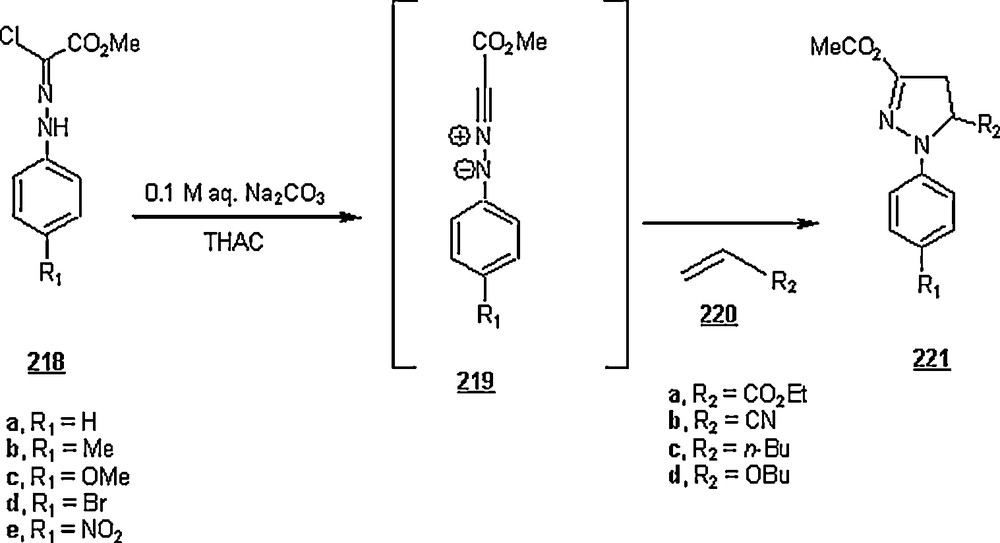
Molteni et al. [112] investigated the aqueous 1,3-dipolar cycloaddition of the in situ generated nitrilimine 220b with a number of dipolarophiles 222 (Scheme 75): (a) ethyl acrylate, (b) ethyl methacrylate, (c) 2,3-dihydropyran, (d) acrylonitrile, (e) sodium acrylate, (f) propargyl alcohol, and (g) CH2C(COOMe)2. No products were isolated when reactions were run only in the presence of 0.1 M NaOH and stirred at room temperature for 24 h. But, addition of additives such as THAC coupled with a magnetic stirring at room temperature for 24 h provoked the reactions to give the pyrazoline cycloadducts 223 in variable yields, depending on the dipolarophile: 223a (100%), 223b (7%), 223c (92%), 223d (28%), 223e (27%), 223f (0%), 223 g (19%). The micellar catalysis displayed by THAC in these reactions was a clear-cut, that is, the hydroxide ion was driven though electrostatic attraction by the cationic surfactant from the bulk aqueous medium to the surface of micellar aggregate.
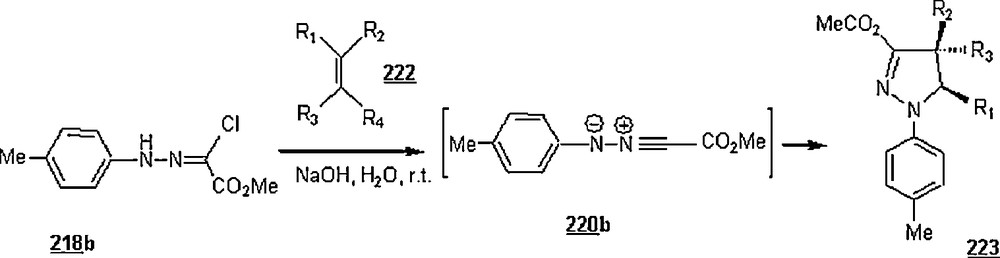
By means of Bambord-Stevens reaction, Pang et al. [113] were able to make 5-perfluorophenyl 4,5-dihydro-1H-pyrazoles (pyrazoline) 226 from 2,4,6-triisopropyl-N′-(perfluorobenzyl)benzenesulfonylhydrazine 224 and and α,β-unsaturated esters or acrylonitrile 225 through an aqueous 1,3-dipolar cycloaddition reaction (Scheme 76). Although the reactions in THF were quantitative (88–98%), the yields of those performed in “on-water” were 99% for all. In this experiment, the work-up was easier and the crystalline products were easily isolated.

3.4 Formation of isoxazoles
Isoxazolines were synthesized via a nitrile oxide intramolecular 1,3-dipolar cycloaddition in water [114], the nitrile oxide being generated in situ by oxidation of the oxime with sodium hypochlorite (bleach). Scheme 77 shows the attempted reactions where benzaldoxime derivatives were the starting materials. The isoxazoline 228a was isolated in 90% yield when the system made of benzaldoxime 227a/sodium hypochlorite/THF-water (7:3) was stirred at room temperature for 18 h. An increasing in water content led to a yield drop. In pure water, the reaction yield was 25 and 90% for a reaction time of 18 and 72 h, respectively, and, performing the reaction in a 90 mM SDS aqueous solution did not enhance the yield (24%). Moreover, no product was detected when the reaction was conducted in a 95:5 dichloromethane–water mixture, because of the poor solubility of 227a in dichloromethane. However, this medium was favorable for benzaldoxime 227b due to its good solubility, giving isoxazoline 228b in 90% yield after 18 h at room temperature. The latter isoxazoline was obtained in 40 and 92% yield in pure water after 18 and 48 h, respectively. The same observation was made as to the effect of SDS surfactant.
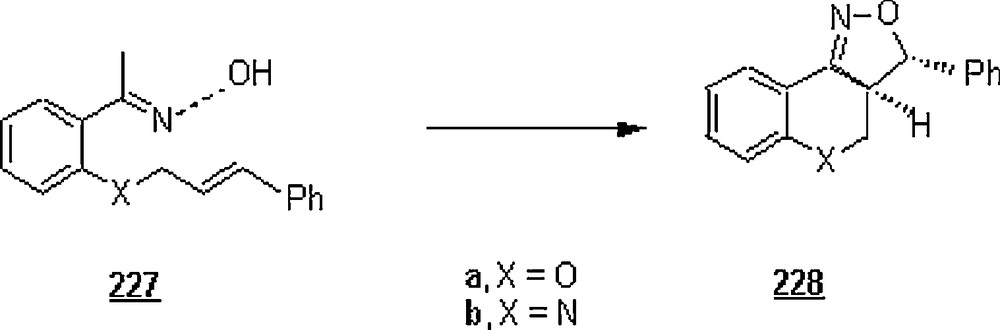
The above experimental procedure when applied to the synthetic benzaldoxime 229 (Scheme 78), yielded diisoxazoline 230, a cyclophane-type molecule, in 97% yield. About the same yield was obtained when the reaction was run in 95:5 DCM/water.

The effect of solvent on the nitrile oxide 1,3-dipolar cycloaddition was soundly undertaken by Rispens and Engberts [115]. The examined reactions involved benzonitrile N-oxide 231 as a dipolar compound and cyclopentene 233 and N-ethyl maleimide 13 as a dipolarophile (Scheme 79); benzonitrile N-oxide is usually generated in situ because it dimerizes quickly. Protic and aprotic solvents, including water, of different ET(30) (Dimroth-Reichardt polarity parameter) were experimented. A general observation was that a decrease in rate constant of the dipolar cycloaddition followed the ET(30) increase for ET(30)s ≤ 40, and a random dependence for ET(30)s > 40. For solvents of ET(30) ≤ 40, the rates were weakly dependent on the solvent, stemming from the fact that a partial disappearance of the 1,3-dipole occurred, leading to only small accelerations, and a decrease in rate on going to a more polar solvent. However, for solvents of ET(30) > 40, the rates depended on both the solvent polarity and the hydrogen bond-making capacity. The more polar the alcohol studied, the lower the rate. Best of all, the rates of reactions run in water were superior, being the most polar solvent with ET(30) = 61.3; compare the rate constant ratios kwater/kTFE: (ET[30] = 59.8 for TFE) for both reactions: 5.74 (Scheme 79a) and 2.60 (Scheme 79b).
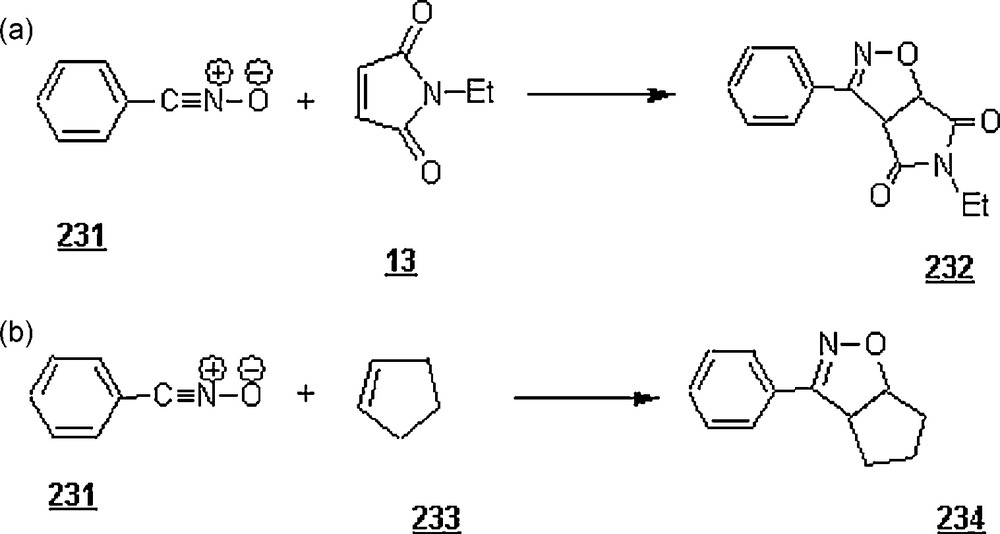
Inoue et al. [116] touched upon the 1,3-dipolar cycloaddition of 2,6-dichlorobenzonitrile N-oxide 235 with 2,5-dimethyl-p-benzoquinone 236a and 2,6-diisopropyl-p-benzoquinone 236b in various organic solvents and water–ethanol mixtures at 20 °C, as illustrated in Scheme 80.
