

1 Introduction
A tremendous amount of experimental work has been devoted to the study of the phosphoenols because of their important biological and pharmacological properties [1–4]. Phosphoenols represent important intermediates in organic chemistry for the synthesis of complex organic molecules [5–8].
The reaction of trialkyl phosphite with α-halogenated ketones [9,10] leads to a competition between the Perkow reaction (phosphoenols formation) and the Arbuzov reaction (β-ketophosphonate) (Fig. 1). However, a great amount of experimental works devoted to the selective syntheses of phosphoenols can be found in the literature [11–21].
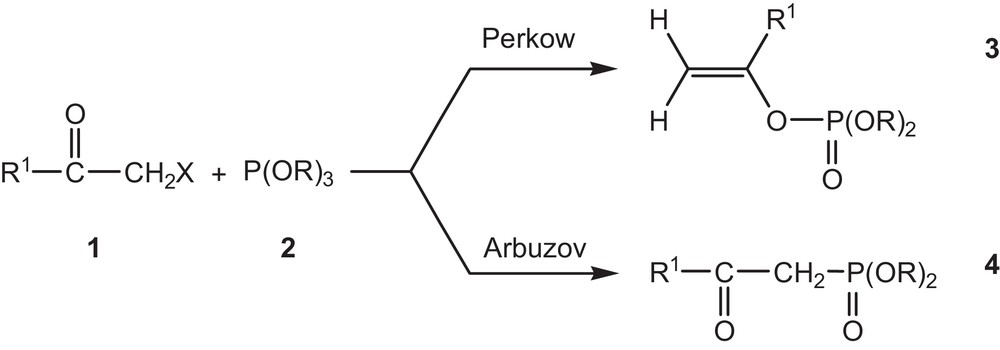
Competition between the Perkow reaction (formation of phosphoenols) and the Arbuzov reaction (β-ketophosphonate).
The aim of this work is first to present a simple and selective method of synthesis of phosphoenol pyruvates (PEP). Based on a preceding work [22], we used the α-chloropyruvates because they present direct precursors of phosphoenol pyruvates and are also easily accessible. We note that, compared with the previous work, our synthesis method has several advantages, such as fairly short reaction times, high yields, mild reaction conditions, absence of catalyst; moreover, it offers ready access to structural variety such as β-substituted phosphoenol pyruvates, not previously available in these series of compounds (Fig. 2).
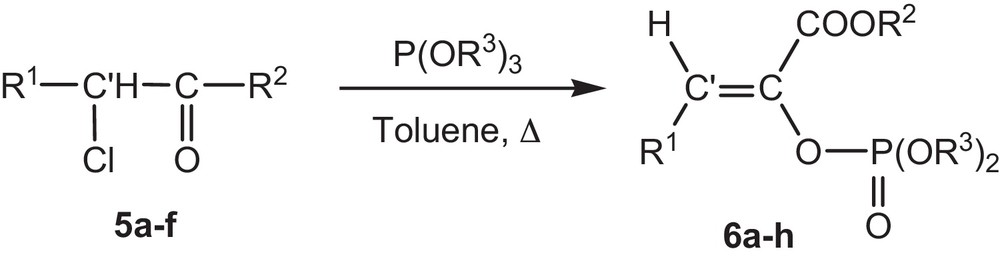
Reaction of α-chloropyruvate 5a-f with trialkylphosphites in toluene.
The second purpose of this study is to evidence the experimental outcomes that state that, in this reaction, the carbon atom of the carbonyl group (CO) of the α-halogenoketone is preferred for the electrophilic attack of the trialkyl phosphites P(OR)3 using theoretical descriptors.
Recently, the popularity and success of density functional theory (DFT) has stimulated many groups to use the Hard and Soft Acids and Bases (HSAB) principle, formulated with DFT [23,24], as a qualitative and quantitative treatment to predict reactivity based upon ground-state properties (density). Furthermore, DFT-based reactivity descriptors, such as condensed Fukui functions [25], local softness [26], local hardness [24b,26], electrophilicity [27], and nucleophilicity indices [28], have proven their utility for the analysis of organic reactions [29–31].
