

1 Introduction
The design of fine chemicals relies on the synthesis of scaffolds enabling wide structural and functional diversity, therefore offering multiple options for targeting specific properties. Looking for easily accessible and novel scaffolds with high level of functionality is therefore an interesting topic, and the Morita–Baylis–Hillman (MBH) reaction is an attractive atom-economical strategy with wide flexibility in scope and conditions [1–23]. In particular, cycloalkenone–MBH adducts shown are useful intermediates toward natural products and analogues (Fig. 1) [22,24–40].
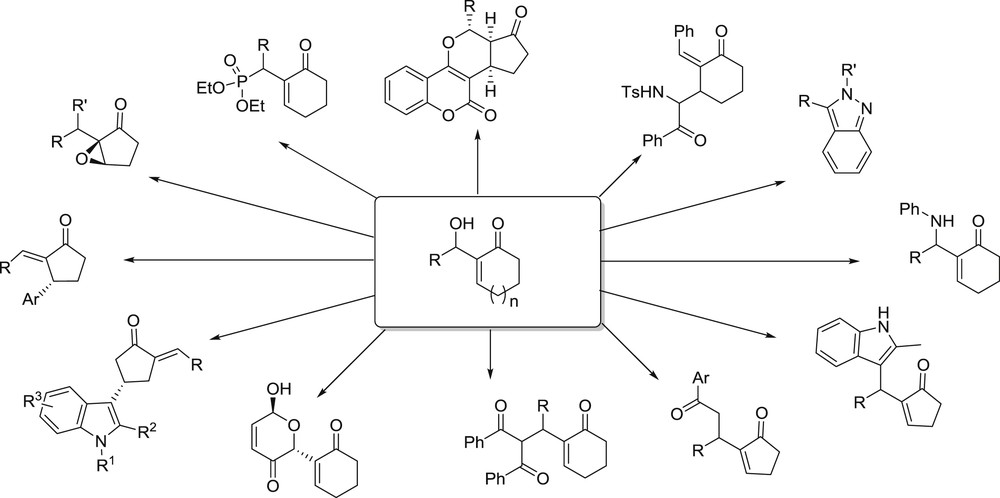
Building blocks and products obtained from cycloalkenone–MBH adducts.
However, none of the reported studies dealing with MBH reactions of cycloalkenones have ever concerned hydroxymethylfurfural (HMF), nor its glucosylated analogue glucosyloxymethylfurfural (GMF), although those substrates are interesting biobased furanic aldehydes able to provide a range of novel fine chemicals including carbohydrate-containing architectures [5,6,41–51]. Furthermore, 2-cycloalkenones can also be obtained via biobased approaches by catalytic [52] or biocatalytic [53] dehydrogenation of cyclopentanone, itself available from furfural [54–62], and several substituted cyclopentenones can be prepared from furfuryl derivatives [51,63–66].
Actually, cyclic enones exhibit specific reactivity in MBH reactions, being less reactive as compared with acyclic enones such as methyl vinyl ketone. Several sets of conditions (promoters/solvents) have been reported for promoting the reaction with a particular focus on cycloalkenones. Some rely on the use of organic solvents such as dichloromethane, tetrahydrofuran (THF), or toluene, and various promoters such as combinations of strong Lewis acids such as titanium tetrachloride and diethylaluminum iodide (TiCl4, Et2AlI) and bases such as sodium methylate (NaOMe), phosphines, tertiary amines, or aminophosphines [67–74]. Using anhydrous THF as solvent, the cooperative action of trialkylphosphines with hydrogen-bond donors such as phenols was also reported [75]. Examples of MBH reactions of cycloalkenones in alcoholic solvents were described, promoted by 1,8-diazabicyclo[5.4.0]undec-7-ene [76], Ba(OH)2 [77], or nanocrystalline MgO [78] in MeOH. For aqueous media, examples include N-methyl-2-pyrrolidone [21], PBu3 [23], imidazole [15–18,22], and 6,7-dihydro-7-hydroxy-5H-pyrrolo[1,2-a]imidazole (DPI) [14,19] as promoters. One additional reported option is the use of proteins (lysozyme C or myoglobin), which promoted the reaction in a phosphate buffer. None of these studies ever included HMF in their structural scope, and actually only few [16,22,74,78] included furfural. Being significantly less available, GMF was logically also totally absent in these earlier reports.
We have thus investigated the MBH reaction of HMF and its glucosylated analogue GMF, specifically applied to 2-cyclopentenone and 2-cyclohexenone for preparing new fully or partially biobased furyl-cycloalkyl-methanols in a very short and clean sequence (Fig. 2).