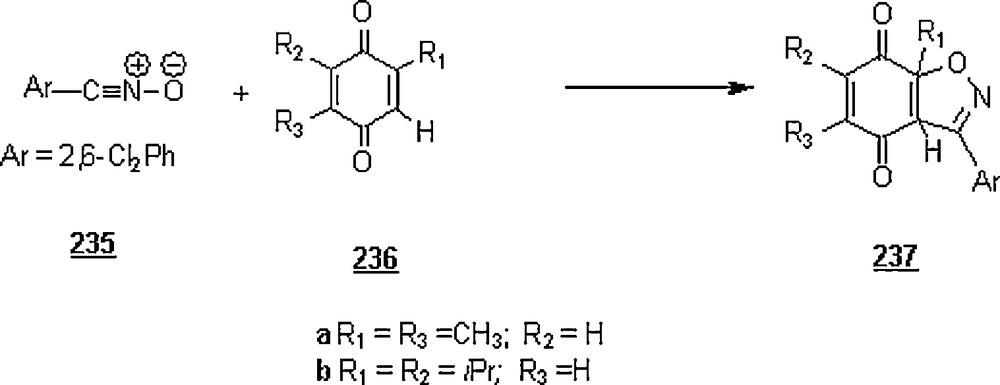
In case of reaction with 236a, the order of increasing reaction rate with solvent was as follows: water–ethanol (40:60) > H2O-EtOH(20:80) > cyclohexane > EtOH > DMSO > DMAc > o-xylene > acetonitrile > benzene ≅ ethylacetate > chloroform. Thus, the reaction was accelerated in highly polarized aqueous ethanol, about 14 times greater than that with chloroform. However, the rates of reaction of 235 with 236b under the same conditions in different solvents were almost twice as large as those of the reaction with 236a, and the rate in H2O-EtOH was always higher than in other solvents. The yield of reaction 235 with 236b was 86% after 5 days when benzene was used as a solvent, but it was 77% after only one hour when water-ethanol (40:60) was the medium. The difference in rates between the two reactions was ascribed to electron-donating character of the isopropyl groups. The cycloadduct from the reaction with 236b could be rearranged to give a isoxazole-fused catechol, which displays barnaclecide activity like 3,4-dihydroxyphenyl-l-alanine (l-DOPA).
Chatterjee et al. [117] reported an efficient strategy for a chemoselective oxidation of aldoximes 238 to nitrile oxides 239 by iodosobenzene in neutral aqueous media. Their in situ intermolecular 1,3-dipolar cycloaddition with olefins in nanometer aqueous micelles (CTAB as surfactant) occurred with improved stereoselectivity and acceleration rate toward synthesis of new chiral synthons of isoxazolines (Scheme 81). The nitrile oxides were formed inside the micellar nanoreactor and trapped by olefins 240 to obtain Δ2-isoxazolines 241. The reactions proceeded at room temperature with a large acceleration of reaction rate (2.5–4.0 h) and with excellent isolated yields (71–82%). The results suggested that the reaction rate and yield were almost independent on the nature of substrates used.
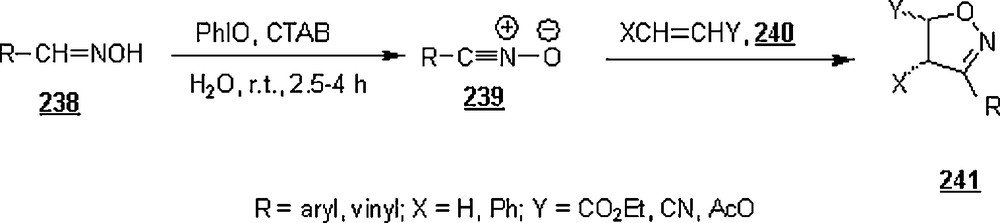
This green protocol was applied to the stereoselective synthesis of 3-(2′-C-3′,4′,6′-tri-O-benzyl glycal)-Δ2-isoxazolines. The reactions for making these isoxazolines using N-chlorosuccinimide (NCS) and 4-dimethylaminopyridine (DMAP) were very slow (∼3 days) and the stereoselectivity was not observed in these cycloaddition reactions. However, by applying this environmentally benign approach to sugar aldoximes, corresponding nitrile oxides were generated in neutral media. Their in situ 1,3-dipolar cycloadditions with olefins produced their chiral corresponding cycloadducts in good yield (57–64%) and with enhanced stereoselectivity.
Engberts'school advanced a research on the influence of electronic nature of the dipolarophiles on their 1,3-dipolar cycloaddition with benzonitrile N-oxide in water, organic solvents, and in aqueous solutions [118]. Two classes of dipolarophiles were examined: electron-rich dipolarophiles (cyclopentene and 2,3-dihydrofuran), and electron-poor dipolarophiles (methyl vinyl ketone, acrylonitrile, and N-methylmaleimide). A rate-accelerating effect of water on the cycloaddition involving the electron-rich dipolarophiles, particularly with 2,3-dihydrofuran, was noticed; kwater/kn-hexane was found to be 3.25 for cyclopentene and 11.10 for 2,3-dihydrofuran at a temperature of 25 °C. However, a rate-retarding effect on the rate constants for cycloadditions with the electron-poor dipolarophiles was observed. These results were rationalized in terms of influence of hydrogen bonding of water to the reactants, that is, positive for electron-rich dipolarophiles, and negative for electron-poor ones. An explanation of these findings based on FMOs (frontier molecular orbital) was also suggested; indeed, electron-withdrawing substituents stabilize FMOs, whereas electron-donating substituents raise the FMO energy. Hydrogen bonding reduced the FMOs’ energies, that is, providing a maximum HOMO-LUMO overlap, and thus promoted 1,3-dipolar cycloadditions.
Profiles of the evolution of rate constant of the reactions held in the water–ethanol mixture as a function of mole fraction of water showed an increasing departure up to a maximum at a mole fraction of 0.9, and a decline beyond this fraction. The accelerating effect seen in the first parts of the profiles indicated an increase of hydrophobic interaction in water, and the retardation effect observed beyond 10 mol % for ethanol was due to a preferential solvation by ethanol of the reactants.
The impact of SDS micelles on the course of cycloaddition of benzonitrile N-oxide with 2,3-dihydrofuran and methyl vinyl ketone was also experimented. Surprisingly, the aqueous SDS solution showed a positive micellar activity, that is, a rate-acceleration even at SDS concentrations higher than CMC value; usually, the rate decreases at higher micelle concentrations. This result was explained by the fact that the binding of benzonitrile N-oxide to the micelles is relatively weak, thus a high concentration of micelles was necessary for a complete binding.
González-Cruz et al. [119] reported the aqueous-processed [3 + 2] cycloadditions between β-phosphonium (or ammonium) allenolates 244 as reactive dipolarophiles with nitrones 242. The allenolates 244, zwitterionic dipolarophile intermediates, were generated in situ as shown in Scheme 82. First, base-catalyzed 1,3-dipolar cycloaddition between phenyl N-benzylnitrone 242a and methyl 2-octynoate 243a in an “on-water” procedure was examined. Only one isomer of 2,3-dihydroisoxazole 245aa (complete regioselectivity) was isolated but in a low yield (10%) yield when an aqueous suspension of nitrone, alkynoate and Ph3P as a catalyst was vigorously stirred at 40 °C for 48 h. The reaction did not occur either in pure water (in the absence of a base), or in organic solvents (toluene, DCM). However, the yields were enhanced, up to 59%, when using amines as catalysts (Et3N, quinine, isoquinoline, DABCO, quinuclidine, DMAP). Also, the addition of LiCl to on-water cycloaddition promoted a better yield (68%).

The above-described protocol was extended to a number of nitrones (242b-e) and alkyonates (242b-e). The yields of corresponding 2,3-dihydroisoxazole depended on the reactivity of the nitrone and the base used as a catalyst. For example, nitrone 242a, which is the less reactive dipolarophile, reacted with alkynoates 243b and 243d at room temperature and in the presence of Ph3P (in the absence of LiCl) to give cycloadducts 245ab and 245ad in good yields, 99 and 61%, respectively. But, the reactions of 242a with alkynoates 243a and 243e required the use of LiCl and a temperature of 40 °C in order to afford the products 245aa and 245ae in acceptable yields, 68 and 71%, respectively. However, nitrone 242b, the most reactive dipolarophile, reacted with alkynoates 243a, 243b and 243d under identical conditions, giving 245ba, 245bb, and 245bd in excellent yields, 81, 95, and 94%, respectively. It was generally remarked that, when quinuclidine was employed in the base catalysis, good yields of the cycloadducts necessitated a temperature of 40 °C. Another attracting observation was that Ph3P was the best catalyst for reactions involving aromatic nitrone, and quinuclidine was for the reactions involving aliphatic ones.
Recently, McKay et al. [120] studied some Kinugasa reactions in micellar water using copper catalysis. As with Chatterjee et al. [117], the sequential reaction involved the micelle-promoted C,N-diphenylnitrones’ formation from substituted benzaldehydes 246 and N-phenylhydroxylamine 247 (Scheme 83). This was followed by the in situ [3 + 2] 1,3-dipolar cycloaddition with Cu(I) phenylacetylide and the rearrangement of resulting 1,2-oxazole to yield β-lactam at room temperature. It was found that Cu(I) loading did not significantly affect the product composition; however, lowering Cu loading resulted in longer reaction times (10 h for 20 mol % vs. 6 h for 100 mol %). On the other hand, the nitrogen-containing ligand was shown to have an effect on the reaction outcome. In addition, the use of excess pyridine (8–10 equiv %) afforded cis- and trans-249a in 46% yield and the reaction time was about 10 h, but, two equivalents of pyridine resulted in a very slow reaction (20 h) and 15% of 249a. One equivalent of 2,2′-bipyridyl as ligand provided cis- and trans-249a and 250 in yields of 42% and 29%, respectively. Tris[(1-benzyl-1H-1,2,3-triazol- 4-yl)methyl]amine (TBTA), a ligand commonly used in azide–alkyne cycloadditions, gave 249a and 250 in 31% and 22% yields after 24 h.

The other results of this work were the effects of base and substitutent on benzaldehyde 246a at the para- and meta-positions. As to the impact of substituents, electron-donating groups (methyl, methoxy) promoted lower yields of the corresponding cis- and trans-β-lactams in slightly shorter reaction times relative to electron-withdrawing groups (CN, CO2Me, NO2), that is, 8–13 h vs. 18 h. As to the base, amines had positive impact on the reaction course. Compared with primary and tertiary amines, secondary amines afforded β-lactam products with higher cis-diastereoselectivity. Also, bulkier amines favored the formation of cis-249a diastereomer.
Engberts et al. [121] worked out the 1,3-dipolar cycloaddition of benzonitrile N-oxide 231 to N-ethylmaleimide 13 in microemulsions of isooctane /AOT /water at 25 °C (AOT denotes sodium bis(2-ethylhexyl)sulfosuccinate) (Scheme 79a). The microemulsions tested ranged from [H2O]/[AOT] = 2 to 40. The results were that varying AOT concentration in the microemulsion had virtually no effect on the apparent second-order rate constant of the reaction, kapp. The cycloaddition reaction was roughly 140 times faster in the microemulsion (5.5 × 10−2 M−1s−1) than that in n-hexane (2.6 × 10−3 M−1s−1), and about 35 times faster than that in pure water (8.5 × 10−3 M−1s−1). The accelerating effect of microemulsion was thought to be the result of an increase in the local concentrations of reactants at the interface and the electrostatic interactions with the head-groups of AOT molecules. The catalytic effect of microemulsion was explained in terms of the frontier molecular orbital (FMO) which is affected by a number of factors, particularly hydrogen bonding; indeed, the latter parameter in cycloaddition reduces the energies of FMOs of reactants. It was rationalized that the interaction between the HOMO of benzonitrile N-oxide (1,2-dipole) and the LUMO of N-ethylmaleimide (dipolarophile) is less intense than that between benzonitrile N-oxide and water, being a protic solvent that promotes hydrogen bonding with dipolarophile. This would increase the Gibbs energy, leading to a retardation of the cycloadditions. Hence, on increasing the water content of microemulsion, the hydrogen bonding increases and would make the reaction rate of benzonitrile N-oxide cycloaddition to N-ethylmaleimide decline.