Very recently, Domingo et al. introduced the electrophilic and nucleophilic Parr functions as local indices and showed that these new descriptors provide useful clues for the characterization of the most electrophilic and nucleophilic centers of molecules, and for the establishment of the regio- and chemoselectivity in polar reactions [32]. The proposed Parr functions were compared with both, the Parr–Yang Fukui functions [33], based on frontier molecular orbitals, and Yang–Mortier condensed Fukui functions [34] based on Mulliken charges. In the present work, we used the electrophilic Parr function as local reactivity indices to justify our experimental finding. To be in agreement with the experimental procedure, the calculations were done in the gas phase then in toluene; the obtained results allow us to discuss the solvent's effect in the determination of the local reactivity indices.
2 Theoretical background
2.1 Global quantities
Assuming the differentiability of the electronic energy, E, with respect to N and v(r), a series of response functions appear, among which the most important are probably electronegativity (χ) [35] and hardness (η) [23,36], have been provided with rigorous definitions within the purview of conceptual DFT [25,37]. Electronegativity is the negative of the chemical potential, defined by:
| (1) |
Hardness (η) is defined as the corresponding second derivative,
| (2) |
Softness (S) is the reciprocal of hardness; S = 1/η.
Using a finite-difference method, the working equations for the calculation of μ and η may be given as [25]:
| (3) |
| (4) |
| (5) |
| (6) |
The electrophilicity index, as defined by Parr et al. [27], is given by:
| (7) |
2.2 Local quantities
As opposed to the global reactivity descriptors described above, the analysis of site selectivity in a molecule demands the knowledge of local descriptors. Taking into account the observations obtained from the atomic spin density ASD analysis performed in polar reactions, Domingo et al. propose a new local reactivity index, named the Parr function P(r), which is obtained from the ASD at the radical cation and at the radical anion of the corresponding reagents, and is given by the following equations [32]:
| (8a) |
| (8b) |
The local electrophilicity index [39], ωk, condensed to atom k, is easily obtained by projecting the global quantity onto any atomic center k in the molecule by using the electrophilic
| (9) |
3 Computational details
The quantum chemistry calculations reported in this work have been performed at the B3LYP/6-311g(d) level of theory using the Gaussian 09 series of programs [40]. All stationary points found were characterized as true minima by frequency calculations. We note that the global indices calculated in this work, namely the chemical potential, μ, the global hardness, η, and the global electrophilicity, ω, are calculated using Eqs. (5), (6), and (7), respectively. The electrophilic Parr function,
4 Results and discussion
4.1 Synthesis of β-substituted-phosphoenol pyruvates
In this study, a series of β-substituted phosphoenol pyruvates derivatives 6a–h are obtained by Perkow reaction of the α-trimethyl phosphite with α-chloropyruvates as shown in (Fig. 2).
The reaction yield ranges from 70 to 95%. The obtained results are presented in Table 1.
Experimental results of the synthesized phosphoenol pyruvates 6a–h.
| Product | R1 | R2 | R3 | Y(%) | Bp/°C/mmHg |
| 6a | H | Et | Me | 80 | 91/1 |
| 6b | Me | Me | Me | 95 | 98/0.5 |
| 6c | Et | Me | Me | 85 | 110/0.1 |
| 6d | i-Pr | Me | Me | 85 | 115/0.5 |
| 6e | t-Bu | Me | Me | 80 | 120/0.5 |
| 6f | Ph | Me | Me | 80 | 180/0.5 |
| 6g | Et | Me | Et | 90 | 117/0.1 |
| 6h | t-Bu | Me | Et | 70 | 110/0.3 |
(1H,13C,31P) NMR confirms the structures of the synthesized phosphoenol pyruvates. The reaction products are obtained as mixtures of Z and E isomers, characterized by 1H NMR, with the proton signal of ethylene. We note that the Z-isomer is obtained mainly in the final mixtures. Our results lead to the same stereoselectivity as that obtained by Gaydou [43].
4.2 Optimized geometries
Optimized geometries are presented in Fig. 3, which shows that for all the optimized systems, the C′C bond lengths range from 1.314 to 1.331 Ǻ and correspond to the double bond (CC) length of ≈1.3 Ǻ. On the other hand, the (R1CCO) dihedral angles D (degree) arise from –5.45 to 1.11°; these results show that, for the studied systems, the equilibrium geometry corresponds to the Z stereoisomer. Our theoretical results are in good agreement with our experimental outcomes.
