

1 Introduction
Aldimines RCH=NR' are Schiff bases well known in coordination chemistry as mixed-donor ligands [1,2]. However, their chemistry found a much larger use for regioselective and stereoselective reactions, for total synthesis [3], and in preparations of pharmaceutically valuable products [4]. Besides a purely synthetic aspect, redox reactions of aldimines have also been studied, especially during the “golden age” of organic electrochemistry, with polarography being then the main method [5–9].
On the other hand, furan and especially thiophene units are present in many drugs [10]; but—with its evidently important role in their bioactivity–the latter evokes a serious concern about the toxicity of reactive metabolites arising from the oxidation of thiophene-containing biomolecules for humans and animals [11]. Oxidation of thiophenes with CYP isoenzymes family—CYP450 (cytochrome 450), CYP2C9, and others—mainly proceeds via S-oxidation and epoxidation, both processes activating the α-position of the heterocycle and usually leading to an α-hydroxythiophene or a thiolactone. Blocking this site with a neutral, oxidation-stable group might improve the oxidative stability of such compounds and decrease their metabolic toxicity. This idea is supported by the fact that such thiophene-containing drugs as duloxetine, rivaroxaban, and others, in which both α-positions of the thiophene unit are substituted, have no toxicity [12], whereas those with one non-substituted α-position (Methapyrilene, Suprofen, Thienilic acid, OSI-920, and others) are toxic [13–15].
Studying thermochemistry and redox properties using DFT (density functional theory) calculations [16] proved efficient providing meaningful leads to bioactivity of thiophene drugs. Redox properties and biological activity of substrates are often correlated. On the other hand, the introduction of a silyl or a germyl substituent might modulate their redox behavior. Thus, we undertook the present study aiming to prepare several new aldimine derivatives of furan and thiophene (1a–e and 2a–e) with silyl and germyl substituents. Their bioactivity will then be considered along with their electrochemical properties and supported by DFT modeling.
2 Experimental
Electrochemical measurements were carried out using a PAR 2373 potentiostat operating under PAR PowerSuite software, using a 5-ml glass electrochemical cell in the three-electrode mode. A glassy carbon (GC) disk (2 mm) sealed in a Pyrex tube was used as the working electrode; a 3 × 50 mm GC rod served as the counter electrode. The working electrode was polished consecutively with Struers 2000 and 4000 paper before each run. All potentials, checked using an Fc+/Fc reversible couple (E0 = 0.31 V vs SCE (saturated calomel electrode) [17]), are given against SCE.
DFT calculations (geometry optimization and frequency analysis) were performed using Gaussian 03W (release B.01) program [18] at the B3LYP/Lanl2DZ//HF/6-31G level. Solvation in CH3CN was accounted for using the Tomasi PCM method [19]. All stationary points were found to be stable global minima as attested by the absence of negative vibration frequencies in harmonic frequency analysis. A scaling factor of 0.98 was applied to B3LYP/Lanl2DZ-calculated IR frequencies [20].
1H and 13C NMR spectra were recorded on a Varian-300 spectrometer (300 and 75 MHz, respectively) in CDCl3, with TMS as the internal standard for 1H and 29Si nuclei and using the residual solvent proton signal (77.05 ppm) for 13C nuclei. Elemental analyses were performed on a CARLO ERBA EA-1108 elemental analyzer. Thin-layer chromatography (TLC) was carried out on Merck silica gel 60 F254 plates. The mass-spectra were recorded under electron impact conditions on a GC-MS Agilent Technologies 7890 GC system with 5975C EI/CI MSD (70 eV) and a capillary column HP-5.
For X-ray analysis, diffraction data were collected at −100 °C on a Bruker-Nonius Kappa CCD diffractometer using graphite monochromatic Mo Kα radiation (λ = 0.71073 Å). The crystal structures of 2b and 2c were solved by direct methods and refined by full-matrix least squares. All non-hydrogen atoms were refined in anisotropic approximation. For further details, see crystallographic data for 2b and 2c deposited at the Cambridge Crystallographic Data Centre as Supplementary Publication Numbers CCDC 1870490 (for 2b) and CCDC 1870491 (for 2c). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK.
For the following syntheses, the hetaryl-substituted 2-carbaldehydes have been prepared according to the Dubac procedure [21] treating furan- and thiophenecarbaldehyde with lithium morpholide (aldehyde protection) and s-BuLi and then with the corresponding Alk3MCl (M = Si, Ge).
2-[2-Furylmethyleneamino]-phenol (1a) [22]. A mixture of furan-2-carbaldehyde (0.25 g, 2.3 mmol), 2-aminophenol (0.22 g, 2.3 mmol), and benzene (2 mL) was refluxed for 4 h. The reaction was monitored by TLC using petroleum ether (PE)/Et2O, 10:0.1. After cooling down, the solvent was evaporated and the residue was dissolved in diethyl ether and dried over MgSO4. The obtained crude product was recrystallized from hexane affording 0.32 g (74%) of pure 1a as a brown solid. M.p. 62–63 °C (lit. m.p. 68 °C [23]; melting with decomposition at 160–175 °C was reported earlier [24]).
2-[(5-Trimethylsilyl-2-furyl)methyleneamino]-phenol (1b). 5-Trimethylsilyl-furan-2-carbaldehyde (0.39 g, 2.3 mmol) was added to a suspension of 2-aminophenol (0.22 g, 2.3 mmol) in ethanol (10 mL) and the resulting solution was stirred for 14 h at room temperature. The reaction was monitored by TLC (PE/Et2O, 10:0.1). After evaporation of ethanol, the residue was dissolved in diethyl ether (ca. 10 mL) and dried over MgSO4. After evaporation of ether, the crude product was recrystallized from hexane providing 0.19 g (71%) of pure 1b as a yellow solid. M.p. 91–92 °C.
