

1 Introduction
Efficient use of biomass as a sustainable feedstock for production of chemicals is an increasingly important topic of academic and industrial research due to uncertainty over long-term accessibility of traditional petrochemical feedstocks and the political drive towards CO2-neutral products and processes, helping to reduce the emissions of greenhouse gases. At the same time, the use of biomass as a feedstock for fuels and chemicals has already caused considerable controversy through unintentional secondary effects, such as volatility in food prices (Mexico), an apparent reduction in biodiversity (Malaysia), as well as an increased use of fertilizers, affecting nitrogen and phosphorus cycles, and the increased demand for irrigation, increasing demand for water. It is prudent to accept that at present we are unable to predict all potential consequences of a biofeedstocks-based economy, especially with regards to system-level environmental, social and economic effects, such as biodiversity, climate change, food and chemicals supply chains, etc. Within this context it is highly attractive to consider non-food biomass and waste biomass as a source of chemical structural diversity for the production of platform molecules.
An increasing number of demonstration projects are being initiated targeting the development of integrated biorefineries (see e.g., www1.eere.energy.gov/biomass/integrated_biorefineries.html for an interactive map of different biorefining projects in the US), and several good reviews and reports were published on the general principles of biorefining [1,2]. There were also earlier examples of large-scale integrated biorefineries. Most notably the acetone-butanol complex in Soviet Union based on fermentation of agricultural waste [3]. All these examples are targeting large-scale processes, aimed at production of platform molecules and high-tonnage products.
In parallel, there exist a well-established industry of manufacturing high-value products from bio-derived feedstocks for specific niche applications, such as bio-pharmaceuticals, flavour and fragrances additives, etc. Within this industry typically either the whole bio-material is used (e.g., micro algae as a health food or component of cosmetic products), or a single compound is obtained from a plant/organism that is either harvested wild or cultivated. Waste from these processes is typically landfilled, used as animal feed or as a fuel in the best case. The future of this industry critically depends on maintaining a sustainable supply of biomass and on the market demand for the bio-derived high-value chemicals. The latter can be affected by many factors, most notably the increasing frequency of extreme weather in many agricultural regions, affecting food commodities prices and availability, an increasing awareness of impact of business and technology on biodiversity and climate, affecting purchasing choices.
In the case of bio-pharmaceuticals, typically the content of the desired metabolite is in the order of 1–30% dry weight at best and thus between 99 and 70%wt of biomass (dry weight) is either being put back into the fields as a source of nutrients, or burned. A specific example considered in this study is the extraction of artemisinin for production of artemisinin-based antimalarial drugs [4]. A typical plant content of artemisinin is between 0.7–1.5%wt. The main process of extraction is using petroleum ether or hexane-ethyl acetate mixed solvent [5]. Residual biomass from this process always contains traces of hydrocarbon solvents even after prolonged evaporation and cannot be put back into soil. Its best use is as a fuel for boilers to reduce the energy intensity of the extraction process, which is done commercially at some artemisinin extraction facilities in China.
The single-use of biomass in production of bio-pharmaceuticals is a significant drawback of the current processes from the point of view of process sustainability. Specifically, in the case of artemisinin a significant price volatility is induced by unpredictable demand driven by the availability of public subsidies. An unstable business environment drives farmers to abandon cultivation of Artemisia annua L., which creates further uncertainty over the future availability of the drug precursor and over its prices. Availability of the semi-synthetic artemisinin produced by Sanofi from 2012 in part resolves this issue. However, there is still a considerable demand for artemisinin obtained through extraction. The hypothesis of this paper is that by increasing the range of high-value products manufactured from such plants as A. annua, a dampening effect on prices due to the increased diversity of the end-products markets will be observed, resulting in an increased profitability of biomass production and processing.
In order to achieve this, it is necessary to identify the entry points for new high- and medium-value bio-derived molecules into chemicals supply chain: what new products could be obtained, what existing products could be replaced or produced starting from complex bio-derived molecules, or what new generic synthetic blocks could be obtained from biomass. This may also require a different extraction technology, amenable to multi-product separation and using cleaner solvents which leave the residual biomass suitable for further fermentation. In the biorefining schemes published to-date [1,2], following the extraction of high-value components, the residual lignocellulose is subjected to fractionation into lignin, cellulose and hemicellulose. Further upgrading of hemicellulose and cellulose is likely to proceed via fermentation. Fermentation of lignin or direct fermentation of lignocellulose is also becoming feasible, given the emergence of high-yielding lignin degraders [6] and intensive work on dissolution and fermentation of cellulose [7,8]. However, fermentation of biomass containing residues of organic solvents, typically used in extraction, may be prohibitively slow due to their toxicity to bio cultures. Thus, the use of clean extraction technologies based on supercritical carbon dioxide and other similar solvent systems is preferred [9].
All interdependent influences described above are brought together schematically into a hypothetical project workflow in Fig. 1. Thus, the development of a high-value biorefinery is a complex interdisciplinary problem requiring involvement of several industries, including some that are not connected at present.
