



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Fluorine is an essential element in pharmaceuticals, with approximately 20% of drugs containing at least one fluorine atom [1, 2, 3, 4]. In many cases, the introduction of fluorine into molecules has significantly improved molecular properties [5, 6, 7]. Fluorination affects several parameters, including the hydrophilicity/lipophilicity balance, the metabolic stability of drugs, and the binding affinity to a target. Due to its small size, the replacement of hydrogen by fluorine leads to minimal steric change while increasing hydrophobicity and polarity. In addition, organofluorinated compounds have significant applications in chemical biology [8]; for example, the so-called fluorous effect can be used to study and manipulate biological systems in an orthogonal manner. In addition, polyfluorinated groups are valuable probes for performing 19F NMR studies and are used as MRI contrast agents in biomedical imaging [9, 10, 11]. Polyfluorinated groups, such as CF3, are often required to observe NMR signals. The incorporation of fluorine into amino acids has therefore proved particularly useful in this context [12, 13]. Fluorinated amino acids have attracted much attention in the drug industry, whether used individually or as components of bioactive peptides or peptidomimetics [3, 14]. Fluorinated analogues of canonical amino acids are of particular interest because they can be incorporated into natural bioactive peptides to modulate their biological properties or to study protein conformation, binding, dynamics, and interaction with membranes using 19F NMR [8].
Recently, efforts have been directed toward the development of synthetic routes to access emerging polyfluorinated motifs [15] that could be highly profitable for the applications mentioned above. However, the widespread use of specific fluorinated motifs depends on their synthetic accessibility, which requires efficient synthetic processes using affordable and safe fluorinated reagents. Our recent research has focused on the introduction of polyfluorinated analogues of the isobutyl group, a hydrophobic branched alkyl chain frequently encountered in medicinal chemistry research programs and present in many drugs, such as tetrabenazine and bromocriptine. In addition, the isobutyl moiety is the side chain of leucine, the most abundant hydrophobic amino acid, making it ubiquitous in proteins. It is typically found in the hydrophobic core of proteins and plays a key role in many protein assemblies, such as leucine zippers [16]. Replacing the isobutyl side chain with a hexafluoroisobutyl group significantly increases hydrophobicity due to the increased side-chain volume while maintaining the overall morphology of the amino acid [17, 18]. This group also exhibits a significant dipole moment, probably comparable to that of a water molecule [19, 20].
Several research groups have investigated the effect of replacing leucine with (S)-5,5,5,5′,5′,5′-hexafluoroleucine (Hfl) in peptides or proteins on folding, self-assembly [21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31], and biological properties [32, 33, 34, 35, 36, 37, 38]. However, the synthesis of the corresponding amino acid typically requires multiple synthetic steps to attach the hexafluoroisopropyl synthon to the β-carbon of the amino acid (Figure 1), which significantly limits its wider application in various fields such as drug discovery and chemical biology [27, 39, 40, 41]. To establish a concise and versatile method for introducing this fluorinated group, we developed an efficient single-step procedure to incorporate the entire fluorinated chain using a non-gaseous fluorinated reagent. This method required overcoming critical chemical hurdles, such as the unwanted elimination of fluoride ions. We have successfully synthesized enantiopure Hfl using this process and have begun to explore its potential in foldamer chemistry and for modulating biologically relevant peptides and proteins. In addition, we have recently reported a cell-free approach to incorporate this highly fluorinated residue into amyloids for high-resolution magic-angle spinning 19F NMR.