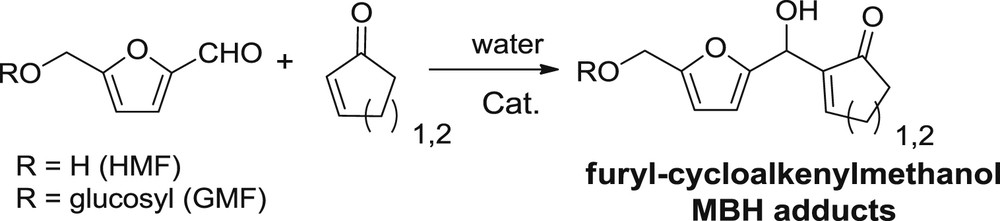
Novel furyl-cycloalkenyl methanol scaffolds based on HMF and GMF by an aqueous MBH reaction.
2 Experimental section
2.1 General methods
The reagents were bought from Aldrich and were used directly without purification. HMF was purchased from Carbosynth, GMF was prepared as previously reported [44,45] from isomaltulose, which was a gift from Cargill. DPI was prepared by refluxing imidazole and acrolein in dioxane in the presence of a catalytic amount of acetic acid as previously reported [79]. The reactions were monitored by thin layer chromatography, on Silica Gel 60 F254 (Merck), and detection was carried out with ultraviolet light (254 nm) and 1% potassium permanganate solution in water. Silica gel (Kieselgel 60, 70–230 mesh ASTM, Merck) was used for column chromatography. The 1H NMR (300 or 400 MHz) and 13C NMR (75 or 100 MHz) spectra were recorded with Bruker ALS300, DRX300, and DRX400 spectrometers. Chemical shifts are given in ppm. The coupling constants are expressed in Hertz and the splitting pattern abbreviations are as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; and br, broad. High-resolution mass spectra were obtained using an electrospray technique in positive mode, with a Thermo-Finnigan MAT 95 XL spectrometer.
2.2 General procedures for MBH reactions
Method A: a solution of DPI (74 mg, 0.6 mmol) and HMF (126 mg, 1.0 mmol) or furfural (96 mg, 1.0 mmol) in water (1.0 mL) was treated with 2-cyclopenten-1-one (164 mg, 2.0 mmol) or 2-cyclohexen-1-one (123 mg, 2.0 mmol) at room temperature for 30 h, extracted with ethyl acetate (3 × 20 mL), dried over Na2SO4, then concentrated under vacuum, purified by silica gel column chromatography to give the desired product. Pure ethyl acetate was used as eluent for 1 and 4; ethyl acetate/pentane: 4:6 for 3 and 6.
Method B: a solution of DPI (74 mg, 0.6 mmol) and GMF (288 mg, 1.0 mmol) in water (1.0 mL) was treated with 2-cyclopenten-1-one (164 mg, 2.0 mmol) or 2-cyclohexen-1-one (123 mg, 2.0 mmol) at room temperature for 30 h, water was removed by evaporation and the crude product was purified by silica gel column chromatography Dichloromethane/methanol (5:1) was used as eluent for 2 and 5.
Recycling of DPI [79]: for HMF products, the reaction mixture was quenched with 0.1 M HCl (60 mL), and extracted with EtOAc. The combined organic phase was dried over Na2SO4 and evaporated for further chromatography to provide product 1. After addition of 6 M NaOH to the aqueous phase up to pH >12, DPI could be extracted with EtOAc. The combined organic phase was dried, filtered, and evaporated, leading to DPI as a solid, which was possible to reuse as such. In the case of the GMF products, which are water-soluble, the reaction mixture was extracted with EtOAc, the organic phase was then dried and evaporated, yielding DPI. The aqueous phase was evaporated and the crude product was purified by silica gel chromatography as described previously in method B.
2.3 Characterization of new products
2.3.1 2-[hydroxy-(5-hydroxymethyl-furan-2-yl)methyl]cyclopent-2-en-1-one (1)
1H NMR (400 MHz, MeOD) δ 7.83 (td, J = 2.8, 1.2 Hz, 1H, H3’), 6.25 (d, J = 3.2 Hz, 1H, H4), 6.19 (d, J = 3.2 Hz, 1H, H3), 5.45 (d, J = 1.6 Hz, 1H, CH–OH), 4.49 (s, 2H, H6), 2.78–2.60 (m, 2H, H4’), 2.53–2.32 (m, 2H, H5’). 13C NMR (100 MHz, MeOD) δ 210.2 (C1′), 162.8 (C3′), 155.8 (C5), 155.5 (C2), 146.9 (C2′), 109.1 (C4), 109.0 (C3), 63.0 (CH–OH), 57.4 (C6), 36.0 (C5′), 27.7 (C4′). MS m/z (ESI) calculated for C11H12NaO4: [M + Na]+ 231.0628; found 231.0630.