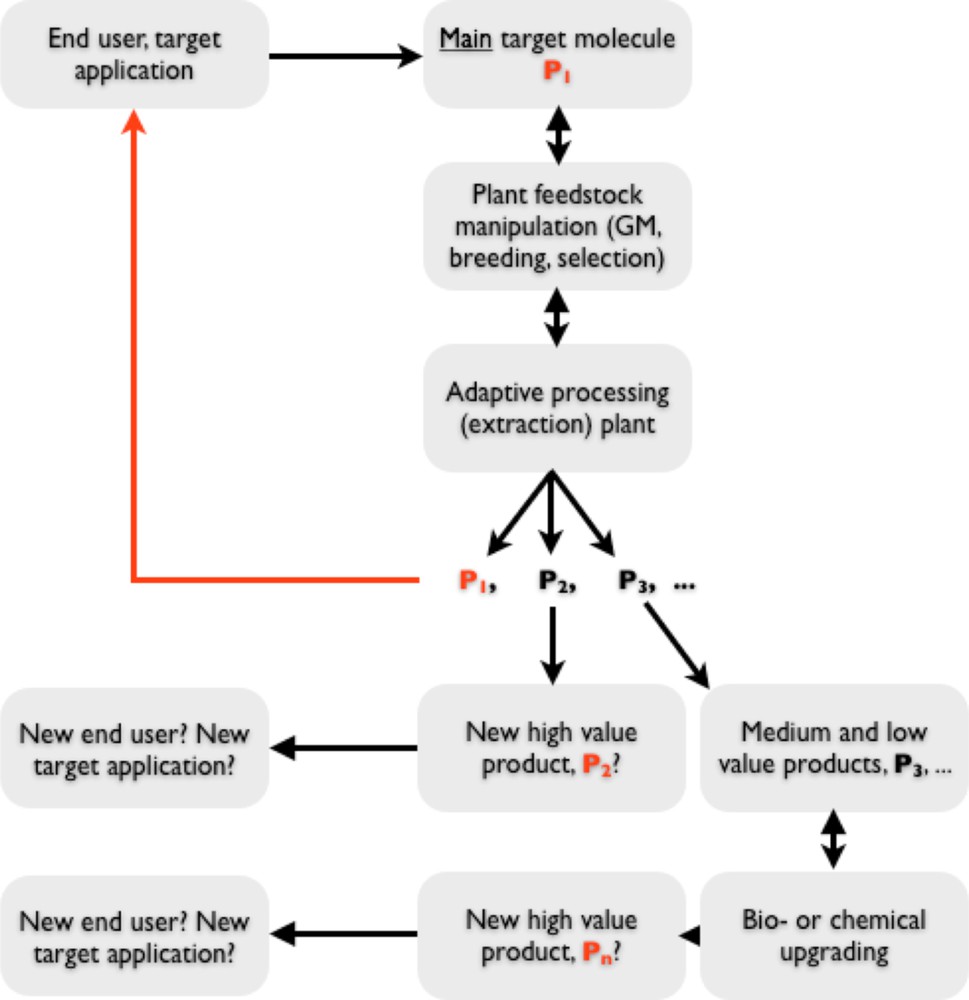
A schematic depiction of a high-value biorefinery project development.
In this paper we describe results of an interdisciplinary project that looked at bringing together some of the pieces of technology required to build a high-value biorefinery. The adopted target was to maximise production of the main target metabolite (P1 in Fig. 1) whilst achieving two more objectives: (i) isolating several more molecules of commercial value and (ii) leaving biomass suitable for downstream fermentation. Thus, the residual biomass from high-value biorefining should be a feedstock for large-scale biorefinery, producing bulk chemical intermediates and fuels. The main target product P1 was artemisinin, whereas additional medium and low-value products of choice were various monomers.
The use of plant metabolites to make monomers, polymers or composite materials is a growing area of research [10,11]. Recent research has focused upon the use of vegetable oils [12], carbohydrates [10] and lignin [13] among others. These natural products may be polymers themselves (e.g. lignin) or molecules which can be made into polymers (e.g. vegetable oils). While some natural oils (e.g. castor oil) can be used directly to produce polymeric materials [14], others often need chemical manipulation before they are suitable. For example, epoxidised soybean oil can be chemically manipulated to hydroxylated monomers useful in the preparation of polyurethanes and polyesters [15]. Similarly, acrylates and methacrylates can be chemically incorporated into vegetable oils to provide useful surface coatings [1].
The use of flavonoids as potential building blocks for new monomers has not been extensively studied. Flavonoids are important secondary metabolites, often exhibiting anti-oxidant activity [16]. Some flavonoids e.g., quercitin, have been suggested to show anti-inflammatory or anti-cancer activity [17]. However, their use as building blocks for fine and speciality chemicals is not well studied. During this study a range of flavonoid derivatives could be obtained from the biorefinery process and one of the simpler flavonoids was used to show the possible derivatization to novel monomers.
The aim of the present work was to evaluate the feasibility of switching from conventional petrochemical extraction of artemisinin to an extraction process using a clean solvent, leaving no residues that could adversely affect downstream lignocellulose fermentation. Also, to evaluate the range of other molecules that could be extracted in a sequential process from A. annua and to show some exemplar chemical transformations allowing to convert some of the new potential chemical precursors to polymer end-products. This approach exemplifies the potential future development of high-value biorefineries, linked with conventional large-scale biorefineries.
2 Experimental
2.1 Chemicals and materials
Hexane, ethyl acetate, acetonitrile were purchased from Sigma Aldrich and used without further purification. Tetrafluoroethane (R134a) was purchased from Ineos Fluor, UK.
2.2 Plant samples
Dried A. annua biomass was obtained from CHEMO (Argentina), GlaxoSmithKline (Tasmania, Australia), REAP (Kenya) and SensaPharm Ltd (UK). These materials were stored in a cool dark condition until required for analysis and extraction. These plant lines are commercial varieties obtained as crushed leaves. The companies hold voucher specimens. However, DNA has also been taken from each plant in order to confirm by genotyping if required at any time.