3.5 Formation of isoxazolidines
Gholami and Yangjeh [122,123] investigated the 1,3-dipolar cycloaddition of C,N-diphenylnitrone 251 with di-n-butyl fumarate 252 and dimethyl fumarate 6 in aqueous solutions and other solvents at 65 °C (Scheme 84) and the results are compiled in Table 6. The rate constants of cycloaddition reaction between 251 and dimethyl fumarate 6 were higher than those with di-n-butyl fumarate 252, a fact imputed to steric effects. Also, it was generally observed that the reaction rate decreased with increasing polarity of the organic solvent; the rates in n-hexane (ETN[30] = 31 Kcal/mol; ETN = normalized Dimroth-Reichardt polarity parameter) were eight- to 10-fold that in ethanol (ETN [30] = 51.9 Kcal/mol). But with water, which has the highest polarity (ETN[30] = 63.1 Kcal/mol), the rates were unexpectedly 13 and 125 times larger than that in ethanol for 6 and 252, respectively, pointing to the fact that the solvent polarity was not an important factor for the reaction rate in aqueous solutions. Indeed, with ethylene glycol, which has a closer polarity (ETN(30) = 56.3 Kcal/mol) to that of ethanol, the rates were about six and 18 times that in ethanol. And as expected, the addition of salts such as LiCl, NaCl or KCl (salting-out effect) enhanced significantly the rates (for di-n-butyl fumarate: kLiCl water/water = 4; k NaCl water/water = 3.9; kKCl water/water = 3.7; for dimethyl fumarate: kLiCl water/water = 2; k NaCl water/water = 1.24; kKCl water/water = 1.3), nearly 1.25 to four times larger than in pure water. Also as expected, the presence of urea (salting-in effect) in water medium diminished the rate by one and half. The increase of reaction rate by addition of alkali metal salts to water in reaction of 251 with 252 was higher than the reaction with 6, because the decrease of alkyl chain length in dialkyl fumarate lowers the hydrophobicity of fumarate, and hence the effect of hydrophobic interactions on the reaction rate decreased. The effect of aqueous alcohol dilution on the reaction rate was demonstrated with aqueous ethanol and aqueous 1-propanol systems; the rate sharply increased beyond 75% mole fraction of water in the first system and beyond 90% mole fraction of water in the second one, suggesting that 1-propanol raised the solubility of reactants in water more than ethanol, and the hydrophobicity of reactants was decreased more in 1-propanol than in ethanol. It was concluded that the solvophobicity Sp played a more important role than the polarity parameter ETN. In this work, the yields were not estimated.
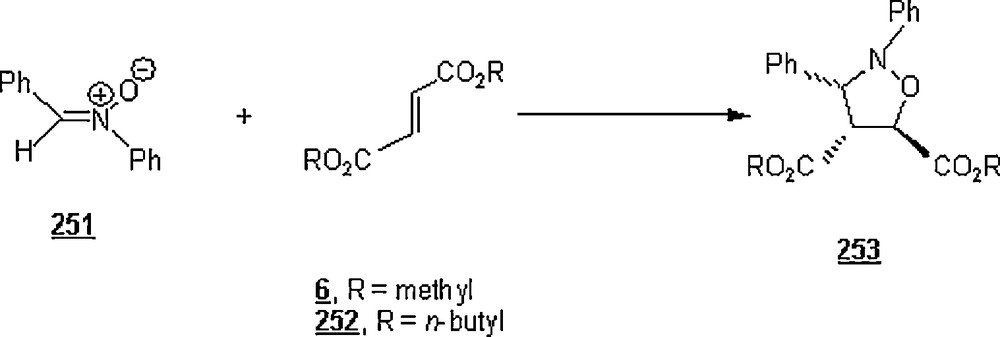
Second order rate constants of reaction between dialkylfulmarate and C,N-diphenylnitrone in various solvents at 65 °C.
| Solvent | 104×k2a | 104×k2b | ETN | Solvent | 104×k2a | 104×k2b | ETN |
| (dm3M−1sec−1) | (Kcal.M−1) | (dm3M−1sec−1) | (Kcal.M−1) | ||||
| n-hexane | 23.0 | 19.4 | 31.0 | EtOH | 2.9 | 1.9 | 51.9 |
| Toluene | 11.2 | 5.8 | 33.9 | Ethylene glycol | 17.0 | 34.9 | 56.3 |
| Benzene | 9.4 | 5.6 | 34.3 | H2O | 379 | 237.0 | 63.1 |
| THF | 12.6 | 5.9 | 37.4 | H2O + 2 MLiCl | 743 | 985.0 | |
| DMF | 5.1 | 2.5 | 43.8 | H2O + 2 M NaCl | 47.1 | 924.0 | |
| 2-PrOH | 4.9 | 3.1 | 48.4 | H2O + 2 M KCl | 42.8 | 876.0 | |
| 1-PrOH | 3.1 | 2.2 | 50.9 | H2O + 2 M Urea | 25.2 | 142.0 |
a Second-order rate constants of the reaction with dimethylfumarate 6.
b Second-order rate constants of the reaction with di-n-butylfumarate 252.
Pandey and Pandey [124] undertook the study of 1,3-dipolar cycloadditions of C,N-diphenylnitrone 251 with methyl acrylate, acrylonitrile and vinyl acetate as dipolarophiles 60, 81, and 254 in aqueous medium at 25 °C, affording the corresponding isoxazolidines 255 (Scheme 85). The yields in the case of methyl acrylate as a dipolarophile were 30 and 95% in benzene and water, respectively, for a reaction time of 3 h. For acrylonitrile, the yields were 15 and 95% after 2.5 h of reaction. However, the yields in the case of vinylacetate were relatively lower, even for a longer time (18 h): 5 and 45%. The appreciable enhancement of the reactions’ outcome in aqueous media was rationalized in terms of hydrophobic effect. The 1,3-dipolar cycloaddition reaction of C,N-diphenylnitrone with methyl acrylate 60 led to a mixture of four diastereoisomeric isoxazolidines. Quite unexpectedly, there was no marked increase in the regio- and/or stereoselectivity. It was observed that there was no significant change in the ratio of diastereoisomers (28:44:16:12) on changing the medium from benzene to water.

A study on 1,3-dipolar cycloaddition reactions of aromatic nitrones 256 to acrylonitrile 81 (ACN), affording isoxazolidines 257, was undertaken in a microemulsion medium made of ACN/water/SDS/n-butanol, ACN being the oily component (Scheme 86) [125]. The results were that the yields were not only enhanced (80–100%), but also the reactions rates were unexpectedly higher, particularly when triflate-based Lewis acids were employed as indicated below. The main feature of this work was temperature-dependence of the reaction kinetics; indeed, a 25 °C temperature rise induced a 90% drop in the reaction time. However, the yields of reactions in toluene, a conventional reaction medium, were rather moderate (31–69%) after refluxing for 12 h.
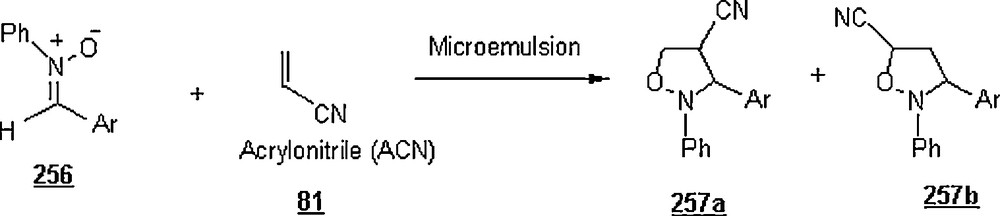
The yield and the reaction rate depended strongly on the type of microemulsion; the acrylonitrile-borne microemulsion afforded better yields and higher rates, because of the higher concentration of ACN. A water-rich microemulsion was a more favored medium than the SDS/n-butanol-rich microemulsion as far as yields and kinetics were concerned. The acrylonitrile-borne microemulsion W/O was in form of water microdroplets dispersed in acrylonitrile, the oily component. The course of reaction in this microemulsion is depicted in Scheme 87 where it is shown that once the nitrone was added, it tended to migrate towards the ACN-water interface. The nitrone molecules would then concentrate within the interfaces and be arranged in such a way that depends on their polarity, making them react faster and more selectively. Hence, the reaction occurred within the interfaces [126]. The formed isoxazolidine molecules, being less polar, would have the tendency to move off, allowing the nitrone molecules to reconcentrate within the interfaces.
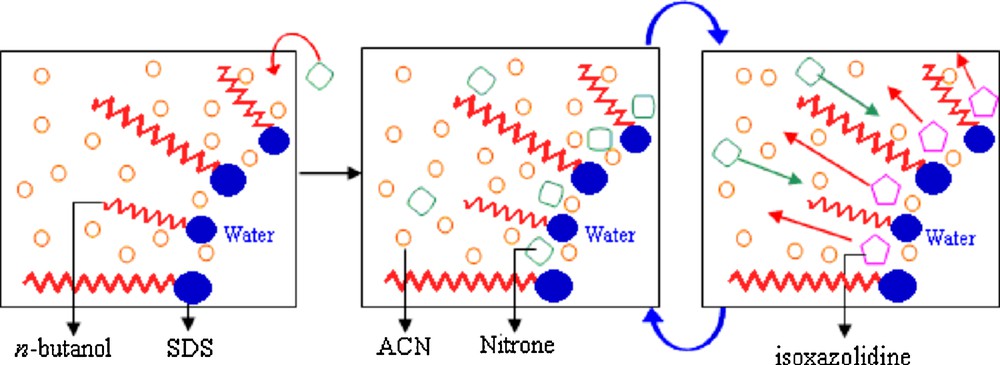
The reactions of nitrone (ArPh) with CAN were conducted in acrylonitrile-rich microemulsion in the presence of a metal triflate Lewis acid acting as a catalyst (La[OTf]3, Yb[OTf]3, Sc[OTf]3, Cu[OTf]2). It was observed that both the reaction rates and the yields were substantially enhanced by using 0.1 equiv of the triflate-based Lewis acids. About 80% yield was realized in the absence of a catalyst after a time longer than 6 h, but this yield was obtained after only 100 min in the presence of Cu(OTf)2. Overall, the efficacy of the catalysts in these reactions within the first 100 min was found to be in the following increasing order: Yb(OTf)3 < La(OTf)3 < Sc(OTf)3 < Cu(OTf)2; the catalytic superiority of Cu2+ observed in Engberts’work as delineated above was confirmed. A hypothesis for the rationale of this finding could be the cation size effect, that is, the better catalytic activity of Cu(OTf)2 owes to the smaller size of the cupric ion Cu2+ (ionic radius rCu = 0.72 Å; rLa = 1.06 Å); the smaller the size, the greater the catalytic activity. The small size of cation allows a facile crossing from the aqueous phase to the organic one via the interfacial film; the cation will then insert within the polar heads of surfactant molecules. Yet, beyond 100 min, the catalytic effect of Cu(OTf)2 started to level off, while those of the other Lewis acids still rose and even surpassed that of cupric triflate. This was quite expected because the latter salt can be partially hydrolyzed with time in an aqueous medium, whereas the other salts are more stable to hydrolysis.