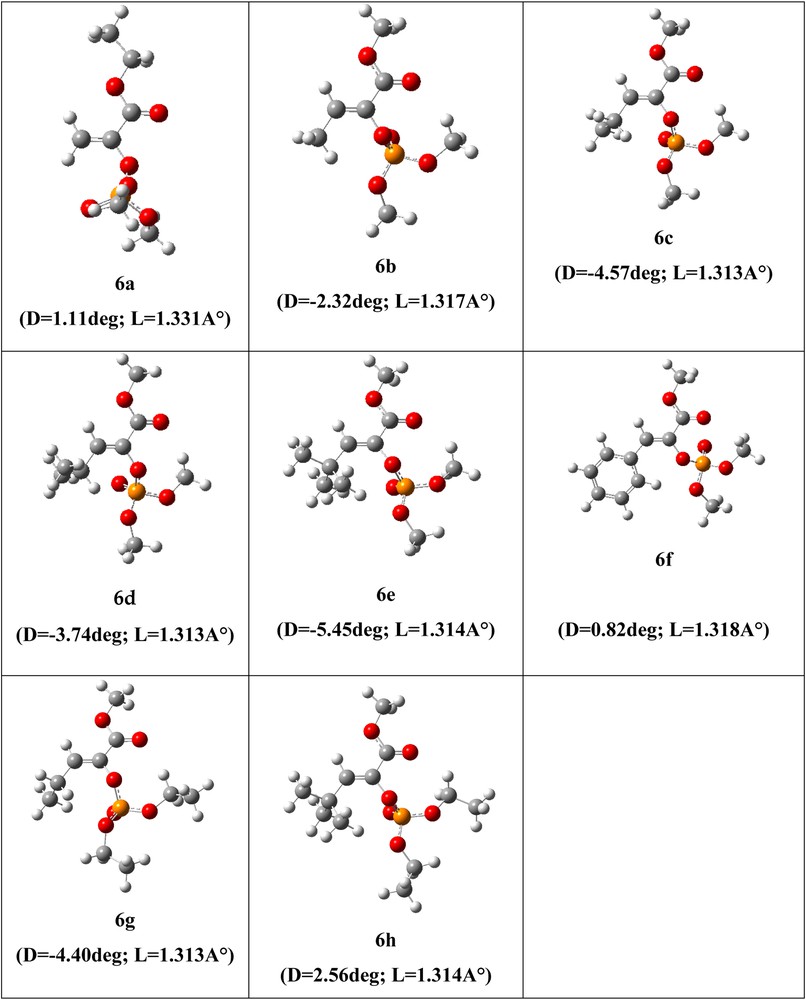
(Color online). Optimized geometries of reaction products (phosphoenol pyruvates) 6a–h, R1C′CO dihedral angle D and the C′C bond length L are given in° (degrees) and Ǻ, respectively.
4.3 Theoretical prediction of the positional selectivity (CO)/(C′Cl) of the nucleophilic attack on α-halogenoketones derivatives
Our experimental results show that the nucleophilic attack will occur preferentially on the C carbon position of the carbonyl (CO) rather than on the C′ position (C′Cl).
In order to rationalize the experimental regioselectivity of the nucleophilic attack of trialkyl phosphites on the α-halogenoketones 5a–f, we have calculated the global and local electrophilicity indices, ω and
Global indices, electronic chemical potentials, μ, global hardness, η, and electrophilicity indices, ω, for compounds 5a–f, calculated in the gas phase.
| Reactant | HOMO | LUMO | μ(ua) | η(ua) | ω(eV) |
| 5a | –0.2749 | –0.0980 | –0.1865 | 0.1769 | 2.67 |
| 5b | –0.2746 | –0.0954 | –0.1850 | 0.1792 | 2.59 |
| 5c | –0.2731 | –0.0940 | –0.1835 | 0.1791 | 2.55 |
| 5d | –0.2712 | –0.0901 | –0.1807 | 0.1811 | 2.45 |
| 5e | –0.2689 | –0.0932 | –0.1811 | 0.1757 | 2.53 |
| 5f | –0.2623 | –0.0971 | –0.1797 | 0.1652 | 2.66 |
Global indices, electronic chemical potentials, μ, global hardness, η, and electrophilicity indices, ω, for compounds 5a–f, calculated in toluene.