1H NMR spectrum (300 MHz, CDCl3), δ, ppm (J, Hz): 0.31 (s, 9H, Si–CH3); 6.78–6.82 (m, 1H); 6.87–6.94 (m, 2H); 7.02–7.13 (m, 2H); 7.16–7.17 (d, 1H); 8.78 (s, 1H); 0.95 (s, 1H, OH). 13C NMR spectrum (75 MHz, CDCl3), δ, ppm: 13C NMR (75 MHz, CDCl3), δ, ppm: −1.77; 115.08; 115.67; 115.73; 119.94; 121.71; 128.67; 135.77; 145.77; 152.34; 156.12; 165.58. 29Si NMR (80 MHz, CDCl3), δ, ppm: −9.55. MS (EI, 70 eV), m/z (Irel, %): 259 [M]+ (58); 231 (100); 216 (24); 186 (21); 150 (18); 73 (34); 28 (17). Anal. Calcd (C14H17NO2Si), %: C 64.82; H 6.60; N 5.40. Found, %: C 64.70; H 6.60; N 5.33.
2-[(5-Trimethylgermyl-2-furyl)methyleneamino]-phenol (1c) was prepared similarly to 1b, reacting 5-trimethylgermyl-furan-2-carbaldehyde (0.49 g, 2.3 mmol) with a suspension of 2-aminophenol (0.22 g, 2.3 mmol) in ethanol (10 mL). After stirring the mixture at room temperature for 4.5 h, the solvent was evaporated and the residue dissolved in diethyl ether to be dried on MgSO4. The crude product obtained after evaporation of ether was recrystallized from hexane affording 0.15 g (66%) of pure 1c as a light yellow solid. M.p. 94–96 °C.
1H NMR (300 MHz, CDCl3), δ, ppm (J, Hz): 0.48 (s, 9H, Ge-CH3); 6.67–6.68 (d, 1H, J=3.3 Hz, H-3); 6.86–6.90 (m, 1H), 6.98–7.02 (m, 2H); 7.14–7.23 (m, 2H); 7.33 (ps, 1H, C–OH); 8.51 (s, 1H). 13C NMR (75 MHz, CDCl3), δ, ppm: −1.88; 114.97; 115.56; 116.09; 119.97; 120.25; 128.59; 135.81; 144.91; 152.34; 155.96; 165.60. MS (EI, 70 eV), m/z ( Irel, %): 305 [M]+ (23); 277 (31); 186 (100); 158 (22); 119 (14); 28 (55). Anal. Calcd (C14H17GeNO2), %: C 55.32; H 5.64; N 4.61. Found, %: C 55.54; H 5.66; N 4.47.
2-[(5-Triethylsilyl-2-furyl)methyleneamino]-phenol (1d) was prepared similarly to 1b from 5-triethylsilylfuran-2-carbaldehyde (0.43 g, 2.0 mmol), with stirring for 4.5 h. The recrystallization of the crude product from hexane gave pure 1d as a brown solid (0.42 g, 69%). M.p. 49–51 °C.
1H NMR (400 MHz, CDCl3, δ, ppm): 0.80–0.89 (m, 6H, Si–CH2), 1.00–1.01 (m, 9H, Si–CH2CH3), 6.86–6.91 (m, 1H), 6.98–7.0 (m, 1H), 7.14–7.18 (m, 2H), 7.65–7.28 (m, 2H), 7.55–7.56 (d, 2H), 8.79 (s, 1H). 13C NMR (75 MHz, CDCl3, δ, ppm): 4.20; 7.26; 114.96; 115.68; 119.99; 128.65; 133.24; 135.21; 135.28; 144.23; 147.29; 149.29; 152.24. 29Si NMR (80 MHz, CDCl3, δ, ppm): 0.98. MS, m/z (I, %): 317 (100), 288 (45), 260 (74), 232 (45), 170 (69), 116 (36), 28(87). Anal. Calcd (C17H23NO2Si), %: C 67.73; H 7.69; N 4.64. Found, %: C 67.46; H 7.69; N 4.57.
2-[(5-Triethylgermyl-2-furyl)methyleneamino]phenol (1e) was synthesized similarly to 1b, using 5-triethylgermylfuran-2-carbaldehyde (0.59 g, 2.3 mmol) and a prolonged (12 h) stirring. Pure 1e crystallized from hexane as a yellow solid (0.19 g, 66%). M.p. 64–65 °C.
1H NMR (400 MHz, CDCl3, δ, ppm): 1.06–1.16 (m, 15H, Si–CH2CH3); 6.83–6.93 (m, 3H); 7.11–7.17 (m, 1H); 7.21–7.23 (m, 1H); 7.38–7.41 (m, 1H); 7.84 (s, 1H); 8.69 (s, 1H). 13C NMR (75 MHz, CDCl3, δ, ppm): 4.44; 8.84; 114.88; 115.45; 115.72; 119.94; 121.41; 128.54; 135.71; 135.73; 144.80; 152.36; 156.16. MS, m/z (I, %): 347 (19), 318 (37), 290 (24), 260 (19), 186 (100), 131 (16), 28(89). Anal. Calcd (C17H23GeNO2), %: C 59.01; H 6.70; N 4.04. Found, %: C 59.13; H 6.71; N 3.97.
2-[2-Thienylmethyleneamino]-phenol (2a) was prepared as described for 1a, using thiophene-2-carbaldehyde (0.26 g, 2.3 mmol). The reaction mixture was refluxed for 4 h. The obtained crude product was recrystallized from hexane affording 0.38 g (55%) of pure 2a as a brown solid. M.p. 78–79 °C (81 °C [25]).
2-[(5-Trimethylsilyl-2-thienyl)methyleneamino]-phenol (2b) was prepared as described above for 1b, using 5-trimethylsilyl-thiophene-2-carbaldehyde (0.42 g, 2.3 mmol). The reaction mixture was stirred in ethanol at room temperature for 10 h. Recrystallization from hexane afforded 0.18 g (69%) of pure 2b as a light yellow solid. M.p. 89–91 °C.
1H NMR spectrum (300 MHz, CDCl3), δ, ppm (J, Hz): 0.37 (s, 9H, Si–CH3); 6.87–6.91 (m, 1H); 6.99–7.01 (m, 2H); 7.16–7.28 (m, 2H); 7.53–7.54 (d, 1H); 8.78 (s, 1H). 13C NMR (75 MHz, CDCl3), δ, ppm: −0.32; 114.96; 115.64; 119.99; 128.69; 133.26; 134.49; 135.18; 147.26; 147.56; 149.24; 152.24. 29Si NMR (80 MHz, CDCl3), δ, ppm: −5.54. MS (EI, 70 eV), m/z (Irel, %): 275 [M]+ (100); 260 (23); 202 (15); 170 (67); 141 (82); 120 (18); 73 (29); 28(87). Anal. Calcd (C14H17NOSSi), %: C 61.14; H 6.23; N 5.09; S 1166. Found, %: C 61.03; H 6.26; N 4.98; S 11.63. Crystal data: monoclinic: a = 6.6387(2), b = 13.4384(5), c = 16.7790(7) Å, β = 91.286(2)°; V = 1496.5(1) Å3, Z = 4, μ = 0.285 mm−1, Dcalc = 1.222 g cm−3; and the space group is P21/n.