2.3 Extraction of artemisinin and co-metabolites
Extraction of artemisinin from A. annua by conventional hexane-ethyl acetate solvent mixture was performed as follows: ratio of solvent to dry biomass was always 10:1 (v:wt), extraction was performed on a 1 L scale in a Buchi double-jacket glass vessel and in 1 L round-bottom flasks–employing either of two methods, involving heating at 40 °C for three hours with mechanical stirring, or extraction in a sonication bath (PUL 125, Kerry Ultrasonics, UK) operated at 220 V, 50 Hz for one hour. The ratio of solvent-to-biomass is taken deliberately large to avoid equilibrium limitation. The temperature of extraction was previously optimized and corresponds to typical industrial conditions [5]. In the US-assisted extraction the duration was optimized.
Exhaustive extraction of artemisinin to determine its amount in biomass was performed as follows. A sample of 10 g biomass was extracted with 100 mL ethyl acetate at 50 °C over 6 hours three times with fresh portions of solvent. Three extracts were combined and evaporated to dryness in a rotary evaporator. The residue was dissolved in 20 mL of acetonitrile. This results in precipitation of waxes. Solution was filtered through a syringe filter (0.2 μm) and injected into HPLC.
Extraction of artemisinin using tetrafluoroethane (R134a) was performed in a pilot rig consisting of a 30 L chilled solvent storage tank, a diaphragm pump equipped with a pressure damper, a Coriolis flow meter, a 5 L extraction vessel equipped with an electrical tape heater, a 45 L jacketed evaporation vessel and two heat exchangers for condensing the solvent into the storage vessel. All equipment was rated to 40 bar. The biomass was packed into the extraction vessel in a nylon mesh bag. Solvent was fed from the storage tank. Extraction vessel was initially cooled to condense liquid R134a solvent prior to starting the pump. Once pumping started the extractor was heated to the required temperature. At the set pressure (between 5–18 bar, depending on the extractor temperature) a throttle valve was open and solvent circulated through the evaporator, heat exchangers and condensed back to the solvent storage vessel. Overall duration of extraction experiments was between 1–8 h. Experiments with the pumped flow were performed at 25 and 40 °C. Due to experimental scatter associated with batch-to-batch variability in the packing density and in the flow rates the data were averaged. Following the extraction and complete evaporation of the solvent, the precipitate in the evaporator vessel was washed with 300 mL of acetonitrile. A 0.2 mL aliquot was made up to 1 mL with acetonitrile and analysed by HPLC. A sample of residual biomass was quickly washed with acetonitrile at ambient temperature and the amount of artemisinin in the wash was determined by HPLC. This comprised the amount of artemisinin extracted, but not transported out of the biomass bed and it was added to the amount of artemisinin found in the evaporator for the purpose of calculating the extraction efficiency. After this wash, a sample of the residual biomass was exhaustively extracted with acetonitrile at 60 °C over 12 h to determine the amount of non-extracted artemisinin in the biomass, and to close the material balance. Material balance was closed within ± 5% of the overall extraction efficiency. Results are expressed as extraction efficiency defined as the percent of artemisinin recovered after extraction from the amount of artemisinin in the biomass. SEM images of the leaves following extraction were obtained using JEOL 6480 instrument.
Fractionation of A. annua total extract was performed as follows. A sample of dry leaves was soaked in ethyl acetate overnight using two portions of fresh solvent (biomass to solvent ratio 10:1 wt:v with each fresh solvent portion). The crude extract was evaporated, re-dissolved in 20% aqueous ethanol and partitioned first with hexane and then with dichloromethane. Each solvent fraction was evaporated to dry residue.
2.4 Analytical procedures
2.4.1 Artemisinins analysis
Artemisinin and related metabolites were quantified using published HPLC protocol [18]. HPLC instrument (Shimadzu Prominence) equipped with a UV-vis diode array (SPD-M20A, DAD) and evaporative light scattering (ELSD, LTII, 350 kPa N2, nebulizer at 40 °C) detectors in line were used. Mobile phase 65:35% v/v acetonitrile:water mobile phase. Column: Betasil C18 5 μm, 250 × 4.6 mm, flow-rate 0.8 mL min−1, column temperature 45 °C.
2.4.2 Flavonoid analysis
An Agilent 1100 series HPLC instrument (Agilent Technologies, UK) equipped with a quaternary pump, auto-sampler, a degasser and a diode-array detector was used. A modification of the method by Bilia et al. [19] was employed. Briefly, the isocratic mobile phase consisted of water adjusted to pH 3.2 by acetic acid (eluent A) and acetonitrile (eluent B) operated at 50% A and 50% B at a flow rate of 1 mL min−1 for 30 minutes. Separation was on a Zobax Eclipse C18 column (150 × 4.6 mm, 5 μm) protected by a Zobax C18 guard column and detection was at 280 nm with an injection volume of 20 μL.
2.4.3 Xanthophylls analysis
Extracts were analysed for their xanthopyll content using a modification of the HPLC method [20]. Briefly, a mobile phase of 5% methanol in acetonitrile (solvent A), 100% methanol (solvent B) and 0.05% triethylamine in ethyl acetate (solvent C) in three separate bottles was used. Separation was achieved on an Eclipse Zobax C18 column (150 × 4.6 mm, 0.5 μm) at a flow rate of 1 mL min−1 over a 30 min run time. Detection was at 470 nm on a photodiode array detector (PAD) attached to an Agilent 1100 series HPLC with a quart-pump, auto-sampler and a degasser.
2.4.4 β-Carotenoid analysis
Extracts were analysed on a Shimadzu Prominence HPLC equipped with an auto-sampler, degasser, and photodiode array detector using a literature method [21]. Separation was achieved on a Phasesep-Partisil C18 (250 × 4.6 mm, 5 μm) column attached to a C18 guard column maintained at 30 °C. A solvent system composed of eluent A (acetonitrile:methanol, 95:5 v/v) and eluent B (acetonitrile:methanol:ethylacetate, 60:20:20 v/v/v). The eluents were modified with 0.1% butylated hydroxytoluene (BHT) and 0.05% triethylamine (TEA) respectively. A flow rate of 1 mL min−1 was used on a gradient elusion which was as follows: 100% A 0-5, 100% B 13 min, 100% B 30 min, 100% A 45 min. Detection was at 450 nm on the PDA.