| Reactant | HOMO | LUMO | μ(ua) | η(ua) | ω(eV) |
| 5a | –0.2662 | –0.0890 | –0.1776 | 0.1771 | 2.42 |
| 5b | –0.2659 | –0.0864 | –0.1761 | 0.1795 | 2.35 |
| 5c | –0.2645 | –0.0851 | –0.1748 | 0.1794 | 2.31 |
| 5d | –0.2626 | –0.0811 | –0.1719 | 0.1814 | 2.21 |
| 5e | –0.2603 | –0.0842 | –0.1723 | 0.1761 | 2.29 |
| 5f | –0.2538 | –0.0882 | –0.1710 | 0.1656 | 2.40 |
Electrophilic Parr functions, Pk,+ and local electrophilicities, ωk, of C and C′ positions of reactants 5a–f. The results were obtained in the gas phase and in toluene.
| Reactant | Gas-phase | Toluene | ||
| Pk+ | ωk | Pk+ | ωk | |
| 5a | ||||
| C′ | 0.0494 | 0.1319 | 0.0354 | 0.0858 |
| C | 0.2894 | 0.7727 | 0.3189 | 0.7717 |
| 5b | ||||
| C′ | 0.0593 | 0.1537 | 0.0422 | 0.0992 |
| C | 0.2978 | 0.7713 | 0.3222 | 0.7520 |
| 5c | ||||
| C′ | 0.0610 | 0.1557 | 0.0423 | 0.0977 |
| C | 0.2987 | 0.7618 | 0.3240 | 0.7485 |
| 5d | ||||
| C′ | 0.0625 | 0.1532 | 0.0415 | 0.0917 |
| C | 0.3208 | 0.7861 | 0.3447 | 0.7618 |
| 5e | ||||
| C′ | 0.0620 | 0.1569 | 0.0441 | 0.1011 |
| C | 0.3150 | 0.7971 | 0.3333 | 0.7634 |
| 5f | ||||
| C′ | 0.0822 | 0.2188 | 0.0696 | 0.1672 |
| C | 0.2320 | 0.6172 | 0.2695 | 0.6469 |
The analysis of the tabulated values shows that the global electrophilicity indices ω range from 2.42 to 2.21 eV in toluene, and from 2.65 to 2.45 eV in vacuo; this result shows that all α-halogenoketones 5a-f are strong electrophiles [44]. Our results also show that the solvent's effect on the electrophilic character of the molecules is moderate, and that the ordering of the studied molecules according to their electrophilic power is the same in the gas phase and in toluene.
The local electrophilicity indices obtained using the electrophilic Parr function (Table 4) show that the carbon position of the carbonyl C is characterized by the highest value of the local electrophilicity indices. Indeed, in all cases,
5 Concluding remarks
In this work, we have theoretically and experimentally examined the reaction of a series of α-halogenoketones compounds with trialkyl phosphite. Global and local DFT-based reactivity descriptors of α-halogenoketones have been used to rationalize the experimental data.
Our calculations show that the experimental regioselectivities are correctly reproduced. Indeed, the local electrophilicity index predicts that the C position is more reactive than the C′ one in all systems. Our calculations also show that the new proposed Parr function correctly reproduces the relative reactivities of the C position. Consequently, we can conclude that the local electrophilicity, as defined by Domingo's group using the electrophilic Parr function, is suitable for the prediction of regioselectivity. Finally, we note that the solvent's effect on the reactivity indices is small, even though the electrophilic power decreases moderately.
6 Experimental
6.1 General procedures and materials
1H NMR and 13C NMR spectra were recorded on a PerkinElmer R12 (300 MHz for 1H NMR and 75 MHz for 13C NMR) spectrometer using CCl4 as the solvent. The chemical shifts (δ) are reported in parts per million (ppm) relative to (TMS) as an internal standard, and coupling constants J are given in Hertz. All the chemicals were obtained from Merck Chemicals and Aldrich, and were used without further purification. FT–IR (KBr) spectra were recorded on a Beckman Acculab 8 spectrophotometer.