2-[(5-Trimethylgermyl-2-thienyl)methyleneamino]-phenol (2c) was prepared as described for 1b, using 5-trimethylgermyl-thiophene-2-carbaldehyde (0.53 g, 2.3 mmol). The reaction mixture was stirred in ethanol at room temperature for 8 h. Pure 2c (0.21 g, 63%) was obtained as a light yellow solid with m.p. 88–89 °C.
1H NMR (300 MHz, CDCl3), δ, ppm (J, Hz): 0.51 (s, 9H, Ge-CH3); 6.86–6.90 (m, 1H); 6.98–7.02 (m, 1H); 7.14–7.28 (m, 3H); 7.53–7.54 (d, 1H); 8.78 (s, 1H). 13C NMR (75 MHz, CDCl3), δ, ppm: −0.59; 114.93; 115.65; 119.99; 128.59; 133.19; 133.39; 135.29; 146.80; 149.18; 149.24; 152.24. MS (EI, 70 eV), m/z (Irel, %): 320 [M]+ (100); 305 (29); 277 (34); 186 (78); 158 (22); 119 (37); 28 (55). Anal. Calcd (C14H17GeNOS), %: C 52.54; H 5.36; N 4.38; S 10.02. Found, %: C 52.53; H 5.35; N 4.38; S 10.19. Crystal data: a = 6.6635(2), b = 13.4717(3), c = 16.8383(4) Å, β = 91.045(1)°; V = 1511.30(7) Å3, Z = 4, μ = 2.153 mm−1, Dcalc = 1.406 g cm−3; and the space group is P21/n.
2-[(5-Triethylsilyl-2-thienyl)methyleneamino]-phenol (2d) was prepared similarly to 1d, using 5-triethylsilylthiophene-2-carbaldehyde (0.52 g, 2.3 mmol) and stirring the mixture for 9 h. After recrystallization from hexane, pure 2d was obtained as a light yellow solid (0.26 g, 71%). M.p. 67–69 °C.
1H NMR (400 MHz, CDCl3, δ, ppm): 0.79–0.87 (m, 6H, Si–CH2), 1.00–1.01 (m, 9H, Si–CH2CH3), 6.76–6.77 (m, 1H), 6.85–6.90 (m, 1H), 6.98–7.02 (m, 2H), 7.14–7.27 (m, 2H), 7.38 (s, 1H), 8.53 (s, 1H). 13C NMR (75 MHz, CDCl3, δ, ppm): 3.05; 7.23; 115.03; 115.36; 115.58; 119.94; 122.79; 128.67; 135.69; 144.99; 152.37; 156.28; 163.78. 29Si NMR (80 MHz, CDCl3, δ, ppm): −2.36. MS, m/z (I, %): 301 (88), 273 (90), 242 (100), 214 (69), 186 (45), 164 (36), 107(61), 59 (42). Anal. Calcd (C17H23NOSSi), %: C 64.30; H 7.30; N 4.41; S 10.10. Found, %: C 64.42; H 7.36; N 4.28; S 10.29.
2-[(5-Triethylgermyl-2-thienyl)methyleneamino]phenol (2e). Using 5-triethylgermylthiophene-2-carbaldehyde (0.62 g, 2.3 mmol) as a starting compound and the same protocol as for 2c and stirring the solution for 14 h, pure 2e was obtained after the recrystallization form hexane as a yellow solid with 69% yield (0.21 g). M.p. 68–69 °C.
1H NMR (400 MHz, CDCl3, δ, ppm): 1.05–1.15 (m, 15H, Si–CH2CH3), 6.85–6.90 (m, 1H), 6.98–7.01 (m, 1H), 7.13–7.19 (m, 3H), 7.24–7.27 (m, 1H), 7.54–7.56 (d, 1H), 8.78 (s, 1H). 13C NMR (75 MHz, CDCl3, δ, ppm): 5.43; 8.82; 114.91; 115.66, 119.99, 128.53; 133.22; 134.15; 135.38; 146.06; 146.85; 149.31; 152.20. MS, m/z (I, %): 363 (59), 334 (97), 306 (21), 276 (17), 202 (18), 170 (100), 139(49), 97 (43), 28 (91). Anal. Calcd (C17H23GeNOS), %: C 56.39; H 6.40; N 3.87; S 8.86. Found, %: C 56.48; H 6.36; N 3.78; S 8.97.
3 Results and discussion
3.1 Synthetic approach
Aldimines 1a–e and 2a–e were prepared by condensation of 2-aminophenol with the corresponding aldehyde in refluxing benzene (Scheme 1). However, refluxing in this solvent (even for 40 h) turned out to be poorly suitable for preparing trimethylsilyl(germyl)-substituted furan and thiophene aldimines: the aldehyde did not react completely and the attempts to recrystallize the product from the reaction mixture led to its decomposition. Changing the reaction conditions (stirring in ethanol at room temperature for 4.5–14 h) allowed us to obtain trimethylsilyl(germyl)-substituted aldimines with 63–71% yields.

Synthesis of aldimines 1a–e and 2a–e. (a) For 1a, 2a: benzene, 80 °C, (b) for 1b–e and 2b–e: EtOH, rt.
3.2 Structural features
The structure of the compounds 2b and 2c was confirmed by X-ray diffraction analysis (Figs. 1 and 2). The crystal structures of 2b and 2c are isomorphous. Their molecular structures are characterized by strong intramolecular hydrogen bonds of the OH⋯N type pointing the OH group toward the azomethine bridge. The corresponding lengths of this contact are 2.617(2) Å (H⋯N = 2.05(2) Å and ∠O–H⋯N = 124(2)°) for 2b and 2.621(2) Å (H⋯N = 2.06(3) Å and ∠O–H⋯N = 128(2)°) for 2c. In the crystal state, these hydrogen bonds are bifurcated forming additional OH⋯O type intermolecular bonds between the neighboring molecules. The lengths of these intermolecular bonds are 2.938(2) Å (H⋯O = 2.48(2) Å and ∠O–H⋯O = 115(2)°) for 2b and 2.934(2) Å (H⋯O = 2.52(2) Å and ∠O–H⋯O = 114(2)°) for 2c.