2.4.5 Mass spectrometry analysis
Metabolites in the extracts were profiled based on our method [22]. Briefly, the system consists of an Acquity liquid chromatograph (LC) (Waters Corp., Milford, MA, USA) coupled to an Acquity tandem quadrupole detector (TQD). The column heater was set at 30 °C and a Genesis® Lightn C18 column (100 × 2.1 mm; 4 μm) (Grace, IL, USA). The mobile phase consisted of A: 0.1% formic acid in water and B: 0.1% formic acid in acetonitrile used in the following gradient: 0–7.0 min, 25–98% B; 7–9.5 min, 98% B; 9.5–10 min, 98–25% B; 10–15 min, 25% B; at a flow rate of 0.4 mL min−1. Weak wash solvent was 10% acetonitrile, strong and needle wash solvent was a mixture of acetonitrile, propan-2-ol, methanol and water (30:30:30:10 v/v/v/v). The TQD was operated with an ESI interface in positive ionization mode (ESI+). The cone and desolvation gas flow rates were set at 45 L h−1 and 800 L h−1, respectively. MS parameters were automatically defined using Waters IntelliStart® software for the tuning and calibration of the TQD and subsequently manually optimized for artemisinin and related metabolites.
2.5 Synthesis of higher-value chemical intermediates
Various simple flavone derivatives were synthesized as potential novel monomers for polyurethane, polyester, epoxy resin and polyglycerol synthesis. One specific example was chrysin methacrylate 6. This was co-polymerized with methyl methacrylate (1:1 molar ratio) in the presence of AIBN in toluene to furnish a novel methacrylate polymer.
3 Results and discussion
3.1 Conventional extraction and optimisation of extraction by tetrafluoroethane (R134a)
Table 1 shows extraction efficiencies obtained with a hexane: ethyl acetate (95:5) solvent mixture using two different extraction methods, conventional soaking with mechanical stirring and ultrasound-assisted extraction. The obtained efficiencies of extraction are very high at 91 and 95% for the two methods used. This is considerably higher than the efficiency of extraction attained normally in industrial-scale operations, when a static biomass bed is soaked in hot solvent, or hot solvent is percolated through a biomass bed [5]. Both, the mechanical agitation and ultrasound provide a considerably higher mass transfer in comparison with soaking or percolation-type extractions. The use of ultrasound marginally increases the efficiency of extraction. The reason for high efficiency of extraction under mechanical stirring conditions and, consequently, only a small improvement due to sonication is due to effective disruption of trichomes containing artemisinin, observed in SEM images of leaves post extraction (Fig. 2, left image). Thus, the poor extraction at larger scales is mainly due to poor access of solvent to leaves and very slow mass transfer through a large bed.
Extraction efficiencies of artemisinin from biomass using hexane-ethyl acetate (95:5) solvent mixture using two extraction methods.
| Method of extraction | Artemisinin in planta biomass (%wt) | Artemisinin extracted (%wt) | Extraction efficiency (%) |
| Heat for three hours (40 °C) with mechanical stirring | 0.78 | 0.71 | 91 |
| Sonication for one hour | 0.78 | 0.74 | 95 |
a Values determined by exhaustive extraction.
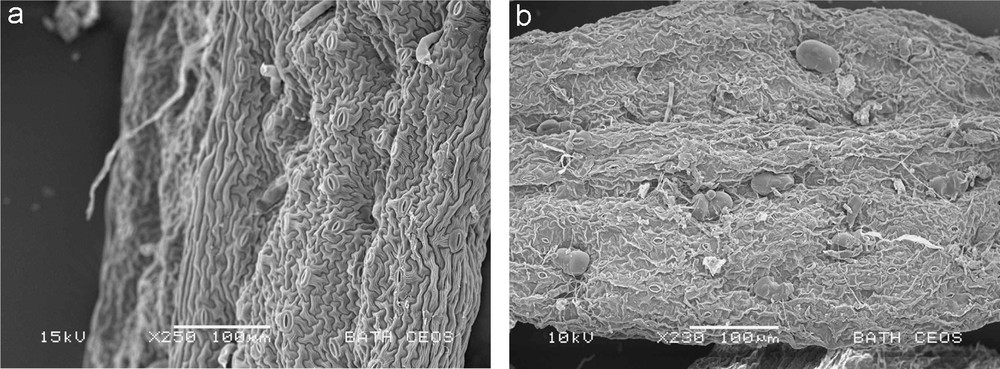
SEM images of A. annua leaves following extraction with hexane:ethyl acetate (left) and with R134a solvents (right).
However, as was mentioned above, extraction by hydrocarbon solvents leaves biomass unsuitable to fermentation processes due to toxicity of the solvent residues to fermentation culture. An earlier study assessed economic and environmental potential of extracting artemisinin by ethanol, scCO2, ionic liquids and tetrafluoroethane, as compared to hexane-ethyl acetate extraction [5]. Extraction by tetrafluoroethane was found to be the most promising technology among the considered new alternative processes.