6.2 General procedure for the synthesis of compounds 6a–6h
To a solution of the appropriate 3-chloro-2-oxoester (0.1 mol) in dry toluene (20 mL), 0.1 mol of trimethylphosphite was added at 70 °C over a period of 15 min. The mixture was heated at 110 °C for 90 min. After removal of the solvent in vacuo, the distillation of the residue gave the yellow oil 6.
6.3 Spectral data for compounds 6a–6h
6.3.1 Ethyl-2-[(dimethoxyphosphoryl)oxy]prop-2-enoate (6a)
Yellow oil, yield 80%; FTIR (KBr, cm−1): 1720 (CO), 1740 (CC), 1266 (PO); 1H NMR (CCl4, 300 MHz): δH (ppm) 3.77 (s, 3H, COOCH3); 3.78 (d, 3H, J = 11.03 Hz, POCH3); 5.6 (dd, 1H, J = 3 Hz, CH2C); 5,9 (dd, 1H, 2 Hz, CH2C); 13C NMR (CCl4, 75 MHz): δC (ppm) 13(COOCH2CH3), 54(POCH3), 60.5(COOCH2CH3), 109(H2CC), 143(H2CC), 160.7(COOC2H5); 31P NMR (H3PO4 85%) δ = –1.70.
6.3.2 Methyl-2-[(dimethoxyphosphoryl)oxy]but-2-enoate (6b)
Yellow oil, yield95%, FTIR (KBr, cm−1): 1720 (CO), 1740 (CC), 1272 (PO); 1H NMR (CCl4, 300 MHz): δH (ppm) 1.92(d, 3H, J = 7.28 Hz, CH3CHC); 3.80 (s, 3H, COOCH3); 4.09 et 4.12 (2d, 6H, J = 11.03 Hz, POCH3); 6.20 (q, 1H, J = 7.28 Hz, CH3CHC); 13C NMR (CCl4, 75 MHz): δC (ppm) 14.6(CH3CHC), 52.2 (COOCH3), 55(POCH3), 120(CH3CHC), 144.8(CH3CHC), 162.7 (COOCH3)); 31P NMR (H3PO4 85%) δ = –3.21.
6.3.3 Methyl-2-[(dimethoxyphosphoryl)oxy]pent-2-enoate (6c)
Yellow oil, yield 85%, FTIR (KBr, cm−1): 1720 (CO), 1740 (CC), 1270 (PO); 1H NMR (CCl4, 300 MHz): δH (ppm) 1.1 (t, 3H, J = 7.80 Hz, CH3CH2CHC); 2.3 (dq, 2H, J = 7.80 Hz et 8.38 Hz, CH3CH2CHC), 3.80 (s, 3H, COOCH3); 4.08 et 4.12 (2d, 6H, J = 11.03 Hz, POCH3); 5.85 (t, 1H, J = 8.38, CH3CH2CHC); 13C NMR (CCl4, 75 MHz): δC (ppm) 11.1 (CH3CH2CHC), 17.9 (CH3CH2CHC), 50.8(COOCH3), 53.5(POCH3), 130.7(CH3CH2CHC), 135.7(CH3CH2CHC), 161.3(COOCH3); 31P NMR (H3PO4 85%) δ = –3.19.
6.3.4 Methyl-2-[(dimethoxyphosphoryl)oxy]-4-methylpent-2-enoate (6d)
Yellow oil, yield85%, FTIR (KBr, cm−1): 1735 (CO), 1740 (CC), 1265 (PO); 1H NMR (CCl4, 300 MHz): δH (ppm) 0.98 (d, 6H, J = 6.80 Hz, CH(CH3)2); 2,44 (m, 1H, CH(CH3)2); 3.79 (s, 3H, COOCH3); 4.04 et 4.11 (2d, 6H, J = 11.03 Hz, POCH3); 5.8 (d, 1H, J = 10.50 Hz, iPrCHC); 13C NMR (CCl4, 75 MHz): δC (ppm) 22(CH(CH3)2), 25.2(CH(CH3)2), 52.3(COOCH3), 55.1(POCH3), 133.1(iPrCHC), 144.8(iPrCHC), 162.7(COOCH3), 31P NMR (H3PO4 85%) δ = –3.22.