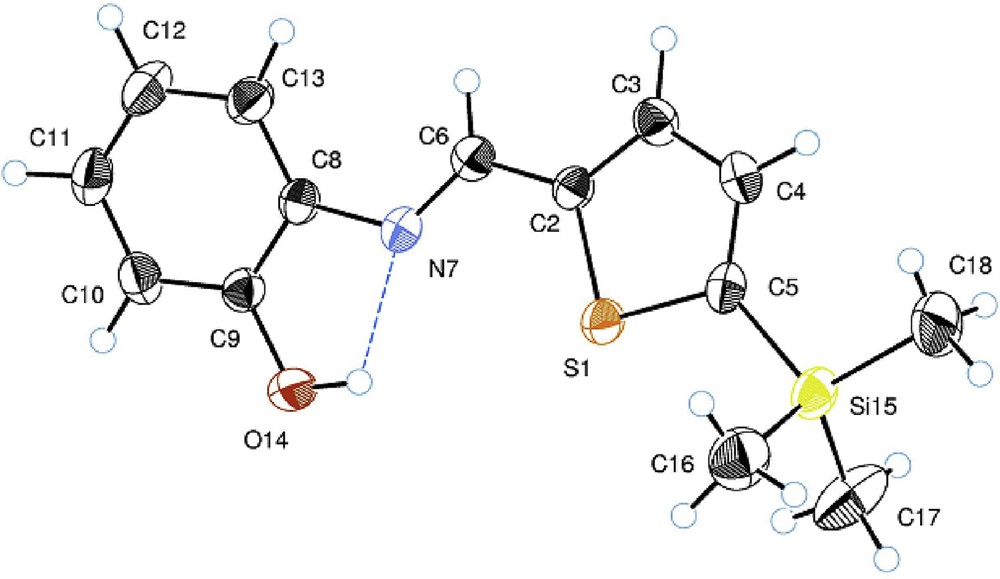
ORTEP structure of 2-[(5-trimethylsilyl-2-thienyl)methyleneamino]phenol (2b). Thermal ellipsoids are shown with 50% probability. The intramolecular OH⋯N hydrogen bond is shown with a dotted line. Selected geometrical parameters: N–C(6) = 1.279(4) Å, N–C(8) = 1.412(6) Å, C(6)–C(2) = 1.442(9) Å, Si–C(18) = 1.851(6) Å, Si–C(16) = 1.858(4)Å, ∠C(8)–N–C(6)–C(2) = 175.61°, ∠N–C(6)–C(2)–S = 1.07°, ∠S–C(5)–Si–C(18) = 179.76°.
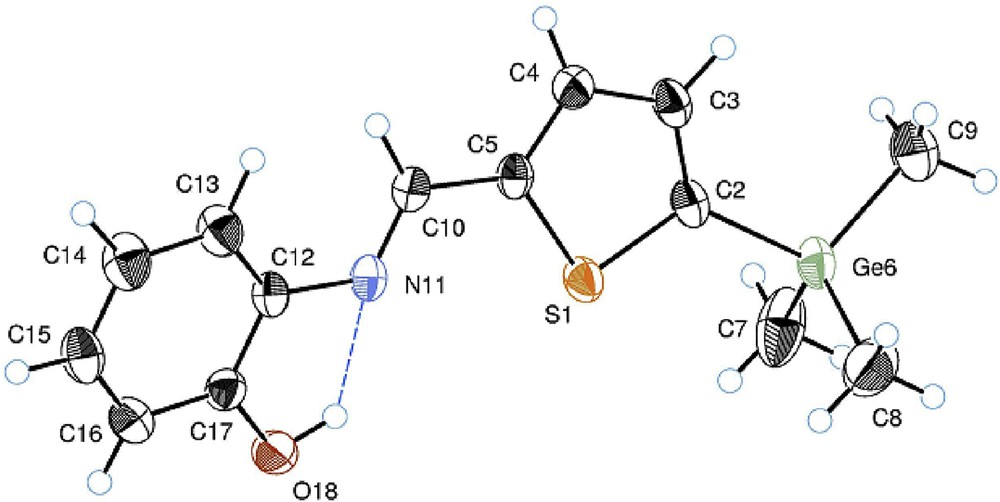
ORTEP drawing of 2-[(5-trimethylgermyl-2-thienyl)methyleneamino]phenol (2c). Thermal ellipsoids are shown with 50% probability. The intramolecular OH⋯N hydrogen bond is shown with a dotted line. Selected geometrical parameters: N–C(12) = 1.415(1) Å, N–C(10) = 1.272(8) Å, C(6)–C(2) = 1.443(6) Å, Ge–C(9) = 1.933(3) Å, Ge–C(7) = 1.937(3)Å, ∠C(12)–N(11)–C(10)–C(5) = 174.93°, ∠N–C(10)–C(5)–S = 1.89°, ∠S–C(2)–Ge–C(9) = 179.98°.
In both structures, the NC azomethine fragment is not distorted (∠NC6C2 = 121.08° and ∠C8NC6 = 122.25° in 2b, whereas in 2c, these angles are 120.85° and 122.28°, correspondingly) and is coplanar to the thiophene unit (dihedral angles ∠N–C–C–S are 1.07° and 1.89° for 2b and 2c, respectively). However, phenyl and thiophene rings are twisted with respect to each other by ca. 18° (the dihedral ∠C13C8NC6 in 2b and its equivalent in 2c are 18.18° and 17.79°, respectively) which somewhat disturbs their ring-to-ring π-conjugation. In the absence of obvious electronic reasons for this, the energy causing this twist visibly arises from ortho-repulsion between the C–H protons of phenyl and azomethine units. In fact, though σ(C3–H) and σ(C6–H) bonds in 2b are eclipsed and practically parallel (same for σ(C10–H) and σ(C4–H) in 2c), they are quite distant (H⋯H = 2.715(2) Å (2b) and 2.681(7) Å (2c)), whereas the phenyl C13–H and methine C6–H protons are closer (H⋯H = 2.379(2) Å and 2.100(8) Å for 2b and 2c, respectively) forming an open six-membered structure (Figs. 1 and 2).