Key disconnections to build or introduce the hexafluoroisobutyl group.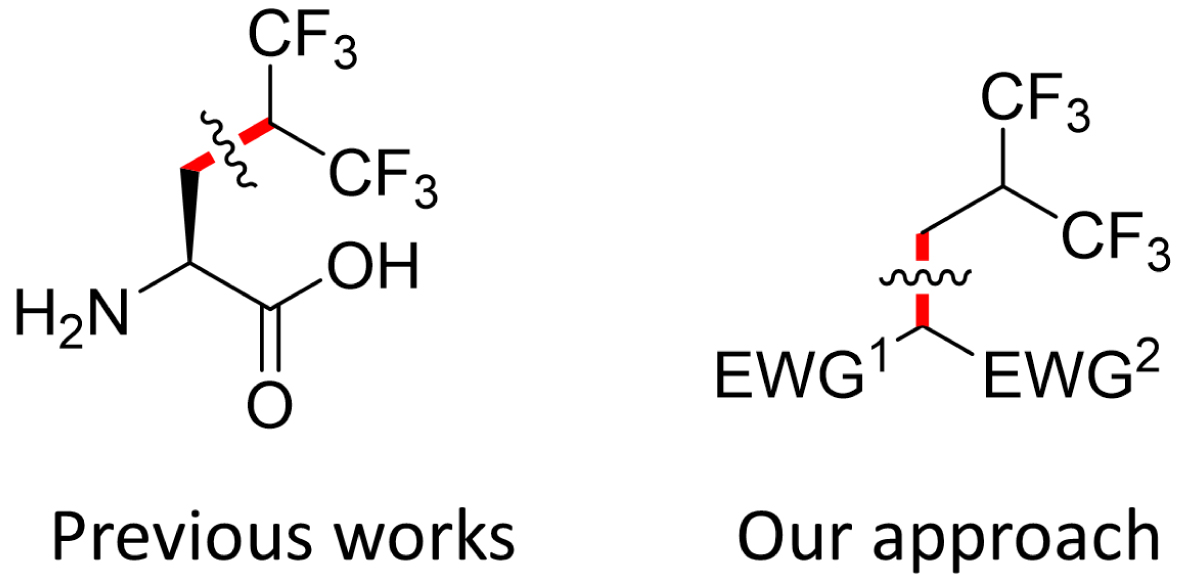
2. A general method to introduce the hexafluoroisobutyl chain in a single step
2.1. Development of the method
We started our investigation using 2-(bromomethyl)-1,1,1,3,3,3-hexafluoropropane as an electrophilic reagent. This reagent is a liquid at room temperature and is easy to handle. Although this reagent is commercially available, its reactivity was almost unknown [42, 43]. In our initial attempts to alkylate substrates with this reagent, we observed that under basic conditions, the reagent undergoes a complete β-elimination of HBr within a few minutes, leading to hexafluoroisobutene (Scheme 1A). The formation of this unsaturated compound was confirmed by NMR. Therefore, this unsaturated fluorinated