Chatterjee et al. [127] succeeded in making substituted isoxazolidine cycloadducts from 1,3-dipolar cycloaddition between ethyl acrylate 259 and a series of nitrones 258 in micellar aqueous systems (SDS and CTAB as surfactants) (Scheme 88). In this work, the nitrones 258 (Table 7) were themselves prepared in situ, that is, in the same pot. This study was featured with the fact that the major products were the trans-5-substituted isoxazolidines, whereas the regioisomers 4-substituted isoxazolidines predominated when the reactions were run in organic solvents. With the exception of the 2,5-dimethoxyphenyl nitrone, all nitrones afforded isoxazolidines 260 in excellent yields (76–91%) at a varying reaction time, 18–76 h; in general, better yields were gained with CTAB.
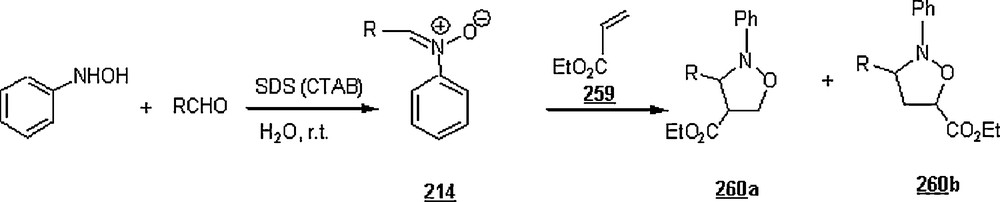
Surfactant catalyzed dehydrative nitrone formation from phenyl hydroxyl Amine and aldehydes followed by cycloaddiction with ethyl acrylate in water at room temperature.
| RCHO (R) | Surfactant | Time (h) | Yield 260 % | 260a exo:endo | 260b cis:trans |
| o-NO2Ph | SDS | 18 | 79 | 32/68 | |
| o-NO2Ph | CTAB | 18 | 85 | 32:68 | |
| m-NO2Ph | SDS | 30 | 81 | 46:54 | |
| m-NO2Ph | CTAB | 26 | 89 | 46:54 | |
| p-NO2Ph | SDS | 16 | 76 | 100:00 | |
| p-NO2Ph | CTAB | 16 | 81 | 100:00 | |
| p-MeOPh | SDS | 76 | 84 | 100:00 | 100:00 |
| p-MeOPh | CTAB | 72 | 89 | 100:00 | 100:00 |
| 2,5-(MeO)2Ph | SDS | 121 | 27 | 0:100 | |
| 2,5-(MeO)2PH | CTAB | 121 | 38 | 0:100 | |
| p-CIPh | SDS | 40 | 90 | 100:0 | 49:51 |
| p-CIPh | CTAB | 40 | 91 | 100:0 | 49:51 |
| 3-pyridyl | SDS | 74 | 89 | 42:54 | |
| 3-pyridyl | CTAB | 70.5 | 91 | 42:54 | |
| PhCH=CH− | SDS | 50 | 87 | 35:65 | 31:69 |
| PhCH-CH− | CTAB | 50 | 90 | 35:65 | 31:69 |
| CH3(CH2)2− | SDS | 48 | 85 | 43:57 | |
| CH3(CH2)2− | CTAB | 48 | 85 | 43:57 |
Later, Chatterjee and Bhattacharya [128] employed CTAB-catalyzed protocol to produce pyran- and oxepane-isoxazolidines 262 and 263 (Scheme 89). The 3-O-allyl carbohydrate nitrone 261 produced in a micellar aqueous system underwent an intramolecular 1,3-dipolar cycloaddition to yield the isoxazolidine cycloadducts in 74 to 85% for reaction times as long as 8–36 h. The nitrones of 3-O-allyl glucofuranose and of the crotyl derivatives produced oxepane isoxazolidines. In contrast, nitrones of prenyl derivatives gave pyran isoxazolidines which may be due to the methyl-methyl steric repulsion restricting the formation of oxepane skeleton. Anionic (SDS, SDBS) and non-ionic (Triton X-100, Triton CF-10, Tween-20) surfactants were also examined in these reactions and it was found that CTAB was the most efficient as far as the yield and the kinetics were concerned.
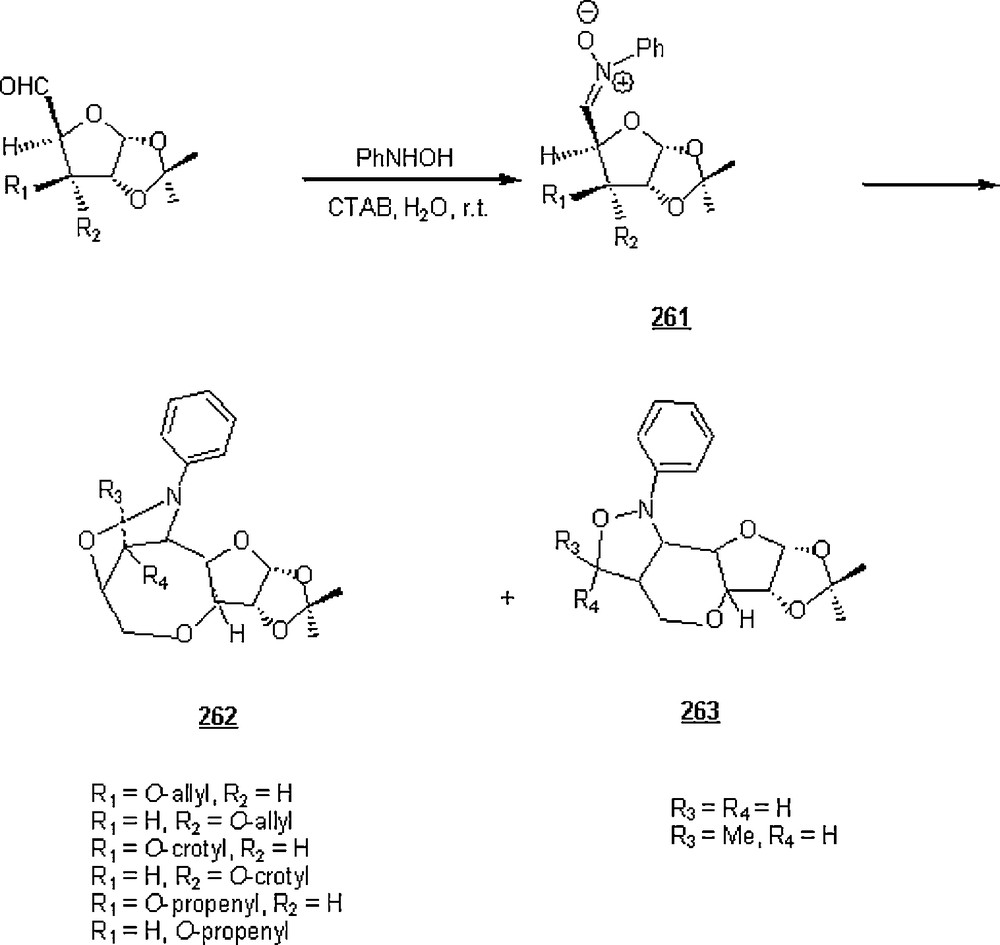
3.6 Formation of pyrroles and pyrrolidines
Butler et al. [129] launched a series of aqueous 1,3-dipolar cycloadditions involving a water–insoluble phthalazinium-2-dicyanomethanide 264, and a water-soluble pyridazinium dicyanomethanide 265, as 1,3-dipoles and a wide range of dipolarophiles, at a temperature of 37 °C, as pictured in Scheme 90. Overall, the rates in water and water-acetonitrile (9:1) were enhanced compared to those measured in acetonitrile. The dipolarophiles were classified according to rate enhancement extents; the ones associated with an enhancement lower than 20-fold were coined ‘water normal-dipolarophiles’, and those with an enhancement higher than 45-fold were called ‘water super-dipolarophiles’. The former species were those having ether, ester, nitrile, phenyl, sulfone, amide groups in the α-positon to alkene or alkyne functionality, and included: methyl acrylate, tert-butyl acrylate, methyl propiolate, 2,3-dihydrofuran, acrylonitrile, phenyl vinyl sulfone, methyl vinyl sulfone, styrene, isoprene, N-methyl maleimide, N-tert-butyl maleimide, N-(p-methoxyphenyl) maleimide, N-(p-tolyl) maleimide, N-(p-ethylphenyl) maleimide, N-phenyl maleimide, N-(p-chlorophenyl) maleimide, N-(p-bromophenyl) maleimide, N-(p-acetylphenyl) maleimide, and N-(p-nitrophenyl) maleimide. The latter ones bear a ketone group conjugated to the alkene or alkyne functionality, including the following: methyl vinyl ketone, ethyl vinyl ketone, but-3-yn-2-one, 1,4-naphthoquinone, cyclopent-2-en-1-one, and cyclohex-2-en-1-one. Examples of reactions involving the super-dipolarophiles were reactions of methyl vinyl ketone, ethyl vinyl ketone and but-3-yn-2-one with pyridazinium dicyanomethanide, which exhibited rate enhancements of 202, 161 and 156, respectively, on changing from acetonitrile to water as a medium. Their reactions with phthalazinium-2-dicyanomethanide presented rate enhancements of 59, 45, and 45, respectively. As expected, these results pointed to the hydrophobic and hydrogen bonding effects. Addition of LiCl (salting-out effect) increased the rate enhancement, whereas the addition of guanidinium chloride GnCl (also called chaotropic agent, salting-in effect), a common denaturant of proteins and nucleic acids, decreased it as typified by the examples shown in Fig. 1. It was noted that both types of dipolarophiles responded in the same way to these salts, implying similar hydrophobic effect. Substantial yields (80–96%) of the cycloadducts from 1,3-dipole 264 were achieved at 80–81 °C in homogeneous CH3CN solutions and in heterogeneous aqueous media, but in most cases better yields were obtained using water as a suspending medium. In addition, heterogeneous conditions seemed to favor the endo/exo selectivity compared to the homogenous ones, results due to the hydrophobic effect and polarity of water medium.