Remarkably, the M–Cthienyl bond in both 2b (M = Si, Si–C5 = 1.874(2) Å) and 2c (M = Ge, Ge–C2 = 1.946(9) Å) is longer than the three other M-C bonds, and one of these three M-C bonds (eclipsed M-Me) is ca. 0.003–0.004 Å shorter than the other two (e.g., Ge–C9 = 1.933(3) Å and Ge–C7 = 1.937(3) Å, Fig. 2). This feature attests to an additional π–σ(C-M)-type hyperconjugation of the Me3M groups with the heterocycle in both 2b and 2c which overcomes the repulsion energy of the eclipsed σ-bonds in the practically planar H–C(3)–C(2)–Ge–C(9) fragment (∠SC2GeC9 = 179.98°).
The structure of the synthesized aldimines was also confirmed by the 1H NMR, 13C NMR, and 29Si NMR (for 1b and 2b) spectroscopy.
3.3 Electrochemical study
The known electrochemical data on aldimines were obtained mostly in acidic or basic media, at pH rarely met in biological systems [7,9,26]. In addition, these compounds are not stable under such conditions. In aqueous media with pH even moderately different form neutral, their redox processes are complicated by acid–base interactions depending, besides their chemical structures, on different factors (concentration, proton activity, supporting electrolyte, etc. [8]). The redox reactions following first electron transfer (ET) usually produce highly reactive intermediates (ions or radical ions very improbably to be formed in biological processes [26]); otherwise they concern secondary stable products resulting from the initial electron release/uptake. When R is a good leaving group, the reduction of the >CN–R unit might result in the N–R bond scission leading to a corresponding nitrile [27,28] and an RH compound so that the immediate biological effect might be anticipated knowing the nature of these products. The oxidation of aldimines in aqueous solutions or in wet organic media involves the NC bond cleavage as well, leading through the formation of a protonated anisidine to the corresponding aldehyde and the quinone imine [29]. Therefore, only first redox steps might be considered as characteristic of biological relevance of aldimines as such.
At a fixed R (o-C6H4OH), electron affinity and ionization of the π-system of aldimines are the prime factors in their redox activity [9], so we considered first redox potentials of the reaction series 1a–e and 2a–e and the energies of the frontier orbitals (FO) corresponding to these processes.
At a Pt electrode in CH3CN/0.1 M Bu4NPF6, the voltammograms of 1a–e and 2a–e are irreversible and poorly reproducible because of electrode passivation. At a glassy carbon (GC) electrode, the curves were better shaped (Fig. 3) albeit some accompanying peaks appeared upon scanning because of the propensity of these compounds to adsorption and electropolymerization [30]. The oxidation potentials are close to those reported (in CH2Cl2/0.1 M Bu4NClO4) for structurally related thiophene(furan) bis-substituted phenyl aldimines [31]. The ip peak currents of both oxidation and reduction steps are diffusion-controlled as follows from their linear dependence on the concentration and scan rate (ip/Cv1/2 = const). The reduction peak half-widths (ΔEp–p/2 = Ep − Ep/2, Table 1) are broadened in both series over 58 mV typical for electrochemically reversible processes [32]. Supposedly, this is not due to a slow ET because the kinetic shift of the Eps of 1a–e and 2a–e with the scan rate (ΔEp/Δln(v) ≅ 35–40 mV) remains close to 30 mV per decade of v, corresponding to a EC mechanism with a fast first-order reaction following ET [32]. This trend is also supported by somewhat larger oxidation peaks (higher ΔEp-p/2, Table 1) because of preferential adsorption of thiophene at anodic potentials [33]. The redox characteristics of 1a–e and 2a–e in reduction and oxidation processes are collected in Table 1.

Voltammograms of (a) reduction of 1c, 2b, and 2c, and (b) oxidation of 2b (first and second cycles) in CH3CN/0.1 M Bu4NPF6 at a GC disk electrode. Scan rate v = 0.2 V s−1. T = 20 °C.
Reduction and oxidation potentials Ep (V vs SCE), peak half-widths (ΔEp-p/2, mV),a frontier orbital energies (ε, Δε, eV),b and dipole moments (μ, D)b of 1a–e and 2a–e.
| Cpd | Reduction | Oxidation | μ | ΔEp c | Δε d | ||||
| Epred | ΔEp-p/2 | −εLUMO | Epox | ΔEp–p/2 | −εHOMO | ||||
| 1a | −2.169 | 75 | 2.327 | 1.879 | 95 | 6.019 | 6.22 | 4.048 | 3.693 |
| 1b | −2.149 | 76 | 2.160 | 1.756 | 88 | 5.722 | 4.06 | 3.905 | 3.562 |
| 1c | −2.140 | 84 | 2.136 | 1.761 | 90 | 5.702 | 4.07 | 3.901 | 3.566 |
| 1d | −2.142 | 80 | 2.128 | 1.753 | 85 | 5.689 | 4.01 | 3.895 | 3.561 |
| 1e | −2.138 | 76 | 2.100 | 1.752 | 80 | 5.664 | 4.02 | 2.890 | 3.563 |
| 2a | −2.185 | 89 | 2.371 | 1.834 | >110 | 5.870 | 4.07 | 4.019 | 3.500 |
| 2b | −2.180 | 80 | 2.309 | 1.782 | >130 | 5.778 | 4.22 | 3.962 | 3.468 |
| 2c | −2.168 | 75 | 2.285 | 1.803 | 90 | 5.759 | 4.29 | 3.971 | 3.474 |
| 2d | −2.168 | 72 | 2.274 | 1.783 | 80 | 5.744 | 4.28 | 3.469 | 3.469 |
| 2e | −2.166 | 78 | 2.248 | 1.767 | 82 | 5.722 | 4.38 | 3.933 | 3.474 |
a At GC electrode in CH3CN/0.1 M Bu4NPF6.
b From DFT B3LYP/Lanl2DZ//HF/6-31G calculations.
c ΔEp = Epox − Epred.
d Δε = εHOMO − εLUMO.