molecule is rapidly generated in solution and acts as an electrophilic reagent. However, the fluoroalkylation reaction on ketoesters with the fluoroalkylating reagent resulted predominantly in a pentafluoroalkene, with the hexafluorinated derivative being formed as a minor product (Scheme 1B) [44]. To circumvent this problem, we found that the use of a large excess of tetrabutylammonium fluoride (TBAF), acting both as a source of nucleophilic fluoride and as a base, provided the desired compound in good to excellent yields for a wide range of carbonylated substrates (Figure 2). The reaction was suitable with substituted ketoesters, malonates, diketones, Schiff base esters, and malononitrile. Interestingly, starting from α-acetylbutyrolactone, the deacetylated product was successfully obtained giving direct access to the α-fluoroalkylated ester (Scheme 2).
(A) Elimination of HBr on 2-(bromomethyl)-1,1,1,3,3,3-hexafluoropropane under basic conditions leads to hexafluoroisobutene. (B) First attempt to introduce the hexafluoroisobutyl group on a ketoester; hexafluorinated compound 1 is formed as a minor product and pentafluorinated compound 2 is obtained predominantly.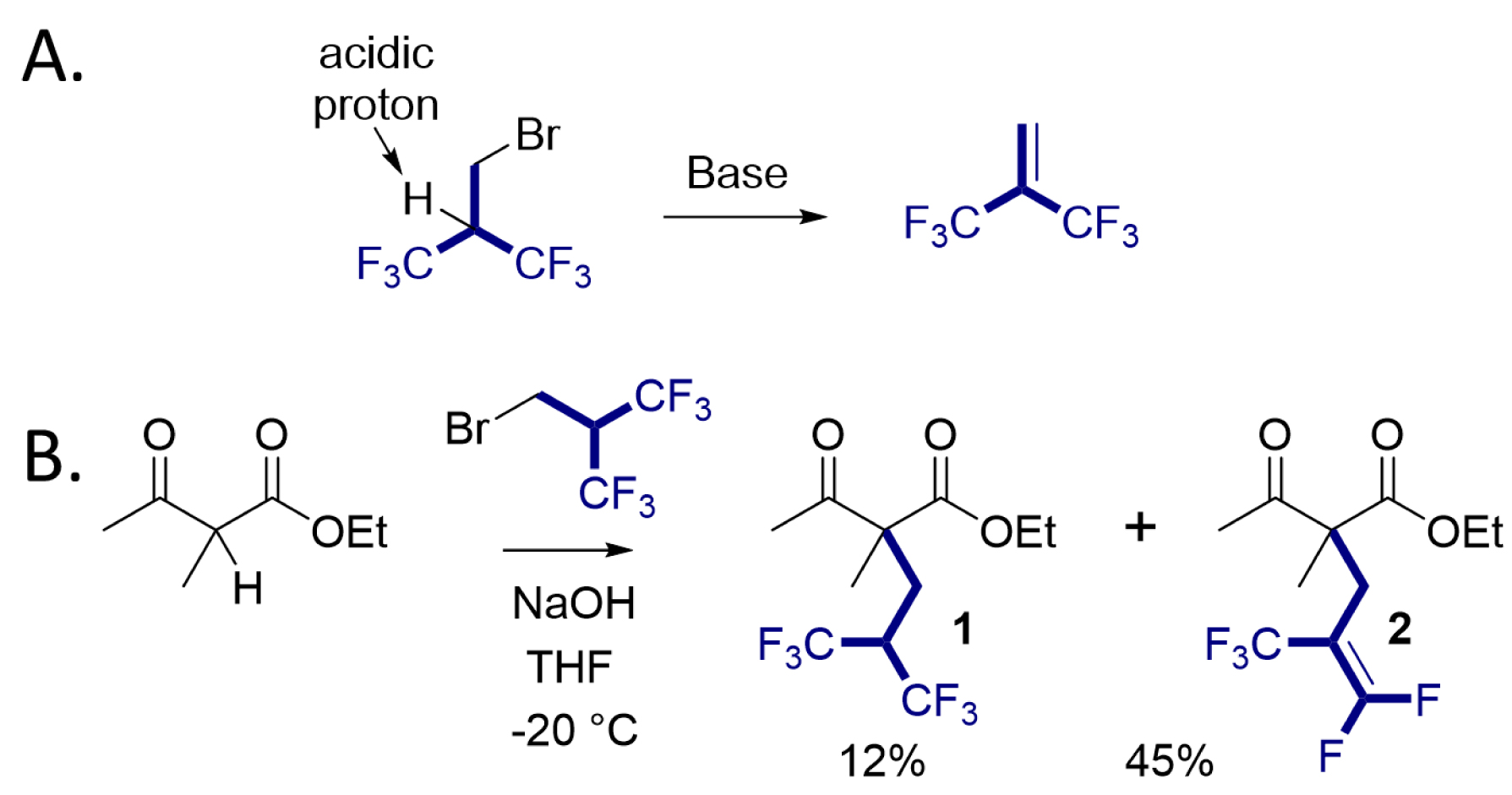
Scope of the hexafluoroisobutylation reaction. a2 equiv of the electrophile was used. bEstimated yield using NMR.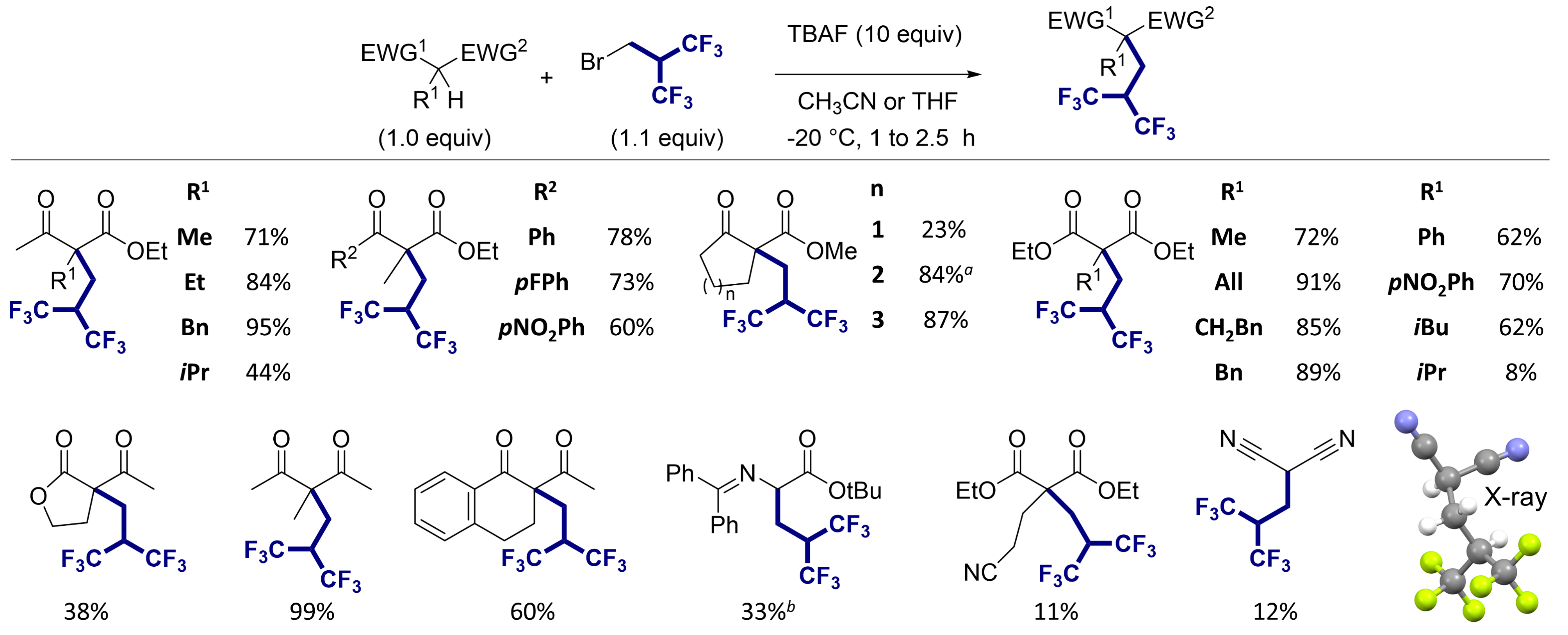
Hexafluoroisobutylation/deacetylation cascade reaction.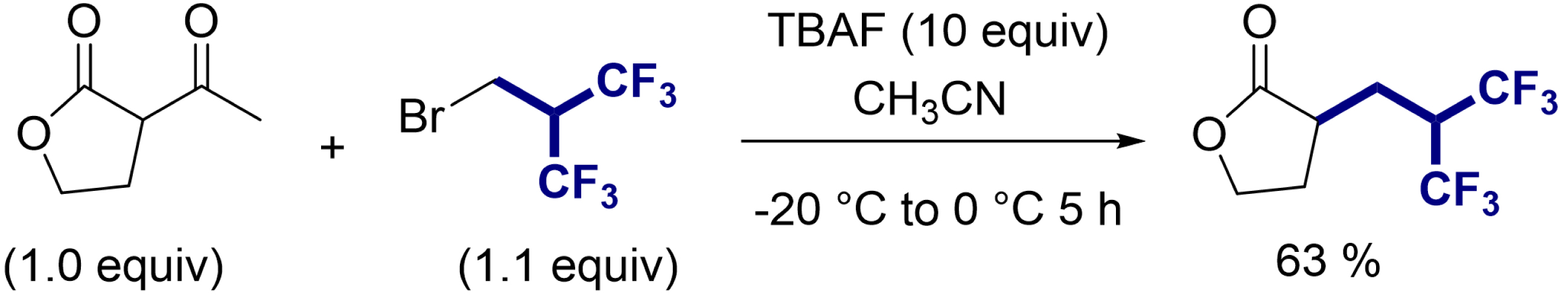
Importantly, hexafluoroisobutene—the key reactive species—is a gas (bp 14.1 °C) but can be easily generated in situ from the liquid brominated precursor with this protocol.