There is a valid concern over the use of tetrafluoroethane as a solvent, due to its significant green house gas potential. The substance is to be withdrawn from the use as propellant in inhalers and as a refrigerant, due to uncontrolled release of the significant green house gas. However, the volume of solvent used in extraction is minuscule compared to the volume used in refrigeration and near-to-complete recovery of used solvent is feasible (300 pm residual concentration in the biomass is attainable with vacuum degassing of spent biomass), which gives a relatively low environmental impact, comparable with that of petrochemical solvents. Low operating temperature and pressure of this process compared to scCO2 extraction make it significantly more attractive in terms of running and capital costs. There remained a question whether sophisticated pumped flow extractor is required for this process or a more basic and energy efficient gravity-flow extraction is sufficient. The latter process has been operated commercially [23]. Thus, tetrafluoroethane as a solvent remains an attractive technical solution.
In this study extraction of artemisinin by R134a was performed in a 5 L scale vessel in three different modes of operation: pumped down- and up-flow, and gravity-driven down-flow. These three cases represent typical potential industrial extraction modes. The observed trend (Table 2) correlates well with the expected improvement in mass transfer. The gravity down-flow is likely to be characterized by incomplete wetting of biomass and slow solvent flow-rate, thus resulting in poor recovery of artemisinin. The maximum attained efficiency of extraction is for pumped up-flow, which offers the most intensive mass transfer within the three flow regimes studied. The obtained efficiency at 84% is well above what is typically obtained in hexane (petroleum ether) extraction on the large scale [5], but is below what we obtained in the small-scale extraction in hexane with intensive mixing (Table 1). We have also observed that many of the trichomes were intact following extraction with R134a (Fig. 2, right image). This may indicate inhomogeneous wetting of biomass by R134a solvent in the static biomass bed and sampling in SEM of a region of a leaf that has seen little contact with the solvent. An alternative explanation would imply a mechanism of extraction that enhances mass transfer of artemisinin out of trichomes without disrupting the walls of trichomes, which we have no evidence for. In any case, there clearly is room for further optimisation of extraction by R134a.
Comparison of extraction efficiency of three different operating regimes of R134a extraction.
| Entry | Extraction mode | Extraction efficiency/% |
| 1 | Gravity down-flow | 31 |
| 2 | Pumped down-flow | 46 |
| 3 | Pumped up-flow | 84 |
3.2 Total extraction and fractionation; profile of co-metabolites
Metabolic profile of A. annua has been extensively studied [4,24]. However, it varies significantly with the specific growth conditions and local climatic and soil conditions. We have investigated this variation by analysing several groups of metabolites either quantitatively or qualitatively in A. annua grown in four different countries with very different soil and climatic conditions: Argentina, UK, East Africa and Tasmania.
A significant variation in the relative amounts of different metabolites in the biomasses grown in different geographic regions is observed (Table 3). Chemical structures of various compounds listed in Table 3 are given in Scheme 1. As far as we have been able to identify, the four biomasses were grown from genetically identical materials. Therefore, the differences in metabolic profiles are due to environmental factors. We did not have access to the same biomasses over several years of cultivation and therefore cannot quantify variability within the same growth region between years. However, we anticipate such variation also to be significant. Based on this variability we may speculate that each biomass would require re-optimization of the extraction-purification process, since particular composition of all the metabolites would influence solubilities and separation effectiveness. Specifically, there are statistically significant variations in the amounts of some flavonoids and pigments.
Major artemisinin related metabolites and other compounds found in Artemisia annua samples of different geographical origin.
| Compounds | Origin of plant sample | |||
| Argentina | UK | East Africa | Tasmania | |
| Sesquiterpenes | ||||
| Artemisinin (mg g−1) | 3.28 | 3.29 | 4.09 | 9.69 |
| Artemisinic acid (mg g−1) | 0.35 | 0.63 | 0.53 | 1.14 |
| Artemisitene (mg g−1) | 0.44 | 0.22 | 0.16 | 0.05 |
| Dihydroartemisinin (mg g−1) | 0.22 | 0.21 | 0.48 | 1.04 |
| Deoxyartemisinin | Present | Present | Present | Present |
| Arteannuin H | Present | Present | Present | Present |
| Arteannuin B | Present | Present | Not detected | Not detected |
| Pigments (μg−1) | ||||
| Xanthophylls | 2.31 | 1.29 | 5.75 | 4.61 |
| Beta-carotene | 4.92 | 4.08 | 30.73 | 149.89 |
| Flavonoids (mg g−1) | ||||
| Para-coumaric acid | Not detected | Present | Not detected | Present |