6.3.5 Methyl-2-[(dimethoxyphosphoryl)oxy]-4,4-dimethylpent-2-enoate (6e)
Yellow oil, yield80%, FTIR (KBr, cm−1): 1735 (CO), 1740 (CC), 1266 (PO); 1H NMR (CCl4, 300 MHz): δH (ppm) 1.20 (s, 9H, C(CH3)3); 3.79 (s, 3H, COOCH3); 4.04 et 4.11 (2d, 6H, J = 11.03 Hz, POCH3); 5.9 (d, 1H, JHP = 2.8, JPH = 0 Hz, tBuCH = C); 13C NMR (CCl4, 75 MHz): δC (ppm) 29.8(C(CH3)3), 33.2(C(CH3)3, 52.4(COOCH3), 55(POCH3), 134.7(tBuCHC), 144.8(tBuCHC), 162.6(COOCH3), 31P NMR (H3PO4 85%) δ = –3.20.
6.3.6 Methyl-2-[(dimethoxyphosphoryl)oxy]-3-phenylprop-2-enoate (6f)
Yellow oil, yield 80%, FTIR (KBr, cm−1):1735 (CO), 1740 (CC), 1265 (P–O); 1H NMR (CCl4, 300 MHz): δH (ppm) 3.77 (s, 3H,COOCH3); 3.79 et 3.79 (2d, 6H, J = 11.03 Hz, POCH3); 6.60(m, 1H, C6H5CHC); 7 (m, 5H, C6H5); 13C NMR (CCl4, 75 MHz): δC (ppm) 52.4(COOCH3), 55(POCH3), 118(C6H5CHC), 128.9(C6H5), 129.3(C6H5), 129.6(C6H5), 130.6(C6H5), 144.4(C6H5CHC), 162.5(COOCH3); 31P NMR: δ = –3.21.
6.3.7 Methyl-2-[(diethoxyphosphoryl)oxy]pent-2-enoate (6g)
Yellow oil, yield70%, FTIR (KBr, cm−1): 1735 (CO), 1740 (CC), 1260 (PO); 1H NMR (CCl4, 300 MHz): δH (ppm) 1.06 (t, 3H, J = Hz, CH3CH2CHC); 1.20 (t, 3H, J = Hz, POCH2CH3), 2.05 (dq, 2H, J = 7.0 Hz, CH3CH2CHC); 3.78 (s, 3H, COOCH3); 4.04 (m, 4H, POCH2CH3); 6.1 (t, 1H, J = 7.8 Hz, CH3CH2CHC); 13C NMR (CCl4, 75 MHz): δC (ppm) 13.2 (CH3CH2CHC), 16 (POCH2CH3), 19.2 (CH3CH2CHC), 52.3 (COOCH3), 64.7 (POCH2CH3), 131.3 (C2H5CHC), 144.8 (C2H5CHC), 162.1 (COOCH3);31P NMR (H3PO4 85%) δ = –5.24.
6.3.8 Methyl-2-[(diethoxyphosphoryl)oxy]-4,4-dimethylpent-2-enoate(6h)
Yellow oil, yield70%, FTIR (KBr, cm−1): 1720 (CO), 1745 (CC), 1274 (PO); 1H NMR (CCl4, 300 MHz): δH (ppm) 1.2 (s, 9H, C(CH3)3); 1.28 (t,6H, J = Hz, POCH2CH3); 3.80 (s, 3H, COOCH3); 4.04 (m, 4H, POCH2CH3); 5.9 (m, 1H); 13C NMR (CCl4, 75 MHz): δC (ppm)16 (POCH2CH3), 29.8 (C(CH3)3), 33.1 (C(CH3)3), 52.3 (COOCH3), 64.7 (POCH2CH3), 134.7 (tBuCHC), 144.8 (tBuCHC), 162.6 (COOCH3); 31P NMR (H3PO4 85%) δ = –5.24.
Acknowledgements
We thank Professor J.-C. Combret (Université de Rouen, France) for the leave during which this work was conducted; we also thank Dr. C. Malhiac for the helpful discussions concerning this synthesis. This work was supported by research funds provided by the Ministry of the Higher Education and Scientific Research of the Algerian Government (CNEPRU E02020130167, MESRS-Algeria).