2.3.2 2-[hydroxy-(5-(α-d-glucopyranosyloxymethyl)-furan-2-yl)methyl]cyclopent-2-en-1-one (2)
1H NMR (300 MHz, MeOD) δ 8.14–7.72 (m, 1H H3″), 6.43 (d, J = 3.2 Hz, 1H, H4), 6.26 (d, J = 3.2 Hz, 1H, H3), 5.51 (s, 1H, CH–OH), 4.97 (dd, J = 5.4, 3.7 Hz, 1H, H1′), 4.70 (dd, J = 13.0, 2.6 Hz, 1H, H6a), 4.60 (dd, J = 12.9, 1.9 Hz, 1H, H6b), 3.86 (ddd, J = 11.8, 4.8, 2.3 Hz, 1H, H6′a), 3.81–3.60 (m, 3H, H6′b, H3′, H5′), 3.49 (ddd, J = 9.8, 3.8, 1.5 Hz, 1H, H2′), 3.37 (d, J = 3.5 Hz, 1H, H4′), 2.77 (dt, J = 4.6, 2.3 Hz, 2H, H4″), 2.53 (dt, J = 6.3, 1.6 Hz, 2H, H5″). 13C NMR (75 MHz, MeOD) δ 210.33 (CO), 163.11 and 163.09 (C2, R&S), 156.01 and 155.98 (C5, R&S), 152.42 and 152.35 (C3″, R&S), 146.66 and 146.62 (C2”, R&S), 111.46 and 111.40 (C4, R&S), 108.94 and 108.88 (C3, R&S), 99.07 and 98.86 (C1′, R&S), 74.84 (C3′), 73.61 (C5′), 73.22 (C2′), 71.47 (C4′), 62.96 (CH–OH, R&S), 62.58 and 62.61 (C6′), 62.19 and 62.05(C6, R&S), 36.05 (C5″), 27.81 (C4″). MS m/z (ESI) calculated for C17H22NaO9: [M + Na]+ 393.1156; found 393.1171.
2.3.3 2-[hydroxy-(5-hydroxymethyl-furan-2-yl)methyl]cyclohex-2-en-1-one (4)
1H NMR (300 MHz, CDCl3): 6.97 (t, 1H, J = 3.9 Hz, H3′), 6.03 (d, 1H, J = 3.0 Hz, H4), 5.98 (d, 1H, J = 3.0 Hz, H3), 5.44 (s, 1H, CH–OH), 4.35 (s, 2H, H6), 2.25–2.5.0 (m, 4H, H4′&H6′),1.75–2.00 (m, 2H, H5′); 13C NMR (75 MHz, CDCl3): 199.6 (C1′), 154.6 (C5), 153.8 (C2), 148.2 (C3′), 138.3 (C2′), 108.3 (C4), 107.6 (C3), 65.1 (CH–OH), 56.9 (C6), 38.3 (C6′), 25.7 (C4′), 22.5 (C5′); MS m/z (ESI) calculated for C12H14NaO4: [M + Na]+ 245.0784; found 245.0790.