| Casticin | 1.47 | 0.42 | 1.45 | 1.80 |
| Artemetin | 0.06 | 0.03 | 0.22 | 0.09 |
| Retusin | 0.17 | 0.09 | 0.49 | 0.39 |
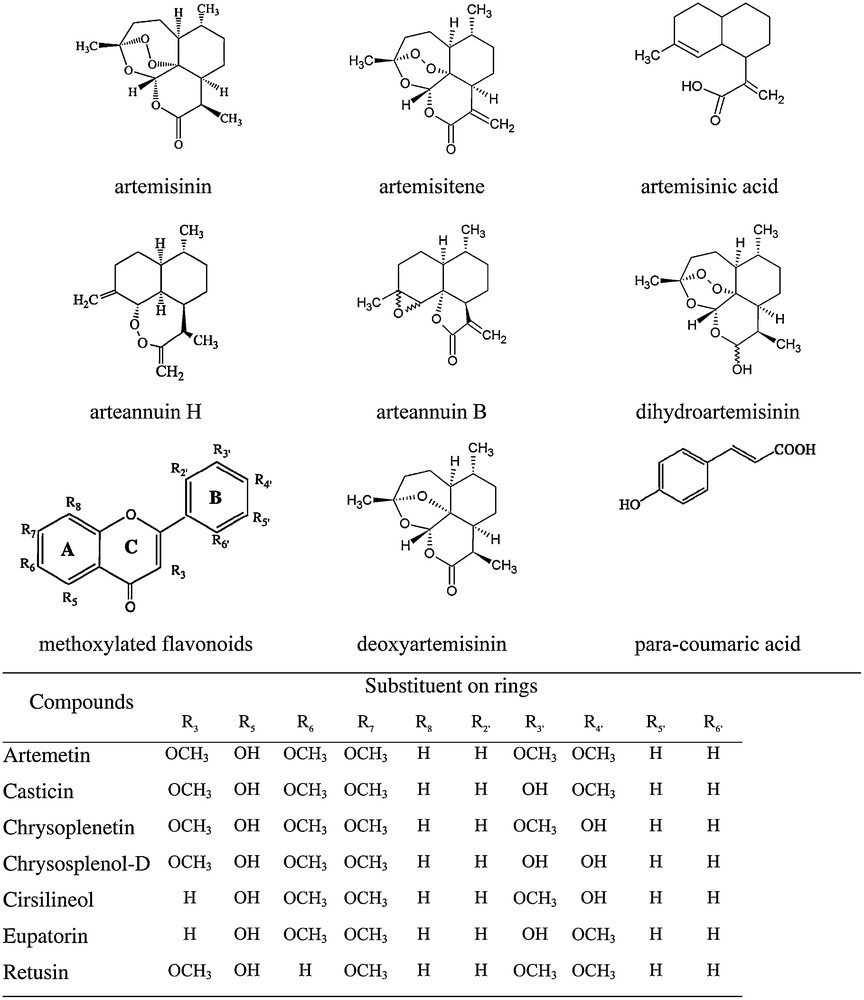
Molecular structures of compounds identified in A. annua extracts.
The data in Table 3 also indicate that some metabolites are either present or not in the plants grown in different countries. This does affect the universal optimization of any high-value biorefining system. Specifically, para-coumaric acid is a potentially useful source of aromatics that could be used in polymerisation and high-value consumer products.
Different metabolites could be grouped according to their polarity and the corresponding type of solvent in which they are preferentially extracted. Thus, the laboratory-scale extraction-fractionation of A. annua was performed with conventional solvents of different polarities. The representative solvents reflect the broad groups of compounds of low, intermediate and high polarity, as shown in Table 4.
The types of compounds identified in different fractions after sequential extraction of A. annua.
| Fraction | Compound category | Specific compounds identified |
| Hexane | Oils, waxes, carotenoids, triterpenoids | Artemisinin, arteannuin B, artemisinic acid |
| Dichloromethane | Flavonoids | Casticin, artemetin, retusin |
| Water soluble | Biopolymers, polyphenols | Tannins |
In a typical extraction sequence, from 329 g of starting dry leaf material (Kenyan biomass was used) 29.7 g of residue was obtained after exhaustive extraction with ethyl acetate. This was re-dissolved in aqueous ethanol and partitioned first with hexane, yielding after evaporation 7.5 g of dry residue, and then with dichloromethane, yielding after evaporation 21.7 g of dry residue. The final water soluble fraction was 1.02 g.
The hexane fraction contains the primary compound of interest, artemisinin, as well as other similar co-metabolites, waxes, oils, carotenoids. The largest group of compounds in this fraction are waxes which are difficult to separate for detailed analysis and further use. However, within artemisinin analogues, artemisinic acid, artemisitene and dihydroartemisinin were identified and quantified (Table 3). Deoxiartemisinin is a known impurity which is believed to be a product of decomposition of artemisinin, whereas arteannuin H and B maybe of interest as bio-actives. Hexane fraction also contains a large amount of pigments.
Within the dichloromethane fraction the class of compounds of interest is flavonoids. A large number of flavonoids were identified in A. annua in the literature [4]. We have quantified three flavonoids due to their potential impact on purification of artemisinin [25].
A potential alternative extraction sequence would involve the initial separation of terpenes by, for example, steam distillation. In this study we have not investigated this option, due to the risk of degrading artemisinin, which is rather unstable in aqueous environment. A rapid vacuum assisted or MW-assisted steam distillation may, however, be a viable option to recover volatile terpenes prior to extraction and fractionation of structurally more complex compounds.
3.3 Potential for new products from A. annua metabolites
For the chemical derivatization study a simple flavonoid chrysin was selected, as it is commercially available in its pure form and it is structurally similar to some of the flavonoids identified in A. annua, enabling a parallel to extraction research programme. Initial studies concentrated on directly polymerizing the flavonoids isolated with di-isocyanates (MDI, TDI and HDI) making use of the reactive phenolic functionality. However, the relative insolubility of the flavonoid derivatives under the polymerization reaction conditions1 and the differential reactivity of the phenolic hydroxyl groups precluded the efficient formation of polyurethane polymers. Consequently, we explored the chemical derivatization of flavonoids to improve both their solubility and reactivity. We illustrate here the chemistry with reference to chrysin. Selective functionalization of the C-7 OH group was possible with a range of C-4 organic fragments. Alkylation with K2CO3/1,4-dibromobutane furnished bromide 1 in 86% yield. Hence, reaction of chrysin (10.1 g) with 1.05 eq of K2CO3 (5.8 g) and 1,4-dibromobutane (9.0 g) in dry DMF (130 mL, 0.3 M) at room temperature for 24 h gave 1 (13.4 g) as a white solid. The X-ray structure of 1 clearly shows the intramolecular H-bond between C9-OH and the C11 C = O which decreases the reactivity of C9-OH respective to the C-7 OH. Spectral details for 1: IR (film)/cm−1 υmax 3061, 2963, 2934, 2865, 1649, 1601, 1586; 1H NMR (300 MHz, CDCl3) δ 12.71 (1H, s), 7.90–7.87 (2H, m), 7.58–7.50 (3H, m), 6.68 (1H, s), 6.49 (1H, d, J = 2.2 Hz), 6.36 (1H, d, J = 2.2 Hz), 4.08 (2H, t, J = 6.0 Hz), 3.50 (2H, t, J = 6.5 Hz), 2.12–2.04 (2H, m), 2.04–1.95 (2H, m); 13C NMR (75.5 MHz, CDCl3) δ 182.5, 164.8, 164.0, 162.2, 157.8, 131.8, 131.3, 129.1, 126.3, 105.9, 105.7, 98.5, 93.1, 67.5, 33.2, 29.3, 27.6; ESI Found (MH)+, 389.0383, C19H17Br79O4 requires (MH)+, 389.0388 (Scheme 2).