The Eps of both processes are clearly affected by the nature of the heteroaromatic unit at the methine carbon and by the substituent at its α-position, though the last effect is modest. Indeed, the GeMe3 group in aromatic systems exerts the same polar substituent effect as H (i.e. σp = 0 [34]), while SiMe3 acts as a weak donor (σp = −0.07) [34]. Of 2-furyl and 2-thienyl substituents, the former is slightly less acceptor (σp = 0.02 and 0.05, respectively [34]) though the whole effect is rather small. A similar trend is true for the additive inductive constants of these groups (σ* = 1.11 and 1.28) [35]. Thus, the resulting influence of R in 1a–c and 2a–c is distinct but rather small to cause a dramatic differentiation in their redox properties.
These molecules are quite polar (dipole moment μ = 4.06…6.22 D), yet this parameter follows different trends in series 1a–c and 2a–c. Interestingly, most polar derivatives 1a and 2c seem to have more pronounced biological activity (see Section 3.5).
3.4 DFT modeling
Experimental electrochemical quantity Epox − Epred (E0ox − E0red in the case of purely reversible processes) reflects the fundamental orbital quantity, the HOMO–LUMO energy difference,∗ which characterizes chemical “hardness” of a molecule [39]. Both quantities are directly linked to its donor–acceptor ability that can be considered in terms of nucleo/electrophilicity or acido-basicity [40,41], the parameters closely related to biological activity [42,43]. However, the comparison of the experimental redox data with orbital energies is only possible using right geometry for the FO energy calculations because the conjugation in planar systems 1a–e and 2a–e is very sensitive to structural factors. The (E)-form of 1a is more favorable over its (Z)-form with the planes of C6H4OH and furan units helix-wise twisted out of the common plane by strong ortho-repulsion, ΔG[1a(E) − 1a(Z)] = −6.44 kcal mol−1 (DFT B3LYP/Lanl2DZ), and this is the form that was actually formed, in agreement with X-ray data. Important intramolecular N•••H interaction in 1a stabilizes this system by 20.4 kcal mol−1 compared to a non-coordinated structure. On the potential energy curve of (E)-2-(furan-2-ylmethyleneamino)phenol, three minima are marked. The form with σ-trans position of N and O(furan) atoms, where all three chelating centers (O, N, O) are pointed to the same direction, while the azomethine C–H bond is turned to the opposite side, corresponds to the global minimum in the gas phase and in CH3CN solution; two other (E)-forms are local minima with higher energy (Fig. 4). A similar configuration is found to be the global minimum for 2a as well.
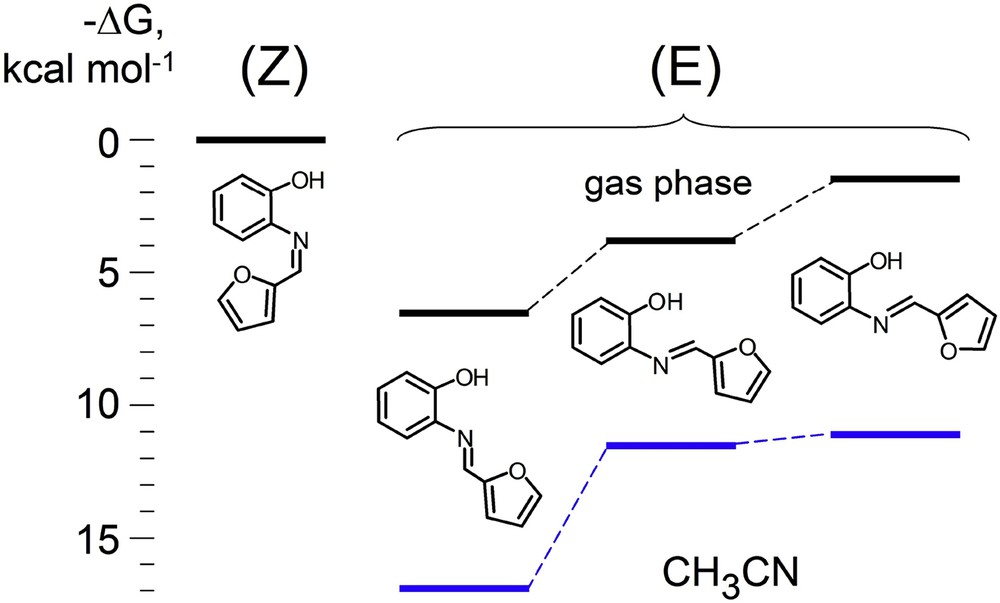
Relative free energies of formation of the different forms of 2-(furan-2-ylmethyleneamino)-phenol 2a. In blue: values in CH3CN. From DFT B3LYP/Lanl2DZ (CPM) calculations.
This configuration is in agreement with the experimental IR spectra of 1a and 2a. A broad band at vO–H = 3000 cm−1 is due to the vibration of the OH hydrogen involved in the intramolecular coordination with N atom [44]. Respectively, the vC–O band now appears at 1472 cm−1, and a characteristic Schiff base vCN band [45] is shifted to lower frequencies (v = 1634 cm−1) because of N•••H coordination (cf. X-ray structures of 2b and 2c, Figs. 1 and 2). The methine C–H bond in 1a and 2a vibrates with a practically similar frequency typical for this conformation (δC–H = 3035 cm−1), which is well reproduced by DFT calculations (Fig. 5).