2.3.4 2-[hydroxy-(5-(α-d-glucopyranosyloxymethyl)-furan-2-yl)methyl]cyclohex-2-en-1-one (5)
1H NMR (400 MHz, MeOD) δ 7.22–7.17 (m, 1H, H3″), 6.30 (d, J = 3.2 Hz, 1H, H4), 6.10 (d, J = 3.2, 1H, H3), 5.59 (s, 1H, CH–OH), 4.86 (t, J = 4.2 Hz, 1H, H1′), 4.65 (dd, J = 12.8, 2.9 Hz, 1H, H6a), 4.55 (dd, J = 12.8, 2.0 Hz, 1H, H6b), 3.77 (dt, J = 11.7, 2.6 Hz, 1H, H6′a), 3.70–3.53 (m, 3H, H6′b, H3′, H5′), 3.36 (ddd, J = 9.7, 3.8, 1.8 Hz, 1H, H2′), 3.29 (dt, J = 3.0, 1.4 Hz, 1H, H4′), 2.57–2.50 (m, 2H, H4″), 2.50–2.44 (m, 2H, H6″). 2.00 (tt, J = 6.7, 5.1 Hz, 2H, H5″). 13C NMR (100 MHz, MeOD) δ 200.3 (CO), 157.3 and 157.2 (C2, R&S), 152.2 and 152.2 (C5, R&S), 149.3 and 149.2 (C3″, R&S), 140.2 and 140.1 (C2″, R&S), 111.5 and 111.4 (C4, R&S), 108.6 and 108.6 (C3, R&S), 99.2 and 99.0 (C1′, R&S), 75.0 (C3′), 73.8 (C5′), 73.4 (C2′), 71.7 (C4′), 64.30 and 64.34 (CH–OH, R&S), 62.59 and 62.57 (C6′, R&S), 62.17 and 62.09 (C6, R&S), 39.2 (C6″), 26.7 (C4″), 23.8 (C5″). MS m/z (ESI) calculated for C18H24NaO9: [M + Na]+ 407.1313; found 407.1311.
3 Results and discussion
On the basis of the existing literature dealing with MBH reactions of cycloalkenones [14–19,21–23,67–78], we first confirmed in a rapid screening that tertiary amines and phosphines in substoichiometric quantities were not appropriate for the reaction of cyclopentenone and HMF in pure water. Promoters such as 1,8-diazabicyclo[5.4.0]undec-7-ene, 3-hydroxyquinuclidine, DABCO, Et3N, PPh3, and P(m-C6H4–SO3Na)3 led to no reaction whereas 4-dimethylaminopyridine and pyridine gave very slow reactions. We then focused on two promoters, imidazole and DPI for our study. Imidazole as BH promoter in 1:1 THF/water mixture was reported independently by Gatri and El Gaïed [15] and Luo et al. [16]. Limiting the protonation of imidazole by addition of base was advantageous [17], and Schwartz et al. [18] demonstrated the interest of adding surfactants such as sodium dodecyl sulfate, cetyl trimethylammonium bromide, or octyl glucoside. In the case of HMF in pure water, we found that imidazole allowed the reaction to proceed, leading to 2-[hydroxy-(5-hydroxymethylfuran-2-yl)methyl]cyclopent-2-en-1-one (1; Fig. 3) after 15 h at room temperature (60% after 30 h) using 2 equiv of cyclopentenone (Table 1, entries 1–4). Although yields were moderate, it was possible to fully identify and characterize the newly formed adduct using 1H, 13C, and heteronuclear multiple bond correlation (HMBC) NMR analyses (see ESI). 1H NMR shows clearly the disappearance of the singlet peak of the aldehyde group of HMF at δ 9.53 ppm to the benefit of the newly formed CHOH at δ 5.50 ppm. The HMBC shows interactions of this CHOH proton with the three carbon atoms of the enone system of cyclopent-2-enone group at δ 146.9 ppm (CHC–CO), 162.7 ppm (CHC–CO), and 210.2 ppm (CO), respectively. The corresponding carbon signal is found at δ 62.9 ppm confirming the structure unambiguously. High-resolution mass spectra also showed the expected molar mass of the adduct.
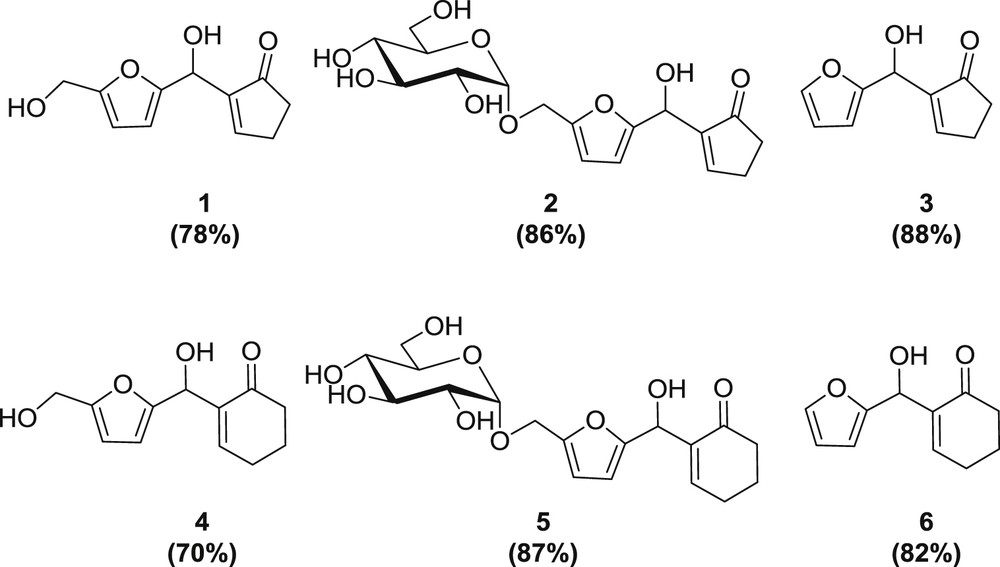
Optimized yields for HMF and extension to furfural and GMF.