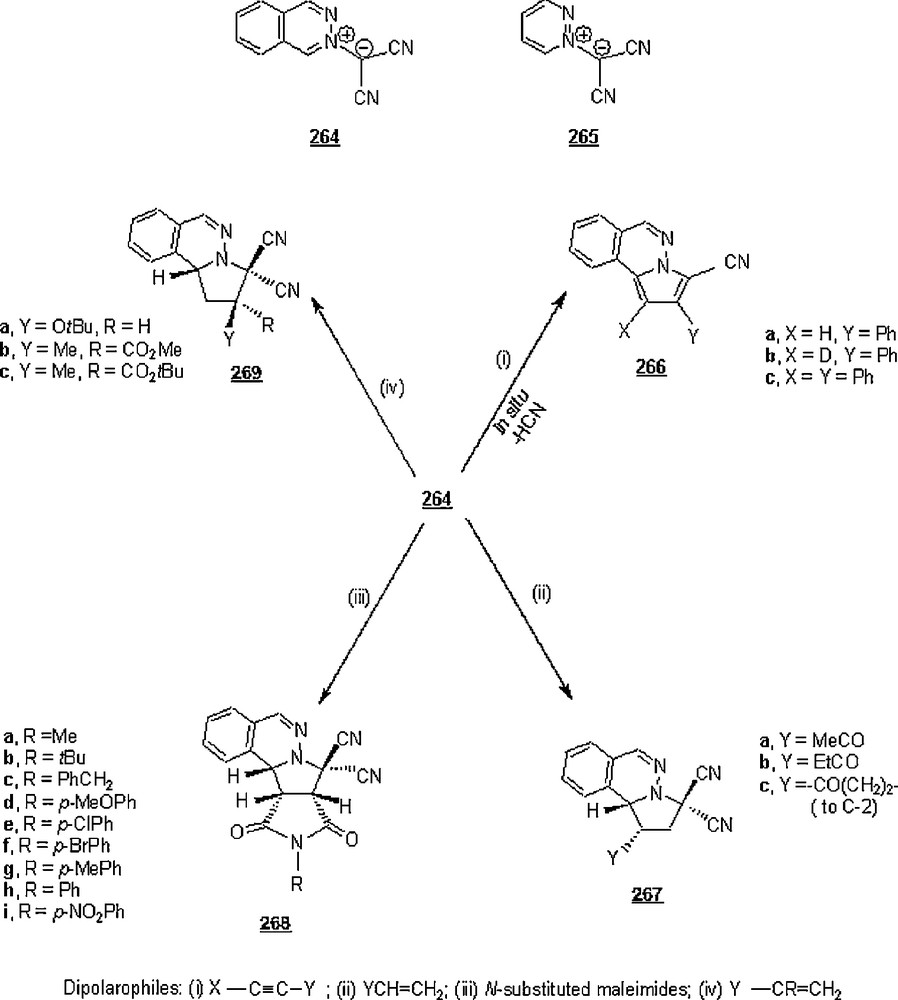
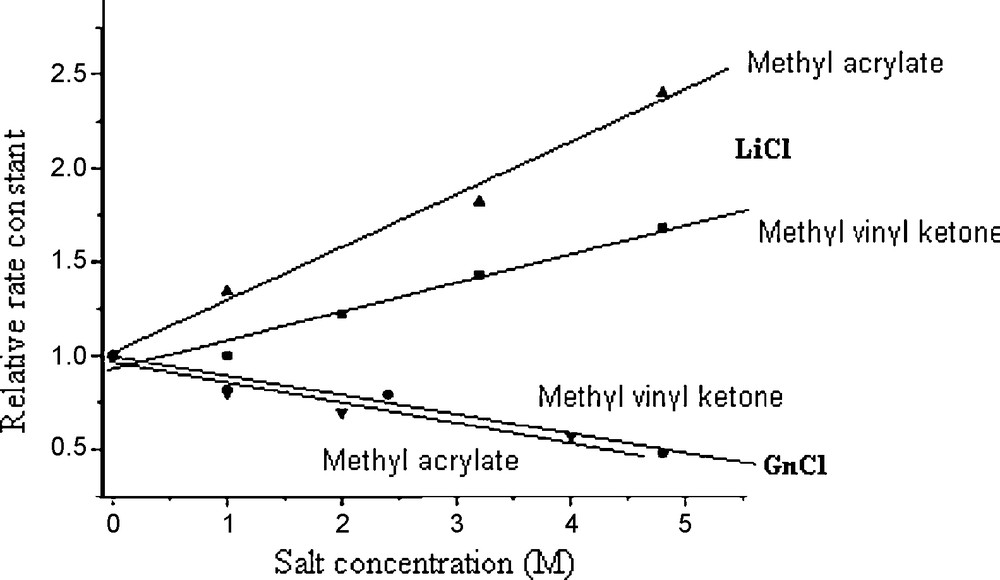
Influence of LiCl and GnCi on methyl acrylate 60 and methyl vinyl ketone 8 from dipole 265 [129]. Reproduced by the permission of The Royal Society of chemistry.
Later, Butler et al. [130] conducted a careful study on the rate enhancements provoked by the extent of water in a solvent mixture for 1,3-dipolar cycloaddition reactions of dicyano(pyridazinium)methanide 265 with ethyl vinyl ketone (EVK) and methyl acrylate (MA) as water-super dipoloraphile and water-normal dipolarophile, respectively. Organic solvents of the water mixtures tested were: acetonitrile, acetone, methanol, and t-butanol. In pure water and at 37 °C, the second-order rate constant was found significantly higher for EVK compared with that for MA, 875 × 104 dm3 M−1 s−1 against 17.1 × 104 dm3 M−1 s−1. In general, the rates for the former dipolarophile in all organic solvents were greater than those for the latter dipolarophile. But, those in water were expectedly higher than those in organic solvents. For both dipolarophiles, significant rate improvement was observed in the different water-organic solvent mixtures starting at nearly 80% of water. Activation energies were measured for the reactions with these dipolarophiles (EVK, MA), and were found to be 28.9 and 55.1 kJ M−1 in water and 51.7 and 59.8 kJ M−1 in acetonitrile. These results would suggest that the reaction course occurred more easily in water than in acetonitrile. As to the influence of temperature on the rate enhancement, the results indicated that the rate ratio kwater/kacetonitrile for the cycloaddition of dicyano(pyridazinium)methanide with EVK fell off dramatically with increase in temperature, but with MA it was merely insensitive. This difference has been rationalized in terms of the ability of hydrogen-bond forming: for EVK (a water-super dipolarophile), the formation of primary hydrogen bonds at room temperature began where a substantial water-structure growth developed around the transition state; no such a hydrogen-bonding occurred for MA (a water-normal dipolarophile).
In a later work on 1,3-dipolar cycloaddition reactions of phthalazinium dicyanomethanide 264, Butler et al. [131] examined the reactions with substituted styrenes (water–normal dipolarophiles) and substituted benzylidene acetones (water–super dipolarophiles) to gain an insight into the transition states in these reactions. With the former dipolarophiles, the second-order rate constants were found to be greater than that of unsubstituted styrene in MeCN and H2O/MeCN (9:1) at 37 °C, and those for electron-donating groups (EDG) were higher than those for electron-withdrawing groups (EWD). Yet, the rate was accelerated in water-acetonitrile mixture by a factor of 5 to 8.5 compared with that in MeCN. Also, the yields in both media were good, and the exo/endo steroselectivity was slightly favored in aqueous medium. The Hammett ρ values were −0.53 and −0.30 in the case of EDGs for MeCN and H2O/MeCN, respectively, inferring to the slight polarity contribution of water in the transition state. For EWGs, they were + 0.48 and + 0.23, indication no such a polarization took place.
The second-order rate constants of reactions of 264 with benzylidene acetone at 37 °C were 7.20 × 10−4 and 2.40 × 10−2 L M−1s−1 for MeCN and H2O/MeCN, respectively. The Hammett ρ values were + 0.62 for EDGs and + 0.31 for EWGs in H2O/MeCN.
Butler et al. [132] extended their work on 1,3-dipolar cycloaddition reactions of 264 in water, to dienopolarophiles that show some extent of insolubility in water. These dienopolarophiles included some N-substituted maleimides, p-chlorobenzylideneacetone, and diphenylacetylene, whose molar water-solubilities were generally in the range of 10−5 to 10−2 M L−1. In case of maleimides, the yields of these “on-water” reactions were excellent, above 90%, at a temperature of 20 °C and for a reaction time of 24 h. For diphenylacetylene and p-chlorobenzylideneacetone, the yields were rather poor (lower than 1%), under the latter conditions, but upon working at temperatures of above 75 °C, the yields were notably enhanced to 71 and 86%, respectively.
Lubineau et al. [133] examined a three-component reaction (Scheme 91), sarcosine 270 /phenylglyoxal 271 /N-ethylmaleimide 13, in different solvents, including water; an azomethine moiety was generated in situ. While the reaction in toluene at 40 °C and for 24 h, the products were the endo- and exo-pyrrolidines 273a and 273b (cycloadducts) in a 67% yield and the oxazoline 274 in 17% yield, the reaction in water afforded a Michael product 272 at the same temperature and for 10 h. However, in a 1:1 (v/v) mixture of water and THF at 40 °C, cycloadducts 273a and 273b were obtained in 21 and 43% yield, respectively.
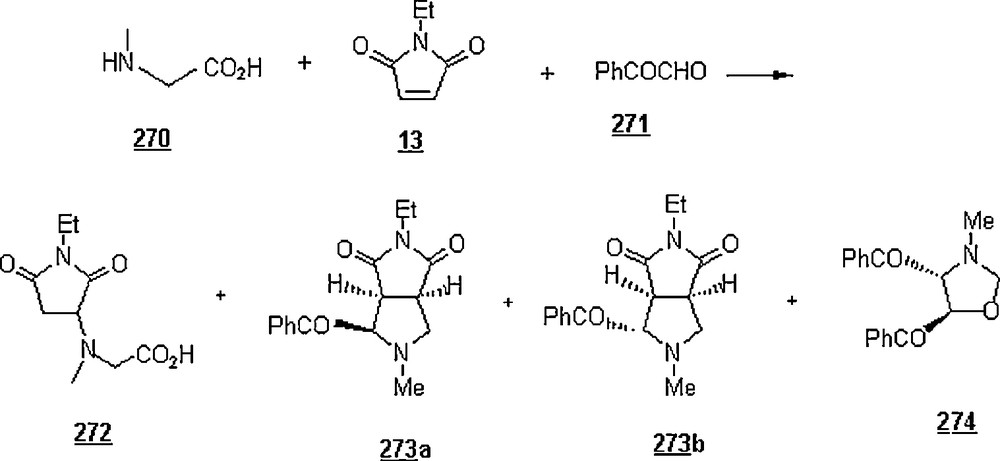
On the other hand, the reaction of sarcosine methyl ester 275 (as hydrochloride) with N-ethylmaleimide 13 and aqueous formaldehyde 276 was studied in various solvents (Scheme 92). The cycloaddition occurred in refluxing toluene (110 °C), giving pyrrolidines 277a and 277b in 48 and 51% yield, respectively. Pure water as solvent was also found to promote the dipolar cycloaddition, providing pyrrolidines 277a and 277b in 18 and 15%, along with succinimide 278 stemming from Michael addition (32%) and oxazolidine 279 only in 3%. The competing Michael reaction was inhibited by running the reaction in THF/H2O and DMF/H2O medium, and only the desired dipolar cycloaddition products were detected. When using diethyl fumarate and diethyl maleate, the reactions in pure water and aqueous THF solutions at 80 °C and after 13 h led to pyrrolidine cycloadducts in acceptable yields (53–91%) as the major products and, in some cases, as the sole products.