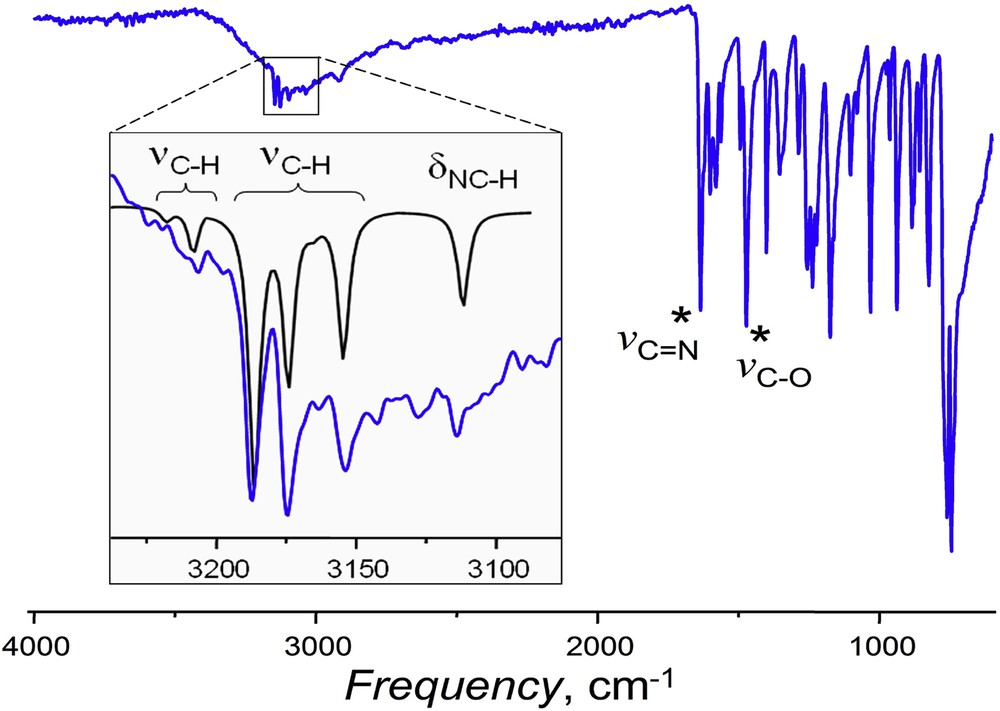
IR spectrum of 1a. In the inset, appearing on the large vO–H band, are DFT-calculated (black) IR frequencies of aromatic C–H bonds in furan (left group) and phenol (right group), and of the methine (N=)C–H carbon–proton bond.
The frontier orbitals (FOs) (Fig. 6) are quite similar in the reaction series 1 and 2, with the main difference arising from the orbital coefficients of O and S in the heteroaromatic rings. Remarkably, Me3Si and Me3Ge substituents do not strongly perturb these orbital features: neither Si nor Ge are directly involved in these FO, the substituents are mostly contributing through hyperconjugation of their M–Csp3 (M = Si, Ge) bonds with the π-electron system (Fig. 6) in agreement with what was observed by X-ray diffractometry (Figs. 1 and 2).
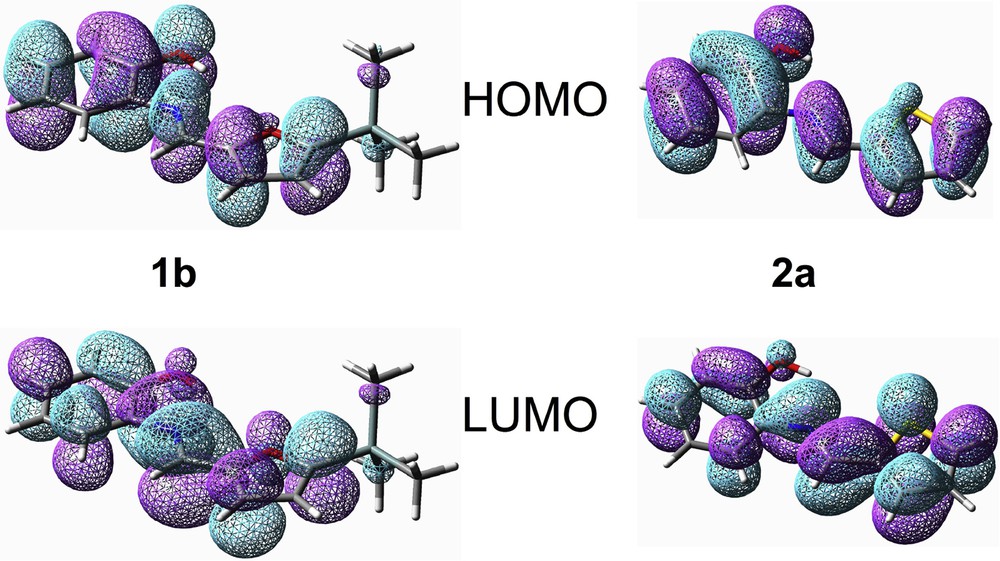
Redox-related frontier orbitals (HOMO and LUMO) of 1b and 2a (from DFT B3LYP/Lanl2DZ calculations).
Fig. 7 thus shows the Epox and Epred plotted against the corresponding FO energies and the correlation of (Epox − Epred) with the εHOMO − εLUMO difference. In both families, 1a–e and 2a–e, Epox − IP and Epred − εLUMO are well correlated, forming common plots, though their less than unity slopes (ΔEpox/ΔIP = 0.38 and ΔEpred/ΔεLUMO = 0.18) indicate a strong influence of the chemical reaction following the first ET on the experimental Eps (large kinetic shift). Quite similar trends were previously found for furyl and thienyl nitrones and nitroethenes [36]. When considered in terms of orbital hardness, furyl- and thienyl-substituted aldimines form two different groups (Fig. 7, c). Interestingly, charge-based isotropic dipole moments μ of 1a–e and 2a–e, reflecting their polarity, are distinctly in line with each of the orbital energies, εHOMO and εLUMO (Table 1), but they loose the selectivity for their difference and are no more specific for properties correlations.
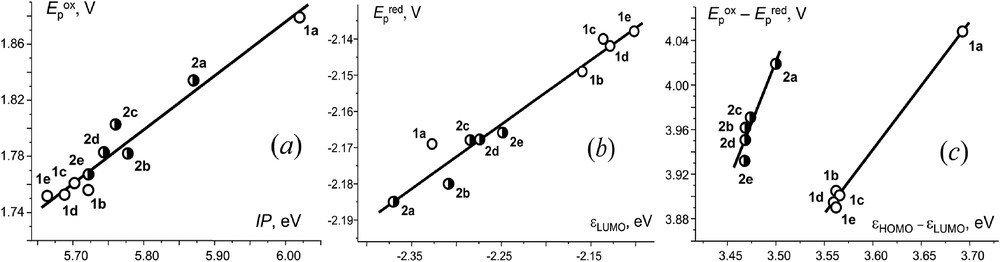
Correlations of (a) Epox with the first ionization potential (IP = -εHOMO); (c) Epred with LUMO energy; (c) the difference in first redox potentials (Epox – Epred) with the εHOMO – εLUMO difference.
