

1 Introduction
Hydrogen fluoride (HF) is a colorless, fuming liquid or gas with a strong irritating odor. It is primarily an industrial raw material used in the separation and purification of uranium isotopes, as a cracking catalyst in oil refineries, and also for etching glass and enamel, removing rust, and cleaning brass and crystal. HF is always produced in uranium enrichment process and it is of paramount importance to remove it from this process, which is carried out under vacuum conditions [1]. Various technologies have been developed by many investigators for HF removal [2,3]. Among all the existing methods, the adsorption of HF by suitable solid adsorbents seems to offer an interesting and more practical alternative. Adsorption by alkaline metal fluorides, such as sodium fluoride and calcium fluoride has been the most popular one [4,5].
Recently, nanostructured materials have aroused considerable interest as gas adsorbents because of their unique properties, especially large surface-to-volume ratio [6–8]. A theoretical study has indicated that the intercalation of polarizable HF into carbon nanotubes is energetically favorable [9]. The calculations were based on a finite length tube of 144 molecules with two HF molecules approaching each other within the tube. It was found that the energy of interaction was lower for the two mentioned molecules compared to the bare interaction of a pair of HF molecules [9]. The adsorption of HF molecules on both intrinsic and Al-doped graphene sheets has been studied by first-principles calculations, showing that the Al-doped graphene has higher adsorption energy (Ead) and shorter connecting distance to the HF molecule than the intrinsic one [10].
AlN nanotubes (AlNNTs) have been successfully synthesized by Tondare et al. and other research groups [11–13], and have been proposed as potential hydrogen storage media by Lim and Lin [14]. They are semiconductors with a wide band gap, exhibiting good dielectric properties, with high thermal conductivity, and low thermal expansion coefficient [15]. AlNNTs have noticeably higher reactivity than carbon or boron nitride nanotubes because of their great polarity. We have recently shown that the pristine type of these tubes is not sensitive toward NH3 [16], CO [17], and H2O [18] molecules; however, it may be used in formaldehyde detection [18]. Our previous study has also suggested that O-doped AlNNT can be a potential candidate for NH3 molecule detection [19]. Herein, the potential possibility of single-walled pristine AlNNT as an adsorbent for HF molecule is investigated using density functional theory (DFT).
2 Computational details
Geometry optimizations, and density of states (DOS) analysis were performed on a (5, 0) zigzag AlNNT (constructed of 40 Al and 40 N atoms), and different HF/AlNNT complexes at B3LYP level of theory with 6-31G (d) basis set as implemented in the GAMESS suite of program [20]. The length and diameter of the optimized pristine AlNNT were computed to be about 21.71 Å and 5.28 Å, respectively. B3LYP is a popular functional that has been commonly used for nanotube structures [21–25]. Transition state (TS) calculations were performed using the same level of theory. Based on the harmonic vibrational frequency calculations, the stationary point was found to be a TS structure with one imaginary frequency whose normal mode corresponds to the reaction coordinate. Atoms at the open ends of the tube were saturated with hydrogen atoms to reduce the boundary effects. Ead of the HF molecule is defined as follows:
| Ead = E(HF/AlNNT) − E(AlNNT) − E(HF) | (1) |
3 Results and discussion
Fig. 1 shows a partial structure of the optimized AlNNT and its DOS plot, indicating that it is considered as a semiconductor with a HOMO/LUMO gap (Eg) of 4.11 eV. Two types of Al–N bonds can be found: one with the bond length of 1.81 Å (Al1–N2 bond, for example) and in parallel with the tube axis, and another with the bond length of 1.83 Å (N2–Al3 bond, for example), but not in parallel with the tube axis. Tomic et al. [26] have shown that the B3LYP provides an efficient and robust basis for the calculations of III–V semiconductors, capable of reliably predicting both the ground-state energies and the electronic structure. It has been already shown that all zigzag AlNNTs are semiconductors [27] with Eg values ranging from 2.84 to 3.95 eV; it has also shown that Eg slightly increases with increasing the diameter of the tube and saturates at a value corresponding to the gap of an AlN hexagonal sheet. The experimental value of Eg has been reported to be about 6.20 eV, which belongs to bulk AlN [28]. It is noteworthy to mention that DFT underestimates the Eg of semiconductors and molecules [29], and this aspect must be kept in mind during the following considerations.