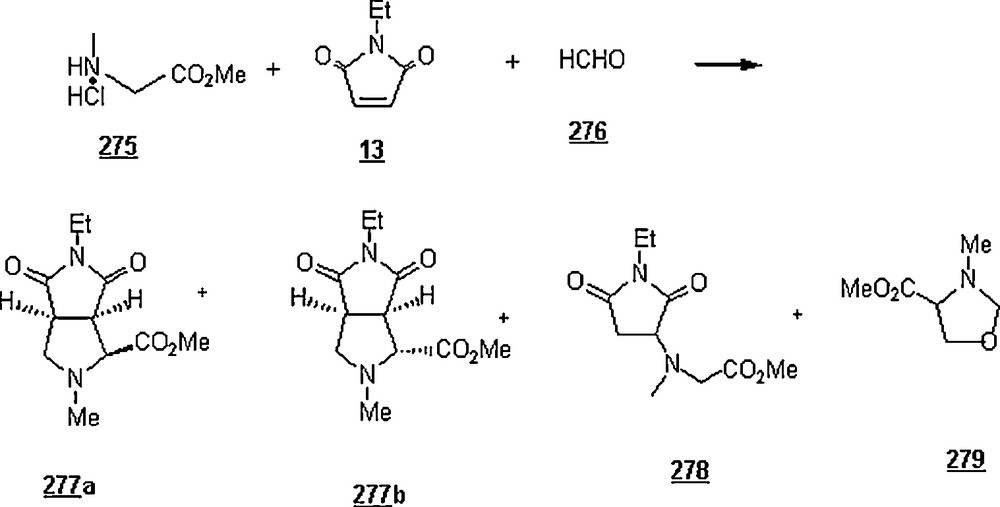
Grigg et al. [134] were able to make 1,8-diazafluorenone (DFO) 280 which was found to be the best reagent for detection of α-amino acids and latent fingerprints on paper. Upon reactiong with α-amino acids and their esters via imine formation, this reagent gave decarboxylated azomethine ylides and ester substituted azomethine ylides, respectively. These ylides may undergo stereospecific cycloaddition with N-methylmaleimide in wet organic solvents (boiling aqueous ethanol, or boiling 20% aqueous DMF) (Scheme 93). The cycloadducts 281 were isolated in 53–79% yields after a reaction time of 0.25–3 h.
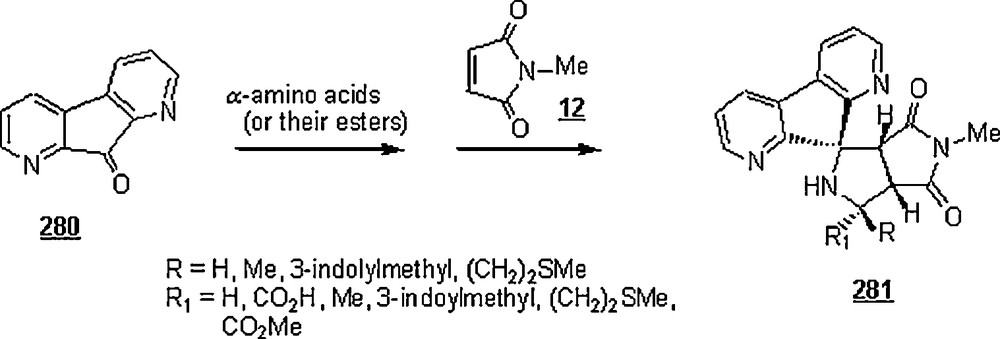
4 Copper (I)-catalyzed azide-alkyne cycloaddition (CuCAAC)
Rostovtsev et al. [135] disclosed the stepwise mechanism of CuCAAC (copper catalysis in [3 + 2] cycloadditions of azides to acetylenes), withdrawing it from its 1,3-dipolar nature. This chemistry is herein treated separately from 1,3-dipolar cycloaddition reactions discussed above. The discovery of CuCAAC, the so-called ‘click chemistry’ [106,136], has boosted the application of 1,2,3-triazoles, especially in medicinal chemistry. Pieters et al. [137] reviewed the synthetic applications of Cu(I)-catalyzed 1,3-dipolar cycloaddition “click” reaction, focusing on multivalent carbohydrates and peptides and peptide-based polymers.
Copper(I) species in aqueous solution are potential catalysts for the formation of 1,2,3-triazoles 284 from organic azides 282 and terminal alkynes 283 usually with high regioselectivity (Scheme 94) [135,138]; 1,4-disubstituted regioisomers were obtained in excellent yields, 82–92%, depressing the 1,5-isomers. Application of microwave assistance has helped reducing dramatically the reaction time to 10–15 min while maintaining higher yield (> 90%) and high regioselectivity (100% of 1,4-disubstituted regioisomer) [139].
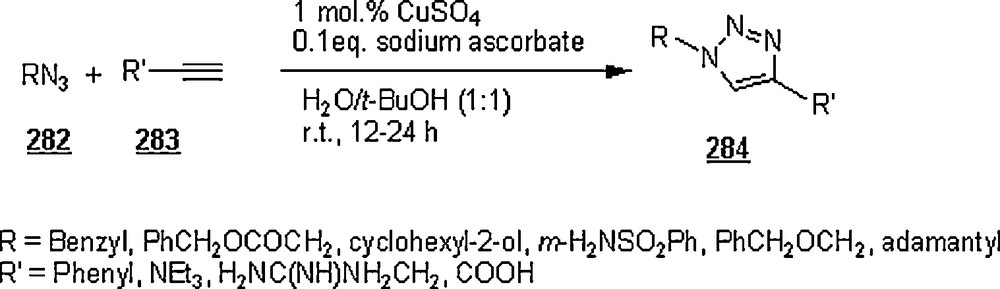
Yap and Weinreb [140] realized a number of N-protected 4-substituted 1,2,3-triazoles 287 from β-tosylethylazide (TSE-N3) 285 with a set of terminal alkynes 286 in water-tert-butanol mixtures (2:1 v:v), β-tosylethylazide being synthesized in 87% from p-tolyl vinyl sulfone and sodium azide. The approach was via copper (I)-catalyzed 1,3-dipolar cycloadditions, exploiting the ‘click chemistry’ strategy, between the TSE-N3 and a series of terminal alkynes as depicted in Scheme 95. The yields of 287 were excellent, 85–97%, under the following conditions: room temperature, 8 h, and a rapid stirring. The removal of the tosylethyl group was effected by potassium tert-butoxide in THF at −78–0 °C.
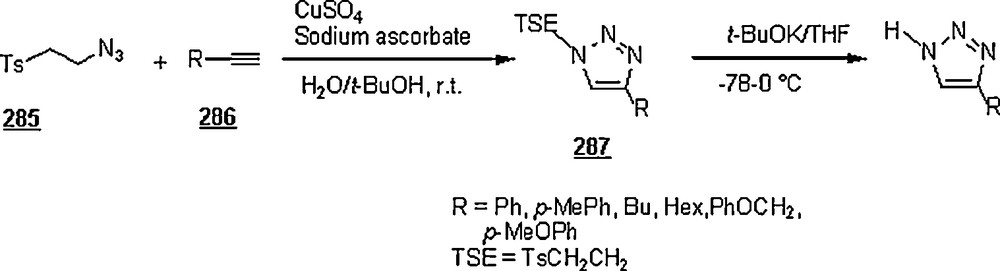
Odlo et al. [141] reported a one-pot procedure for the direct conversion of α-bromo esters 288 to 1,4-disubstituted 1,2,3-triazoles 290 in H2O–t-BuOH (1:1), via a Huisgen 1,3 dipolar cycloaddition (Scheme 96). The feature of this work was that the organic azide, a potential hazard, was formed in situ from reaction of α-bromo ethylacetate with sodium azide. The one-pot Cu(I)-catalyzed reaction proceeded at room temperature and for 12 h. The yields of products 290 were excellent, 80–94%.
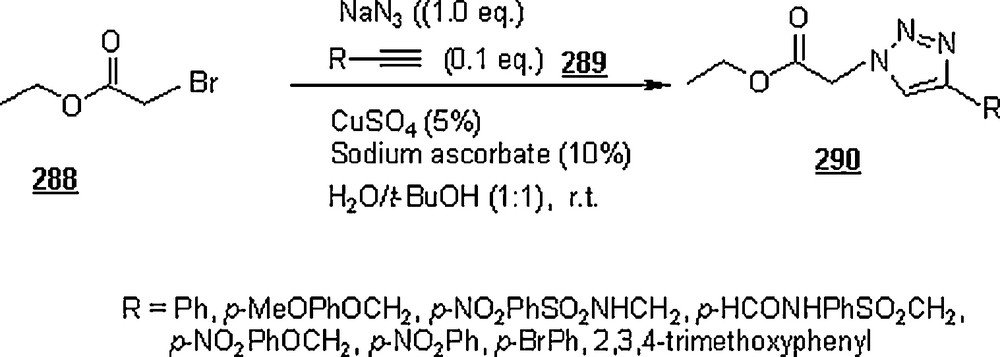
Ozçubukçu et al. [142] were able to prepare a tris(1-benzyl-1H-1,2,3-triazol-4-yl)methanol ligand 291 via a triple Cu(I)-catalyzed alkyne-azide 1,3-dipolar cycloaddition. Copper (I) complex, 291–CuCl, by reacting CuCl with this ligand in dioxane at 60 °C. The new catalyst was tested in the reaction of azides with acetylenic compounds in water at room temperature as shown in Scheme 97. For most cases, the excellent yields (92–99%) of corresponding 1,2,3-triazole compounds for a reaction time of about 4 h demonstrated the high efficiency of this new copper(I)-based catalyst. This catalyst was cleverly exploited using the one-pot strategy, involving the alkyl bromide 295 and sodium azide, thus avoiding handling organoazides; indeed, as illustrated in Scheme 98, the reactions were microwave-promoted and were run in a 1:1 mixture of acetonitrile/water at a temperature of 100 °C. Under these conditions, reaction times as short as 40 min were sufficient to allow isolation of the cycloaddition products in very satisfactory yields (80–99%).