The MBH reaction of HMF with cyclopent-2-enone.a
| Entry | Catalyst (equiv) | Cyclopentenone (equiv) | Concentration of HMF (M) | Time (h) | (1) Yieldb (%) |
| 1 | Imidazole (0.6) | 2.0 | 1 | 15 | 50 |
| 2 | Imidazole (0.6) | 2.0 | 2 | 15 | 51 |
| 3 | Imidazole (0.6) | 2.0 | 1 | 30 | 60 |
| 4 | Imidazole (0.6) | 2.0 | 2 | 30 | 57 |
| 5 | DPI (0.6) | 2.0 | 1 | 15 | 71 |
| 6 | DPI (0.6) | 2.0 | 1 | 30 | 78 |
| 7 | DPI (0.8) | 2.0 | 1 | 30 | 79 |
| 8 | DPI (1.0) | 2.0 | 1 | 30 | 77 |
| 9 | DPI (0.4) | 2.0 | 1 | 30 | 75 |
| 10 | DPI (0.2) | 2.0 | 1 | 30 | 67 |
| 11 | DPI (0.1) | 2.0 | 1 | 30 | 48 |
| 12 | DPI (0.05) | 2.0 | 1 | 30 | 26 |
| 13 | DPI (0.6) | 1.5 | 1 | 30 | 75 |
| 14 | DPI (0.6) | 1.2 | 1 | 30 | 70 |
| 15 | DPI (0.6) | 1.0 | 1 | 30 | 61 |
| 16 | DPI (0.6) | 0.8 | 1 | 30 | 70c |
| 17 | DPI (0.6) | 0.5 | 1 | 30 | 64c |
| 18 | DPI (0.4) | 2.0 | 1 | 30 | 75 |
| 19 | DPI (0.4) | 2.0 | 2 | 30 | 79 |
| 20 | DPI (0.4) | 2.0 | 4 | 30 | 78 |
| 21 | DPI (0.6) | 2.0 | 1 | 7.5 | 56 |
| 22 | DPI (0.6) | 2.0 | 2 | 7.5 | 62 |
| 23 | DPI (0.6) | 2.0 | 4 | 7.5 | 58 |
| 24 | DPI (0.6) | 2.0 | 2 | 15 | 78 |
| 25 | DPI (0.6) | 2.0 | 4 | 15 | 75 |
| 26 | DPI (0.6) | 2.0 | 2 | 30 | 78 |
| 27 | DPI (0.6) | 2.0 | 4 | 30 | 75 |
| 28 | DPI (0.6) | 2.0 | 1 | 30 | 44d |
| 29 | DPI (0.6) | 2.0 | 1 | 30 | 1e |
| 30 | DPI (0.6) | 2.0 | 1 | 30 | 13f |
a Standard conditions: HMF (1 mmol), cyclopent-2-enone (2 mmol), catalyst (60%), H2O (1 mL), r.t; otherwise noted.
b Yield of isolated product.
c Yields were calculated based on cyclopentenone.
d Solvent H2O/EtOH = 1:1 total 1 mL, 50% HMF was recovered.
e EtOH was used as solvent, 89% HMF was recovered.
f No solvent condition, 86% HMF was recovered.