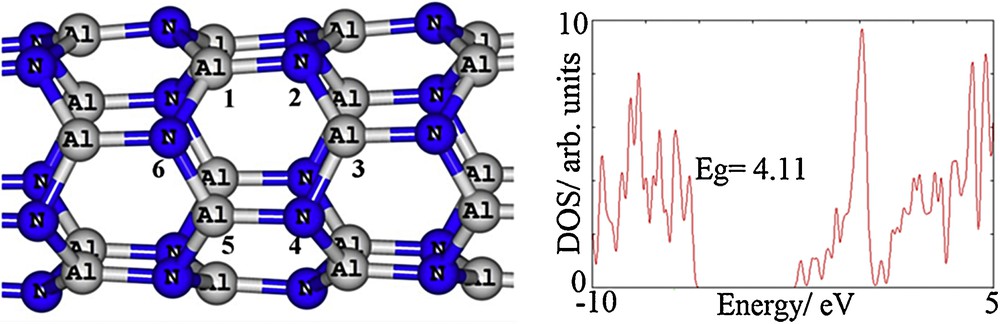
Partial structural model and density of states (DOSs) of the studied zigzag AlN nanotube. Color available on the web.
In order to find energetically stable configurations of a single HF adsorption on the tube, several initial adsorption geometries have been considered, including H or F atom of HF located on an Al or an N atom (perpendicular to the tube surface) and above the center of a hexagonal ring in the tube surface. Interestingly, after optimization, it was found that the molecule becomes parallel to the tube surface and dissociates into H and F atoms. Having gained an understanding of the adsorption process, we then considered the thermodynamic and kinetic possibility of the HF dissociation on the tube via different pathways.
In order to consider the favorability of HF dissociation on the AlNNT, we first investigated the energetic possibility of the hypothetical HF dissociation into –H and –F species on various Al–N bonds of the tube surface, including Al1–N2, Al3–N2, Al1–Al3, Al1–Al5, N2–N6, Al1–N4, Al3–N6 and N2–N4 (Fig. 1). Only four energetically stable states with negative Ead values have been obtained after full relax optimization, and Ead of the others was positive. These four configurations, namely A, B, C and D, are depicted in Fig. 2, in which the HF molecule is dissociated on the Al1–N4, Al3–N6, Al1–N2, and Al3–N2, respectively. In all configurations, two new H–N and F–Al bonds are formed, and their lengths are shown in Fig. 2.
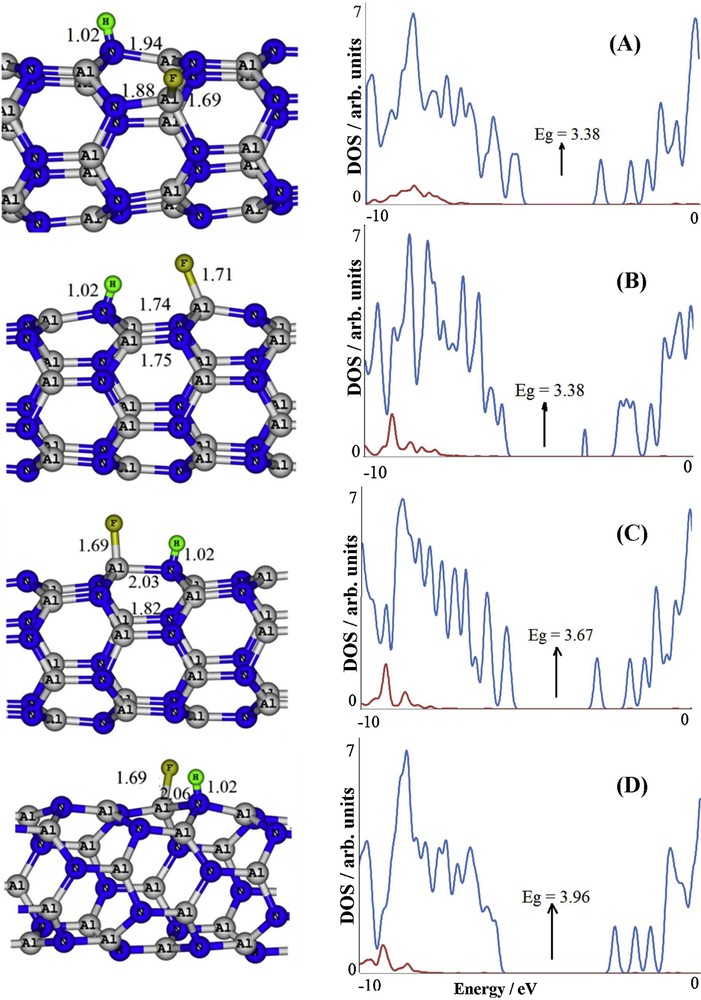
Partial geometrical structures for HF dissociated on the AlN nanotube and their partial density of states (DOSs) plots. Distances are in Å. In DOS plots, red (bottom) and blue (top) lines indicate the HF molecule and the AlNNT, respectively. For interpretation of references to color, see the web version of this article.