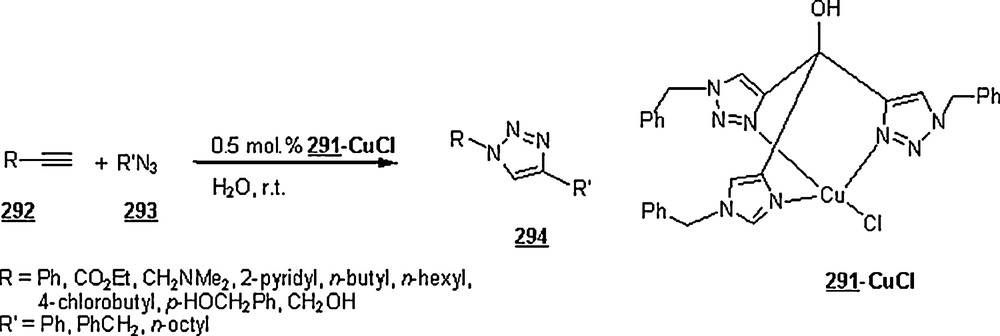
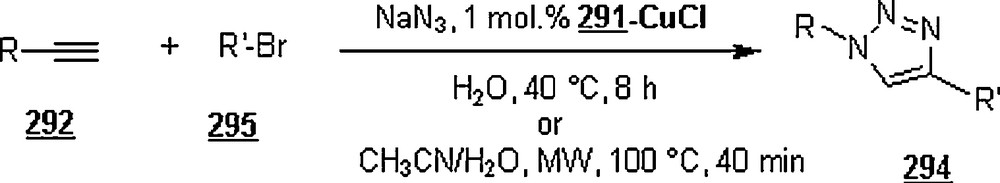
Sivakumar et al. [143] also explored the methodology of Cu(I)-catalyzed Huisgen 1,3-dipolar cycloaddition of azides to alkynes for making fluorescent 1,2,3-triazole products. The study dealt with aqueous ethanolic reactions of nine non-fluorescent azidocoumarins 296 with twenty-four alkynes 297 with different nearby functionalities catalyzed by Cu(I) species at room temperature (Scheme 99). Triazolylcoumarins 298 were isolated in good yields, 62 to 86%, under the reaction conditions shown in the scheme. It was observed that the reactions with aromatic azides went to completion even at 0 °C, reflecting their high reactivity. All the triazolylcoumarins showed intense fluorescence due to the triazole linkage (λmaxAbs = 298–445 nm; λmaxEm = 388–521). Their quantum yields were around 0.6–0.7. Electron-releasing substituents on the 3-azidocoumarins displayed a strong enhancement of fluorescence.
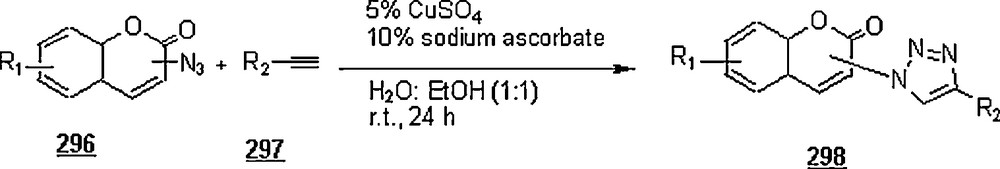
Wang et al. [144] coupled a Cowpea mosaic virus (CPMV), a biomolecular scaffold having a structurally rigid assembly of 60 identical copies of a two-protein asymmetric unit around a single-stranded RNA genome, to fluorescein dye 305 by means of Huisgen cycloaddition in aqueous t-butyl alcohol medium (Scheme 100). The presence of Cu(I) is imperative, otherwise no reaction occurred, and sometimes, a reducing agent such as tris(carboxyethyl)phosphine was necessary to protect the cysteine residues in proteins from oxidative coupling. Yields of the first reaction of the scheme fluctuated between 86 to 100%, and were 75 to 96% for the second reaction, at 4 °C and 16 h.
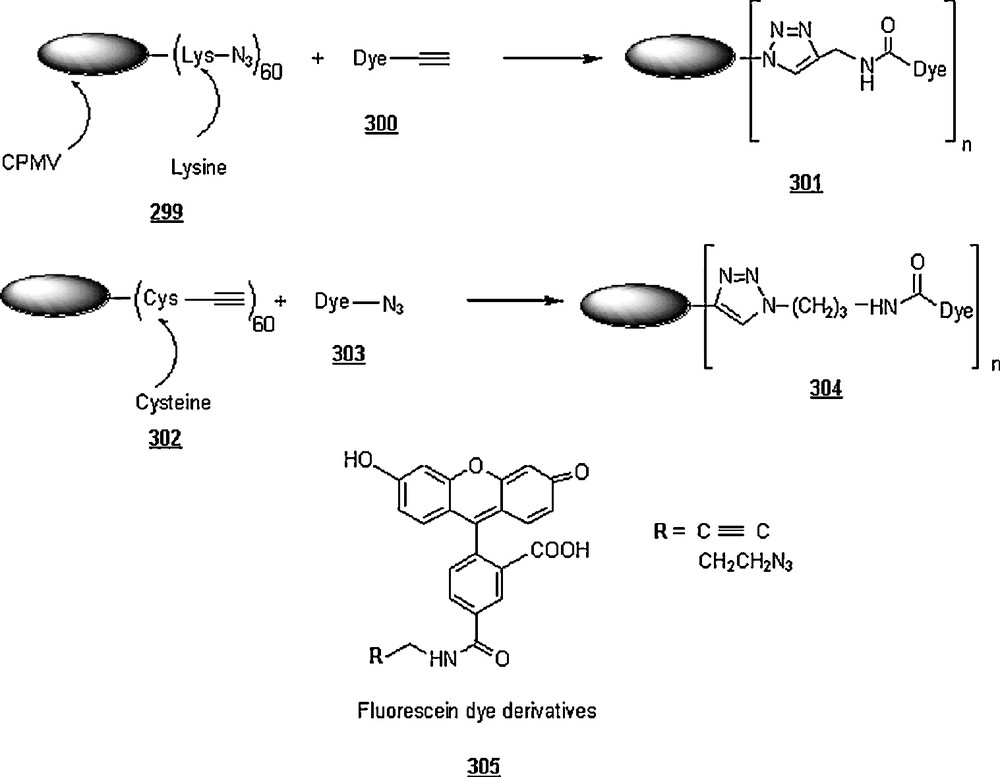
Wan et al. [145] exploited the CuI-catalyzed Huisgen 1,3-dipolar cycloaddition of azides and terminal alkynes, to link oligosaccharides 306–308 to polypeptides and proteins (Scheme 101). The cycloadduct 309 was obtained in quantitative yield after 40 h when the corresponding azido-carbohydrate and alkynyl polypeptides were subjected to “click chemistry” in water and acetonitrile (1:1, v/v), employing polished copper wire and aqueous solution of CuSO4. However, when sodium ascorbate was used as the reducing agent instead of copper wire, the reaction was found to be markedly less efficient. On the other hand, the reaction provided the desired tris-triazole adduct after 3 days when run with nanosized copper powder and in water/t-BuOH (4:1 v/v); in addition, a decreasing effect of CuSO4 on the reaction rate was noticed.

Sommer and Weck [146] grafted 1,2,3-triazole moieties onto gold nanoparticles 310 by means of click chemistry of microwave-assisted Huisgen 1,3-dipolar cycoaddition in THF and aqueous dioxane/t-butanol/water (1:1:0.5) (Scheme 102). The yields of reactions shown in the scheme ranged from 45 to 88% for dioxane/n-butanol/water and 55 to 100% for pure THF as solvent. The reaction times were 3–5 min for the first solvent system and 10–20 min for the second one. The gold nanoparticles were employed in catalysis, imaging, disease diagnostics, and gene expression. Moreover, in biotechnology, the grafting of carcinoembryonic antigen antibodies onto gold nanoparticles followed by immobilization of the functionalized particles onto a gold electrode enhances the selectivity of immunoassay electrodes.
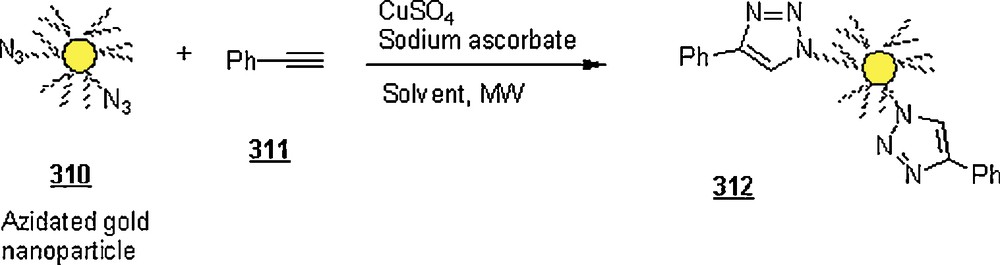
Lee and Kim [147] explored the benefits of the strategy of click chemistry, the Cu(I)-catalyzed Huisgen [2 + 3] dipolar cycloaddition in water, for the realization of triazole dendrimers. They adopted the convergent synthesis approach through the trimerization reaction of Fréchet-type azido-dendrons and via click chemistry using trispropagyl amine as a tripodal core (Scheme 103). At a temperature of 50–60 °C, the reaction of tripropargylamine 313 with 2-D1 in 0.09 M solution provided the triazole dendrimer 3-G1 having just 1,4-disubstituted 1,2,3-triazole units in yield of 93% after 12 h. For the second generation dendrimer, the reaction of 313 with 2-D2 in 0.06 M solution gave the triazole dendrimer 3-G2 in 81% yield after 12 h, which was enhanced to 88% by prolonging the reaction time to 24 h. But, the reaction of 313 with 2-D3 in 0.02 M solution afforded the triazole dendrimer 3-G3 in 40–42% only after 24 h.

Recently, Kumar et al. [148] synthesized 16 1,4-disubstituted 1,2,3-triazoles 315 by one-pot condensation of α-tosyloxy ketones 314, sodium azide, and terminal acetylenes in aqueous PEG 400 and in the presence of CuI as a catalyst of ‘click’ azide-alkyne cycloaddition reaction (Scheme 104). The feature of this work was that the reactions were one-pot systems and smoothly run, and afforded exclusively 1,4-disubsituted triazoles in good yields, 75 to 86%; the reaction conditions were: H2O/PEG400 (1:1, v/v) as reaction medium, 10 mol % Cu (I), ambient temperature, and 30 min. Also, it was found that copper (I) catalyst and aqueous PEG could be reused for three times in a row without loss of activity and product yields.
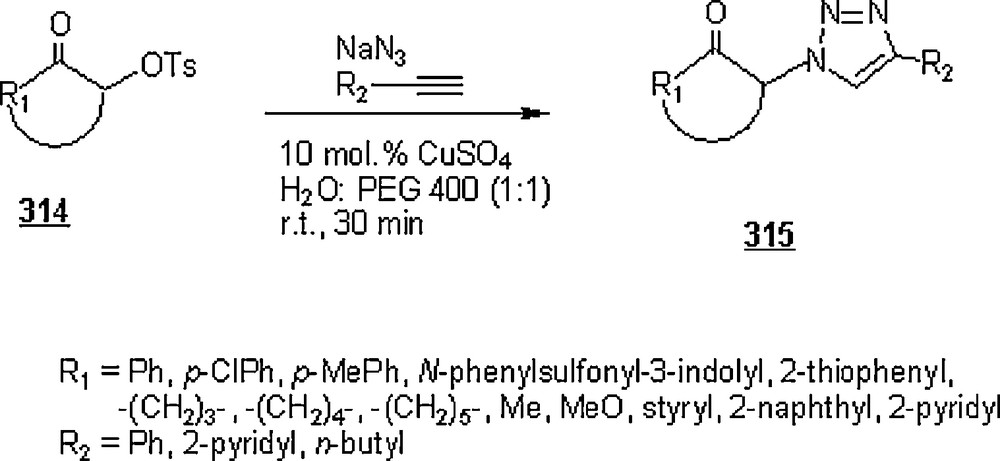
Inspired from Sharpless’ observation of using Cu(0) coiled metal turnings in Huisgen cycloaddition [135], Orgueira et al. [149] employed Cu(0) in form of nanosize powder in the synthesis of several 1,4-disubstituted 1,2,3-triazoles from 1,3-dipolar cycloaddition of organoazides with terminal alkynes in water/t-butanol (1:1, v/v) at room temperature and after 2 h. Attempt with activated copper powder failed, but the addition of triethylamine hydrochloride salt provoked the reaction success, promoting the formation of Cu(I) catalytic species. Accordingly, nine 1,2,3-triazoles were prepared in quantitative yields (>90%). The reactions worked well regardless the chemical nature of groups on azides and terminal alkynes. It was observed that the amine hydrochloride salt enhanced the dissolution of Cu(0) and the facile generation of catalytic Cu(I) species; the use of free amine did not bring the reactions to success. Also, under the same conditions, the reaction of propargyl amine hydrochloride and benzyl azide in an equimolar amounts gave the corresponding 1,4-disubstituted triazole with high regioselectivity.