Turning to DPI led to even better results. Gomes et al. reported DPI-promoted MBH reactions of aromatic aldehydes with 2-cycloalkenone in water in the presence of a surfactant such as sodium dodecyl sulfate, and suggested that the conjugated bicyclic structure and the hydrogen-bond donor group of DPI contribute to its higher efficiency as compared with imidazole, and that the hydroxyl group may stabilize the betaine intermediate resulting from the initial Michael addition of the nucleophilic catalyst to the activated alkene [14]. The same group later reported purely aqueous MBH reaction of cyclic enones with unprotected ketoamides (isatins) to afford 3-substituted 3-hydroxy-2-oxindoles [19]. Apart from MBH, DPI [80] was only reported in a couple of other reactions [79,81]. Applied for the first time to HMF, we found these conditions to be very efficient, providing the desired MBH adduct 1 in very good yields. The DPI-promoted MBH reaction of cyclopentenone with HMF gave 71% yield after 15 h and 78% yield after 30 h (Table 1, entries 5–6). Increasing the DPI quantity up to 1.0 equiv did not improve the yield (entries 7–8). Interestingly, lower amounts, as low as 0.05 equiv are able to trigger the reaction, although in lower yields after the same 30 h reaction time (entries 9–12). The influence of the stoichiometry for HMF and cyclopentenone was then studied (entries 13–17), showing that it is possible to reduce the excess of Michael acceptor to 1.5 equiv without significant loss of performance in the reaction (entry 13). We verified that the promoter DPI and excess cycloalkenone could be recovered and reused, using a similar protocol as what was reported for the use of DPI in asymmetric Steglich rearrangement [79], based on acidification of the reaction mixture using 0.1 M HCl and extraction of excess enone and BH adduct with EtOAc. Further rebasification of the aqueous phase allows us to recover DPI by extraction using EtOAc. Decreasing the amount of solvent from 1 mL to 0.5 and 2.5 mL results in increase in HMF concentrations of 2 and 4 M, which was found to exhibit little influence with similar yields and rates for whatever the reaction time and DPI amount (entries 18–27). This can be ascribed to the specific kinetics of the MBH reaction in which one of the rate-determining steps is the proton shift in the aldol-intermediate [12], which is an intramolecular process, therefore not or less depending on concentration. Finally, we confirmed that pure water was the best choice, as water/EtOH mixture in a ratio of 1:1 led to a slower reaction (entry 28), whereas pure ethanol or no-solvent conditions, although appropriate for acrylates or acrylamide in earlier studies [6,82], were here unsuccessful with cycloalkenones (entries 29–30).
The optimized conditions, in pure water, room temperature, 30 h, 0.6 equiv of DPI, and 2 equiv of cyclopentenone, were finally also applied to the two other aldehydes, furfural and GMF. GMF is available in one step from the commercially available disaccharide isomaltulose (a glucosyl fructose isomer of sucrose) [44,45], and furfural is obtained by dehydration of pentose-containing resources. As either a more or a less polar analogue of HMF, the scope of functionalization in the MBH adducts is thus extended. From cyclopentenone, the corresponding furyl-cyclopentenyl-methanol scaffolds 2 and 3 [22] were obtained in high yields. Good results were also obtained using cyclohexenone, which led to adducts 4, 5, and 6 [22] (Fig. 3). We confirmed again in these cases that the promoter DPI and excess cycloalkenone could be fully recovered. For the GMF products, being water soluble, the recovery of DPI and excess enones is even simpler being possible by extraction with EtOAc from the reaction mixture. For GMF adduct 2, the newly formed CHOH signal appears at δ 5.40 ppm as a singlet in the 1H NMR and δ 63.0 ppm in the 13C NMR. GMF being a chiral molecule, two diastereoisomers are produced in a ca. 1:1 mixture. The signals of the two epimers differ slightly and show equivalent height for the carbon atoms close to the chiral center. This is notably seen for carbon C2 at δ 163.09 and δ 163.11 ppm, carbon C3 at δ 108.88 and δ 108.94 ppm, carbon C2″ at δ 146.62 and δ 146.66 ppm, among others (see ESI). The same observations were made for the C-6 adduct 5, for which clear patterns for the newly created CHOH are seen at 5.586 ppm, and slightly different shifts for both diastereoisomers, notably around the furan ring. The HMBC spectrum shows also the clear relationship between the new CHOH and the furan and cycloalkenyl systems (see ESI).
4 Conclusions
New furyl-cycloalkenyl-methanol scaffolds have been prepared based on the HMF and GMF structures used for the first time in such strategies, leading to adducts with enriched functional patterns and with significant level of renewable carbon content in the targets. Using the efficient MBH reaction, the target compounds can be obtained in one-step reaction, in pure water at room temperature. The present study confirms first that the MBH route is an interesting option when straightforwardness and mildness and cleanliness of reaction conditions are specific requirements, and second that HMF, which is more readily available nowadays, can be considered as a valuable building block in strategies toward fine chemicals.
Acknowledgments
The authors thank the Ministère de l’Enseignement supérieur et de la Recherche (MESR) and CNRS for their financial support, as well as the China Scholarship Council (UT-INSA-CSC call) for the fellowship to L.W. and J.N.T.