Based on the obtained results, configuration D, in which the HF molecule is dissociated on a diagonal Al–N bond, is energetically the most stable configuration, with Ead = −70.10 kcal/mol. In this configuration, the length of the adsorbing Al–N bond is elongated by about 0.25 Å (increased from 1.81 Å in pristine bare AlNNT to 2.06 Å in configuration D). In addition, the Al and N atoms bonded to the fragments are both projected out, and the geometry around them goes from triangular to tetrahedral. The NBO analysis suggests that the orbitals of the atoms are hybridized in order to form four bonds instead of three.
Subsequently, the kinetic favorability of these energetically possible configurations was investigated. As shown in Table 1, an energy barrier must be overcome to get into a final configuration in all pathways. The geometrical parameters representing the TS structures are shown in Fig. 3. In the case of configuration D, while getting the adsorption process to the TS structure, the H–F bond becomes so weak that its length increases from 0.93 Å in free HF to 1.63 Å in TS, and two new bonds, including F–Al and H–N are formed, with lengths of 1.77 and 1.04 Å, respectively. A similar trend is also observed in the case of configurations A, B and C (Fig. 3). It can be concluded that the energy barriers for HF dissociation on a bond (diagonal or parallel) are significantly smaller than the others. It can be also inferred that the activation energies in configurations C and D are so small that the dissociation process may overcome them, even at room temperature.
Adsorption energy of HF on the AlNNT (Ead, kcal/mol), activation energy (Eact, kcal/mol), and the HOMO/LUMO gap (Eg), energies of HOMO, LUMO, and Fermi level of different HF/AlNNT complexes in eV.
| Complexa | Ead | Eact | ELUMO | EF | EHOMO | Eg | ΔEgb (%) |
| AlNNT | – | – | −2.20 | −4.25 | −6.31 | 4.11 | – |
| A | −52.57 | 52.30 | −2.45 | −4.14 | −5.83 | 3.38 | −17.76 |
| B | −57.65 | 51.42 | −2.50 | −4.19 | −5.88 | 3.38 | −17.76 |
| C | −68.49 | 27.90 | −2.33 | −4.16 | −6.00 | 3.67 | −10.7 |
| D | −70.10 | 28.36 | −2.25 | −4.23 | −6.21 | 3.96 | −3.65 |
a For the studied systems, Fig. 2
b Change of Eg of the AlNNT after HF adsorption
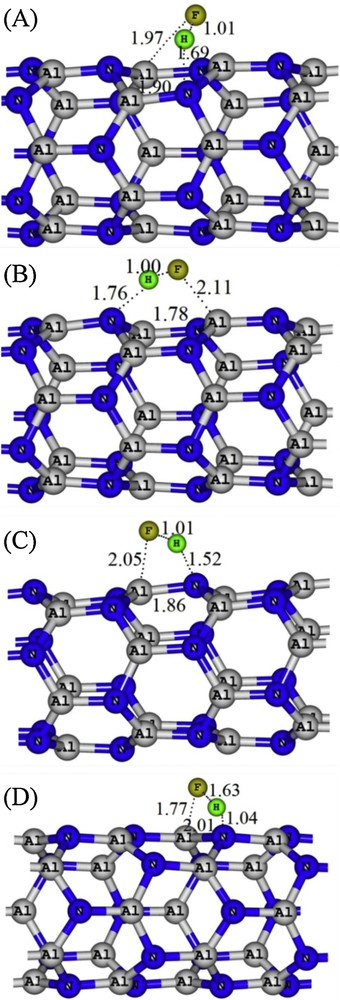
Geometrical parameters of transition state structures (a partial view) for HF dissociation on the AlN nanotube. Distances are in Å. Color available on the web.