The product of functionalization of chrysin via C7 position.
Repeating the reaction with only 0.5 eq of dibromobutane and K2CO3 furnished the dimer 2. After three hours a precipitate was formed which was re-dissolved in THF and the solution left overnight. This solution was neutralized with 2 N HCl and the solid precipitate washed with water to give 2 [1H NMR (300 MHz, CDCl3) δ 12.71 (2H, br s), 7.89–7.86 (4H, m), 7.55–7.49 (6H, m), 6.66 (2H, s), 6.50 (2H, d, J = 2.2 Hz), 6.38 (2H, d, J = 2.2 Hz), 4.15 (4H, m), 2.05 (4H, m)]. A range of other C-4 fragments ultimately derived from succinic or maleic acids (themselves renewable chemicals derived from fermentation of carbohydrates [26], potentially part of this biorefinery) were incorporated. In this way it was possible to prepare a range of symmetrical diol derivatives as potential monomers for polyurethane or polyester synthesis 2-4. Reaction of chrysin with succinic anhydride (1.0 eq) in a 1:1 v/v THF/Et3N mixture furnishes 5, a potential monomer for polyester synthesis (Scheme 3) [27].
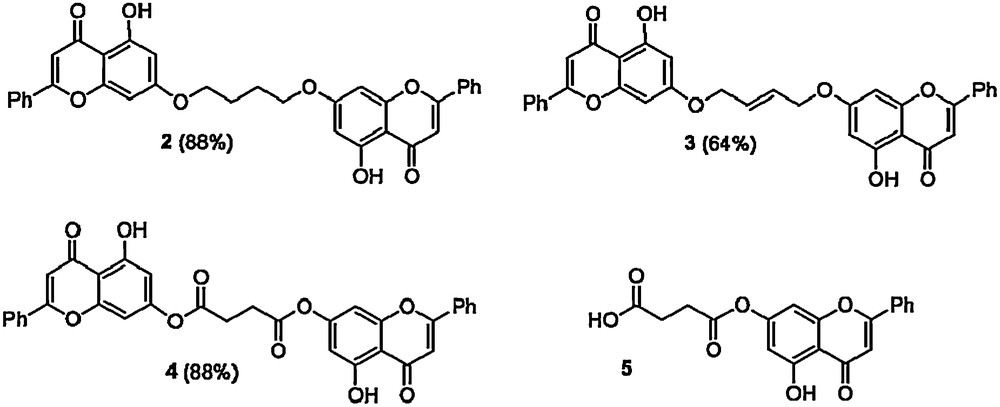
Potential monomers for polyurethane and polyester synthesis.
We also explored the feasibility of incorporating methacrylate 6, 10 and glycerol groups 7 and 8 into the flavonoid skeleton. Methacrylate derivatives can be polymerized via a free radical process [28], while polyglycerols have attracted significant attention in recent years [29]. Methacrylate 6 was prepared by acylation of chrysin with 1.0 eq of methacryloyl chloride in pyridine overnight. After acidification (pH 2) the mixture was extracted with ethyl acetate to furnish 6 in 89% yield [1H NMR (300 MHz, CDCl3) δ 12.73 (1H, s), 7.90 (2H, m), 7.59–7.51 (3H, m), 6.91 (1H, d, J = 1.1 Hz), 6.74 (1H, s), 6.62 (1H, d, J = 1.1 Hz), 6.39 (1H, s), 5.83 (1H, s), 2.08 (3H, s)]. Derivative 7 was prepared from chrysin in 98% yield by selective deprotonation with K2CO3 and alkylation using commercially available tosylate 9 (derived from glycerol). [1H NMR (400 MHz, CDCl3) δ 12.72 (1H, s), 7.89 (2H, dd, J = 14.3, 7.7 Hz), 7.60–7.50 (3H, m), 6.67 (1H, s), 6.53 (1H, d, J = 2.2 Hz), 6.39 (1H, d, J = 2.2 Hz), 4.51 (1H, app quin, J = 5.8 Hz), 4.19 (1H, dd, J = 8.5, 5.8 Hz), 4.12 (1H, dd, J = 9.6, 5.8 Hz), 4.03 (1H, dd, J = 9.6, 5.8 Hz), 3.92 (1H, dd, J = 8.5, 5.6 Hz) 51.48 (3H, s), 1.42 (3H, s)]. Simple acid catalysed deprotection of 7 with acetic acid furnished 8 in 87% overall yield from chrysin. While the other derivatives 2-7 were soluble in chloroform, glycerol derivative was less soluble but dissolved readily in methanol or ethanol. Selective methacryloylation of 8 at the primary hydroxyl was possible using the method for formation of 6. Finally, epoxide 11, a potential monomer useful for polyether synthesis was prepared from tosylated glycidol. It was found that the thermal stability of the functionalised monomers was lower than chrysin itself with thermal gravimetric analysis indicating that the temperature at which 10% of the monomer decomposed to be 1 (382 °C), 2 (330 °C), 3 (303 °C), 4 (320 °C), 6 (333 °C), 7 (350 °C), 8 (309 °C), 9 (304 °C) respectively (Scheme 4).