3.5 Cytotoxicity and toxicity
Aldimines and their derivatives possess a broad spectrum of biological activity, such as anti-bacterial, anti-cancer [46], anti-fungal (Candida albicans and Aspergillus niger) [4], anti-inflammatory, anti-virus, anti-pyretic [47], and a significant activity against Staphylococcus aureus and epidermidis, Bacillus subtilis and cereus, Micrococcus luteus, and Escherichia coli. Their complexes with metals (e.g., Cu, Ni, Zn) showed very good activity against leukemia cells (CEM-SS) [4].
The in vitro cytotoxicity of heteroaryl-substituted aldimines 1a–e and 2a–e was investigated for two tumor lines, HT-1080 (human fibrosarcoma) and MG-22A (mouse hepatoma), and on normal mouse fibroblasts NIH 3T3 to determine the effect of the substituent at the fifth position and of the type of heterocycle on antitumor activity. The results of experimental evaluation of the cytotoxic properties are presented in Table 2.
In vitro cytotoxicity (IC50) and toxicity (LD50) of aldimines 1a–e and 2a–e.
| Compound | IC50, μg ml−1 | LD50, mg·kg−1 | ||
| HT-1080 | MG-22A | NIH 3T3 | ||
| MTTa | MTT | NR | ||
| 1a | 2 | 1 | 9 | 187 |
| 1b | >100 | 30 | 11 | 259 |
| 1c | 3 | 8 | 1 | 91 |
| 1d | 0.2 | 2 | <0.3 | <60 |
| 1e | 24 | 21 | 5 | 232 |
| 2a | 10 | 8 | 12 | 144 |
| 2b | 26 | 6 | 193 | 4 |
| 2c | 18 | 12 | 15 | 192 |
| 2d | 56 | 12 | 43 | 483 |
| 2e | 21 | 25 | 17 | 398 |
a Coloration indicators: MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide, NR = neutral red.
The aldimines 1a and 1c have high cytotoxic activity (IC50 1–8 μg ml−1) on both cancer cell lines and also have high cytotoxic effect on NIH 3T3 cells (IC50 1–9 μg ml−1). The introduction of the Me3M substituent to the heterocycle has a special effect in both O and S series. In the furan series, the silylated aldimine 1b generally shows a decrease in both cytotoxicity and toxicity. Its thiophene analogue 2b shows better cytoselectivity: it has higher cytotoxicity on cancer cell line MG-22A (IC50 6 μg ml−1) than on HT-1080 (IC50 26 μg ml−1) but unlike other aldimines of this family, this compound is very toxic (LD50 4 mg kg−1). Me3Ge-substituted 2c is more promising here as it has practically similar cytotoxicity as non-substituted 2a but with reduced toxicity. Aldimine 1d has most pronounced cytotoxicity in these series for both HT-1080 and NIH 3T3 cells but at the same time, it is quite toxic, LD50 < 60 mg kg−1. In contrast, its thiophene analogue 2d is the less toxic (Table 1), although also less efficient against the two tumor lines and NIH 3T3. Et3Ge-substituted furan derivative 1e has a good cytotoxicity toward NIH 3T3 at rather high LD50, but it is less selective for HT-1080 and MG-22A tumor cells.
Remarkably, in the corresponding series, the oxidation Eps of both Me3Si derivatives, 1b and 2b, are lower than those of non-substituted 1a and 2a (Table 1). However, while the α-Me3Si group promotes (for the Me3Si neighboring group effect, see, e.g., Ref. [48]) the S-oxidation, similar to the oxidation by cytochrome CYP450 [16], the oxidation of the furan derivative is not O-centered and follows a different way. We venture to suppose that the acute toxicity of 2b might be related to its easier metabolic S-oxidation leading to toxic products [11,16]. The parent α-Me3Ge group in 2c is less conjugated with the π-system of the heterocycle because of the size mismatch of its more diffuse σ(C–Ge) orbitals responsible for the electronic effects, so probably its overall action results from different factors (solubility, coordination of Ge with biological phosphates, etc.). Interestingly, when the Me3Si group increases the toxicity, the Me3Ge group induces the opposite trend.
Overall, aldimines 1a, 2a, and 2c seem to have the most advantageous combination of biological properties, thus providing good leads for the further development in these reaction series.
4 Conclusion
To conclude, new aldimine derivatives of o-aminophenol, with furanyl and thiophenyl substituents (C4H4X, X = H, SiAlk3, GeAlk3, with Alk = Me, Et) were synthesized through a simple procedure condensing the aniline with the corresponding precursor aldehyde. The structure of these new products as well as redox properties in CH3CN/0.1 M Bu4NPF6, their frontier orbital energies, cytotoxicity, and toxicity have been considered. Their electrochemical redox potentials Ep show good correlation with the related orbital energies and the difference Epox − Epred corresponds well to the orbital hardness of these compounds. Me3Si and Me3Ge substitution of the heterocycle facilitates the oxidation of the corresponding molecules (promoting their oxidative metabolism), and—though not being dramatical—alters their redox properties. However, remarkably reducing their orbital hardness, Me3M substitution can also substantially affect solubility and hydro(phobicity/philicity) and hydrogen-bonding ability of these molecules. The steric hindrance of both Me3M groups is relatively small because of longer C–Si and C–Ge bonds compared to C–C; this same feature accounts for attenuating their electronic interaction (hyperconjugation) with the aldimine π-system, especially for the Me3Ge group. To the overall effect of this latter, additional factors add such as more diffuse conjugating σ(C–Ge) orbital and longer Ge–C(H3) bonds, making Ge more available for external coordination and nucleophilic interactions.
The new aldimines have shown a pronounced cytotoxicity toward cancer cells of human fibrosarcoma HT-1080 (with IC50 as low as 0.2–3 μg ml−1) and of mouse hepatoma MG-22A (IC50 ≅ 1–8 μg ml−1). Their cytotoxicity can be largely modulated by the Me3M substituent to the fifth position of the heterocycle, up to 50 times in furan series and to 3–5 times for thiophene derivatives. For electronic reasons, the α-Me3Si substituent exerts a strongest neighboring group effect when attached to the thiophene unit. As a consequence, it enhances the oxidation on the S atom thus altering the oxidative metabolism of this compound, which is translated by its acute toxicity. In other cases, toxicity generally decreases upon R3M substitution, though Et3Si and Me3Ge derivatives in the furan series fall out of this trend. Further works in this field are in progress.
Acknowledgments
The authors gratefully acknowledge the support of this research by the Latvian Council for Science (Project 225/2012) and from PHC OSMOSE #39669QC.