Calculated partial DOS plots for the stable configurations are shown in Fig. 2, indicating that both configurations A and B attain an Eg value of 3.38 eV, which is significantly smaller than that of the pristine AlNNT (4.11 eV). For example, by comparing the DOS of configuration A with that of an intrinsic tube, we can see that there is a strong interaction between the HF molecule and the intrinsic AlNNT, and the tube states are significantly altered by the adsorption of the HF molecule. The Fermi level is slightly changed from −4.25 eV in pristine tube to −4.14 eV in configuration D (Table 1). The canonical assumption for the Fermi level is that in a molecule (at T = 0 K), it lies approximately in the middle of the Eg. It should be noted that, in fact, what lies in the middle of the Eg is the chemical potential, and because the chemical potential of a free gas of electrons is equal to its Fermi level as traditionally defined, herein, the Fermi level of the considered systems is at the center of the Eg.
The change of the Fermi level can lead to a change in the work function, which is important in field emission applications. The work function can be found using the standard procedure by calculating the potential energy difference between the vacuum level and the Fermi level, which is the minimum energy required for one electron to be removed from the Fermi level to the vacuum. The change in the work function will change the field emission properties of the nanotube. As it can be seen in Table 1, the Fermi level of the nanotube is not notably changed after HF adsorption in all configurations, indicating that the field electron emission current density may not be significantly altered in the presence of HF molecules. AlNNTs are potential candidates to be used as electron field emission sources because of their ability to support large current densities, their mechanical strength, chemical stability, and their high aspect ratio [12].
As shown in DOS plots of Fig. 2, contrary to configurations A and B, the Eg of the nanotube is not significantly changed in configurations C and D, while comparing the DOS of the most stable configuration (D) with that of the pristine tube indicates that there is also a strong interaction between the HF and the tube, and the tube states are considerably changed after HF adsorption, similar to both configurations A and B. Herein, the Fermi level is also slightly changed; therefore, the work function and the electron field emission will not be significantly altered. Generally, we think that the Fermi level and Eg of the AlNNT will not be influenced by the presence of HF at room temperature because it seems that the complexes of A and B cannot be formed because of a large amount of energy barriers. Partial DOS plots suggest that the effect of the HF molecule is mainly on the occupied states of the tube (red lines in the plots of Fig. 2). Thus, one can conclude that the changes in Eg and the Fermi level are mostly due to the presence of local structural deformations caused by the presence of fluorine and hydrogen atoms of the HF molecule.
Therefore, it can be concluded that the presence of the HF molecule cannot essentially change the electrical conductance of AlNNT, suggesting that AlNNT has no capability of generating an effective response to the HF molecule after these adsorption states through the four stable configurations. It may be interesting from the gas sensing standpoint. It has been recently revealed that the pristine zigzag AlNNT is not sensitive toward NH3 [16], CO [17], N2 [30], CO2 [30], H2S [31], and H2O [18] molecules, while they may be used in formaldehyde [18], SO2 [32], O2 [33], and NO2 [34] detection. Moreover, it can be concluded that the gas detection may also be feasible in the presence of HF gaseous molecules. In other words, pristine AlNNTs can be considered as a potential resource for serving as chemical sensors of some toxic gases in the presence of the others.
4 Conclusion
DFT calculations were employed to investigate the dissociative adsorption of a HF molecule on the exterior surface of AlNNT. The HF molecule preferentially tends to be dissociated into –H and –F species on a diagonal Al–N bond of the tube wall. An energy of 70.10 kcal/mol is released after this process and kinetically, it has to overcome a barrier energy of 28.36 kcal/mol. We believe that the activation energy is so small that the dissociation process may overcome them, even at the room temperature. The chemical modification of the AlNNT with HF molecule has negligible effect on the electronic and field emission properties of the tube.