Potential monomers for polymethacrylate, polyglycerol and polyether synthesis.
Most methacrylates can be polymerized under free radical conditions. Flavonoids however are known to be anti-oxidants and will inhibit free radical reactions [30]. It was therefore not clear that monomers such as 6 would readily undergo polymerization. Using the TEAC (Trolox equivalent anti-oxidant activity) ABTS•+ assay [31] it was possible to measure the relative anti-oxidant abilities of a range of C-7 functionalised derivatives with respect to chrysin itself (23.6 μM). As expected, removal of one of the phenolic groups lowered the anti-oxidant activity significantly, [7 (15.9 μM), 8 (14.2 μM) and 12 (14.5 μM)] and it was possible to polymerise methacrylate 6. Hence, reaction of a 1:1 molar ratio of methyl methacrylate and monomer 6 with AIBN in toluene at 70 °C furnished a polymethylmethacrylate 12 (Mn 7.3 kDa, Mw 14.8 kDa, PDI = 2.03) incorporating the renewable derived monomer. Fig. 3 shows the traces using both ultraviolet (UV) and infrared (IR) detection. The UV trace indicates that monomer 6 has been incorporated into the polymer.
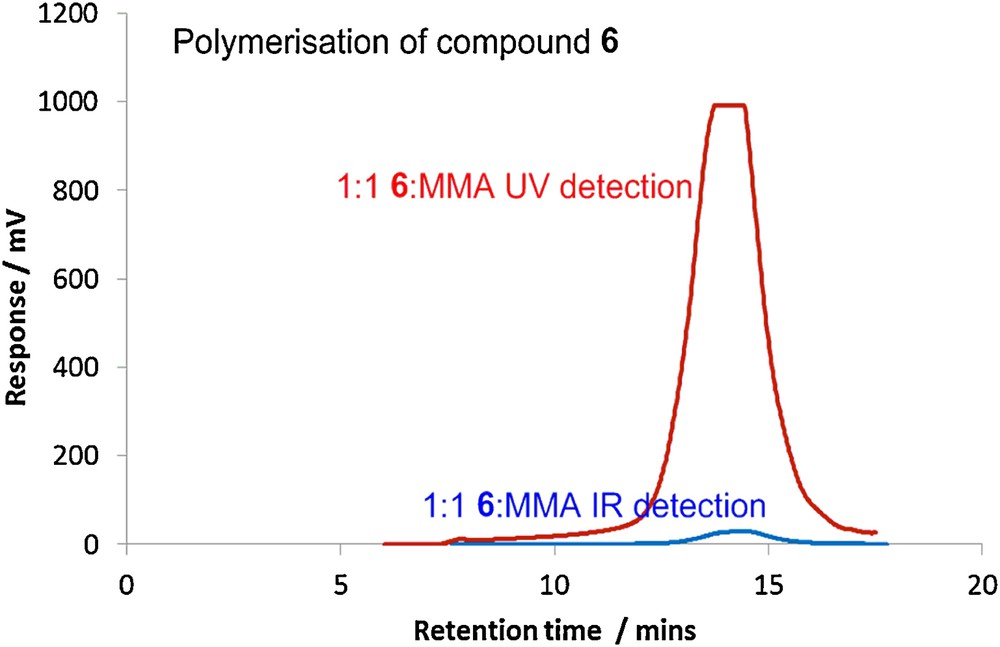
GPC traces for the polymerisation of 6 with MMA at 70 °C.
4 Conclusions
In conclusion, we have shown that a conventional extraction of a high-value bio-pharmaceutical could be effectively performed using a clean solvent extraction, leaving residual biomass suitable for downstream fermentation in bulk biorefineries. The metabolic profile of the chosen example biomass was highly variable depending on the geographic origin of the biomass. This affects optimization of each extraction-separation process as well as target compounds for isolation in a potential high-value biorefinery. We have identified a number of co-metabolites of potential commercial interests and have shown that one flavonoid output from the biorefinery can be transformed into a large number of novel building blocks. These new feedstocks are potential monomers for polyurethane, polyester, polymethylmethacrylate, polyglycerol and polyether synthesis. Despite the known anti-oxidant, and radical inhibiting properties of flavonoids, it was possible to polymerise one derivative using a free radical strategy.
Acknowledgements
This project was funded by Engineering and Physical Sciences Research Council (EPSRC) via a Sandpit initiative “Processing Industries 2020”. The authors are grateful for excellent collaboration within this project with the groups of Prof. Ian Thompson (University of Oxford), Prof. Yulong Ding (University of Leeds) and Prof. Adam Harvey (Newcastle University). J.S. was funded via EPSRC CASE studentship jointly with Sensapharm Ltd (UK). A. annua biomass for this project was kindly provided by Fundacion Mundo Sano (Argentina), Sensapharm Ltd (UK), GSK (Tasmania) and REAP (Kenya). A.L. is grateful to Medicine for Malaria Ventures (MMV) for funding the HPLC instrument and extraction plant, and to Ineos Fluor for technical advice on tetrafluoroethane extraction. Specifically, the pilot plant study would not have been possible without the support by late Dr Ian Bathurst (MMV) to whose memory this paper is dedicated.
1 Polymerisation was attempted by heating the flavanoids at 110 °C with the isocyanates in a mixed solvent system of toluene and dry DMF. Addition of either pyridine or triethylamine as catalyst was also attempted.