Kumaraswamy et al. [150] took advantage of the click chemistry of azide functionality to promote epoxide or aziridine ring opening. The one-pot epoxide azide-click reaction employing phenoxy methyloxirane 316, sodium azide, and terminal alkyne using polyethylene glycol (PEG-400) as the reaction medium in the presence of 5 mol % of CuI. After 16 h of stirring at room temperature, the desired product 317 was obtained in 91% isolated yield (Scheme 105). Unexpectedly, the reaction in water did not proceed even after prolonged stirring at room temperature. However, the mixtures of water/PEG-400 (8:2) and water:18-crown-6 as reaction media provided low yields of product 317 after 48 h of stirring at ambient temperature: 45 and 35%, respectively.
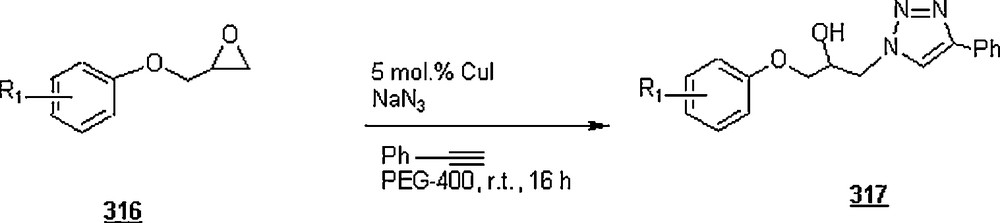
Zhang et al. [151] applied the CuCAAC protocol to ynamides and bis-ynamides. The cycloaddition reaction of ynamide 318 with benzyl azide 319 (BnN3) led to chiral amide-substituted triazole 320 in 82% yield as the only regioisomer (Schemes 106). This synthetic methodology of 1,2,3-triazoles were expanded for a number of yanimides and alkylazides which were in situ produced from the reaction of the correponding alkylbromides with sodium azide. The results were that the yields were moderate to good (50–85%). A one-pot sysnthesis of bis-triazole was realizable via the CuCAAC strategy as shown in Scheme 107; the bis-triazoles 324 were obtained in modest yields (53%).
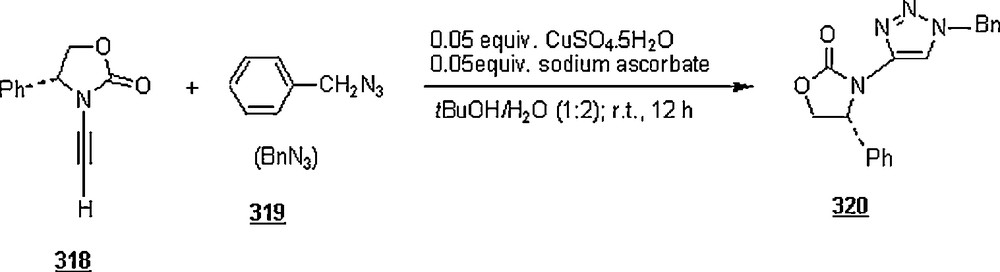
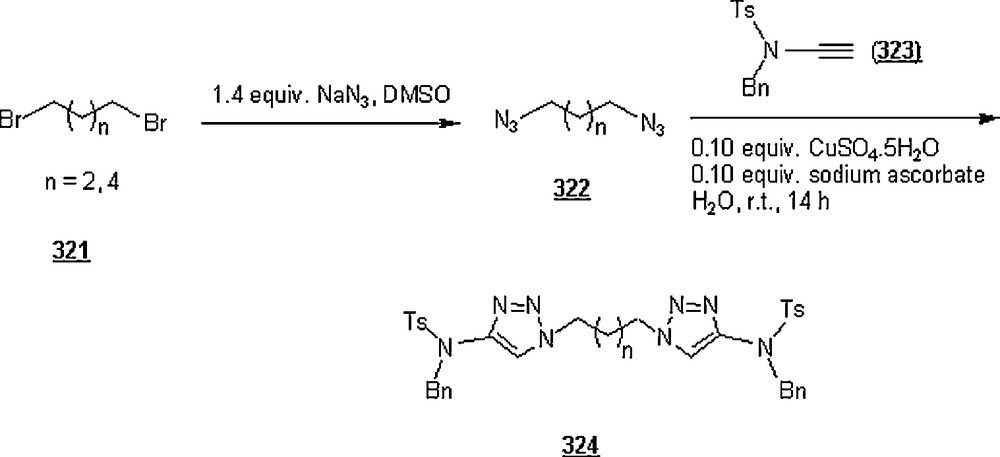
5 Miscellaneous cycloaddition-related reactions
In water-borne cycloaddition reactions, the existence of hydrogen bond sites responsible for high rates is highly demanded. For example, the [2π + 2π] cycloaddition of cyclohexene with nitrosobenzene had a kwater/ktoluene of only 44.3, because of the absence of hydrogen bond-activating sites in nitrosobenzene [152]. Also, the hydrogen bond effect is manifested in the reaction shown in Scheme 108 [153]. For this reaction, the acceleration by water of the reaction of 325a with 1 was modest (kwater/kacetonitrile = 8.95), as can be expected in the absence of activation by hydrogen bonds. But, upon introduction of an acetyl group, a hydrogen-bond accepting substituent (325b), the aqueous acceleration was significantly enhanced (kwater/kacetonitrile = 102).
Because the cycloadduct 327 of the cycloaddition between nitrosobenzene 328 and CP 1 or cyclohexa-1,3-diene 16 is unstable, the cycloreversion reaction is actually an equilibrium as shown in Scheme 109. Engberts analyzed this hetero retro Diels–Alder (HRDA) reaction in organic solvents and in water at 25 °C [152]. The equilibrium constant K was larger for polar and protic solvents. The equilibrium constant K in TFE was the highest among the organic solvents screened, KTFE/n-hexane ≅ 93.60; K in DMF was also importantly larger, KDMF/n-hexane ≅ 33.20. However, the equilibrium constant in water was spectacularly greater than in any other solvent, as suggested by Kwater/n-hexane ≅ 875.
When the reaction was run in water–alcohol mixtures, the equilibrium constant K was found to drop with increasing alcohol content, to the same degree as the second-order rate constant. The decreasing order of K was as follows: MeOH > EtOH > 1-PrOH > t-BuOH, reflecting that hydrophobic alcohols induced a reduction of K at low mole fractions.
A quite featuring point was that even the retro Diels–Alder reactions (RDA) were found to proceed much faster in water than in classical media. Wijnen and Engberts [154] investigated the cycloreversion of the anthracenediones 329 into naphthaquinone derivatives (75, 53c) and CP 1 in conventional solvents and in water (Scheme 110) at 40 °C. The first-order reaction rate constant k increased with polar and protic solvents as observed for the normal DA reactions: 2.6 × 10−8s−1 (n-hexane), 6.6 × 10−8s−1 (benzene), 23 × 10−8s−1 (DMSO), 27 × 10−8s−1 (2-PrOH), 62 × 10−8s−1 (acetic acid). Conspicuously, the rate constant was much higher in fluoroalcohols and water: 161 × 10−8s−1 (TFE), 359 × 10−8s−1 (water), 469 × 10−8s−1 (HFIP). These k values and the decreasing Gibbs energy of activation ΔG≠ paralleled the polarity factors ETN(30) of the media experimented. These data confirmed further the efficiency of water as a hydrogen bond donating solvent (HBD) and its potency to stabilize the polar “activated complex” in DA and RDA reactions.
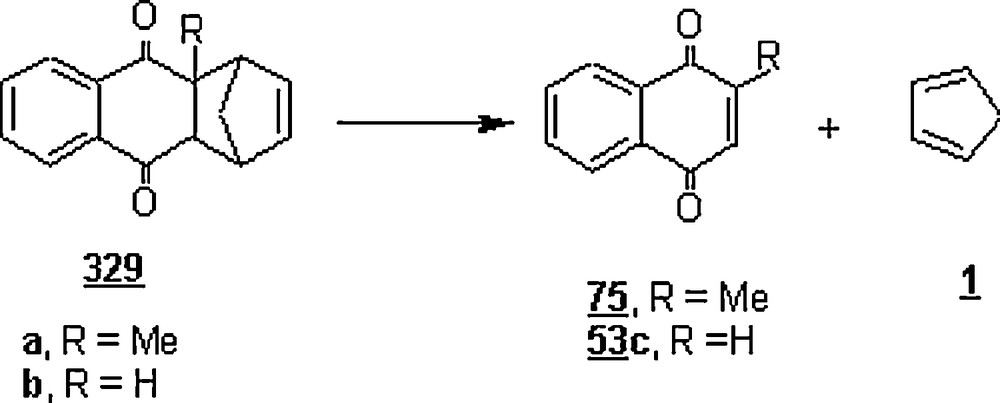
Incorporating an alcohol to water medium slowed down the RDA reaction compared to the rate in pure water. The extent of reduced rate constant k varied with alcohol proportion; the lower the alcohol content, the higher the rate. The rate decreased in the following order of alcohols: MeOH > EtOH > 1-PrOH > t-BuOH. The addition of 5 mol % of urea slightly decreased the rate, whereas 5 mol % of glucose accelerated the reaction. The effect of SDS surfactant on the reaction rate depended on its concentration. Below its CMC (8.56 mM at 40 °C), there was an accelerating effect, and above it a retardation was noted. The greater its concentration above CMC, the more pronounced the decelerating effect.
1 Fairly noting is the fact that D. C. Rideout, the co-author, has been scarcely mentioned in such papers.
2 Organic chemistry is a discipline within chemistry which involves the science study of the structure, properties, composition, chemical reaction, and preparation of chemical compounds that contain carbon…
3 A chemical reaction is a process that always results in the interconversion of chemical substances. The substance or substances initially involved in a chemical reaction are called reactants…
4 A cycloaddition is a pericyclic chemical reaction, in which two pi bond are lost and two sigma bond are gained. The resulting reaction is a cyclization reaction…
5 The Huisgen 1,3-dipolar cycloaddition is the frontrunner of click chemistry that has been intensively explored in the field of chemistry and pharmaceutical discipline [96,97].