2.2. Access to (S)-5,5,5,5′,5′,5′-hexafluoroleucine
We then decided to apply this methodology to the stereoselective synthesis of Hfl. We used the robust strategy originally developed by Belokon et al., based on the stereoselective homologation of a glycine Schiff base complex [45, 46], and further developed by others, to access fluorinated amino acids, including Soloshonok [47, 48, 49], Larionov [50, 51], and Koksch [52], or other non-canonical amino acids [53]. We found that the reaction is compatible with the chiral Ni(II) complex 3, which provides the desired alkylated complex 4 in 66% yield (Table 1). In contrast, none of the alkali metal bases commonly used with this nickel complex (NaOH, LiOH, KOH, t-BuOK) were suitable for the reaction, which predominantly gave the elimination product 5, similar to the results observed with ketoesters. Remarkably, we showed that the organic base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) also favors the formation of the hexafluorinated compound, with a 84% yield [54]. This study highlights the crucial role of the nature of the base and its pKa in determining the outcome of the reaction.
Influence of the base on the hexafluoroisobutylation reaction of a chiral Ni(II) complex
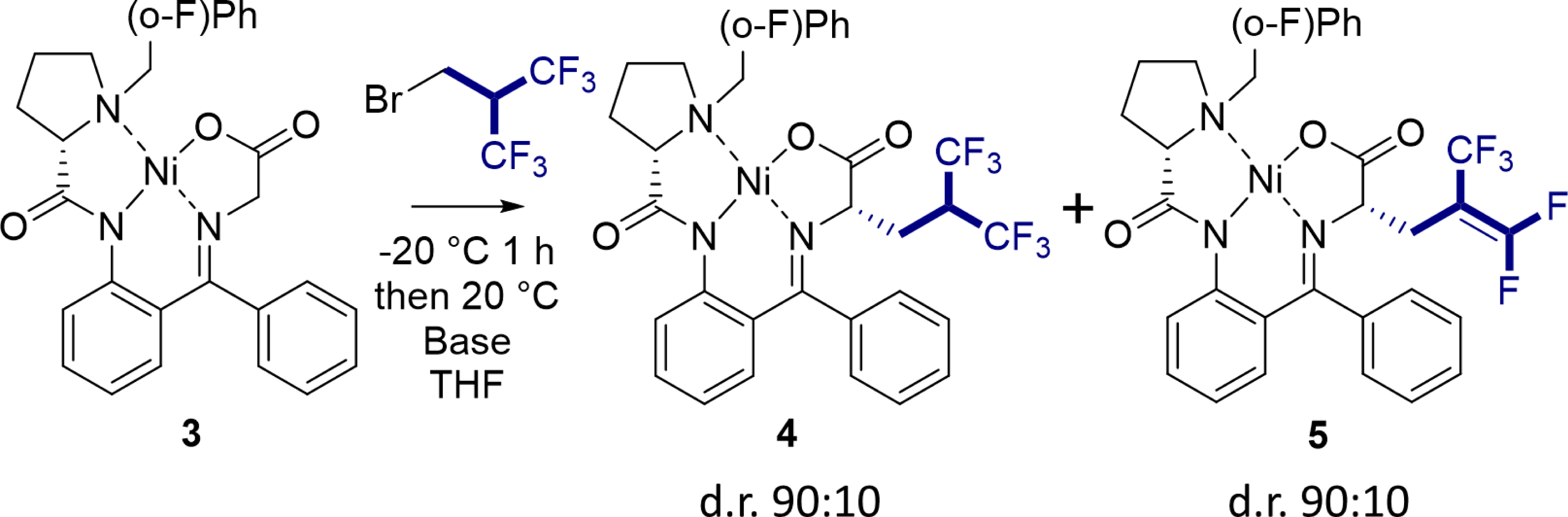
| Base (equiv) | Time (h) | Yield | ||
|---|---|---|---|---|
| 3 | 4 | 5 | ||
| TBAF (10) | 3 | - | 66% | - |
| NaOH (4) | 4 | 20% | 6% | 55% |
| LiOH (4) | 4 | 40% | 15% | 38% |
| KOH (4) | 4 | 32% | 6% | 44% |
| t-BuOK (4) | 4 | 20% | 13% | 47% |
| DBU (4) | 3 | - | 84% | - |
| Et3N (4) | 23 | >95% | trace | - |
The procedure is suitable for large-scale synthesis, and the hydrolysis of the Ni(II) complex produced free enantiopure Hfl as well as its N-Fmoc and N-Boc derivatives (Scheme 3). This route provides easy access to this building block for use in a wide range of applications.
Hydrolysis of the fluoroalkylated Ni(II) complex to synthesize Fmoc- and Boc-protected Hfl and the unprotected form.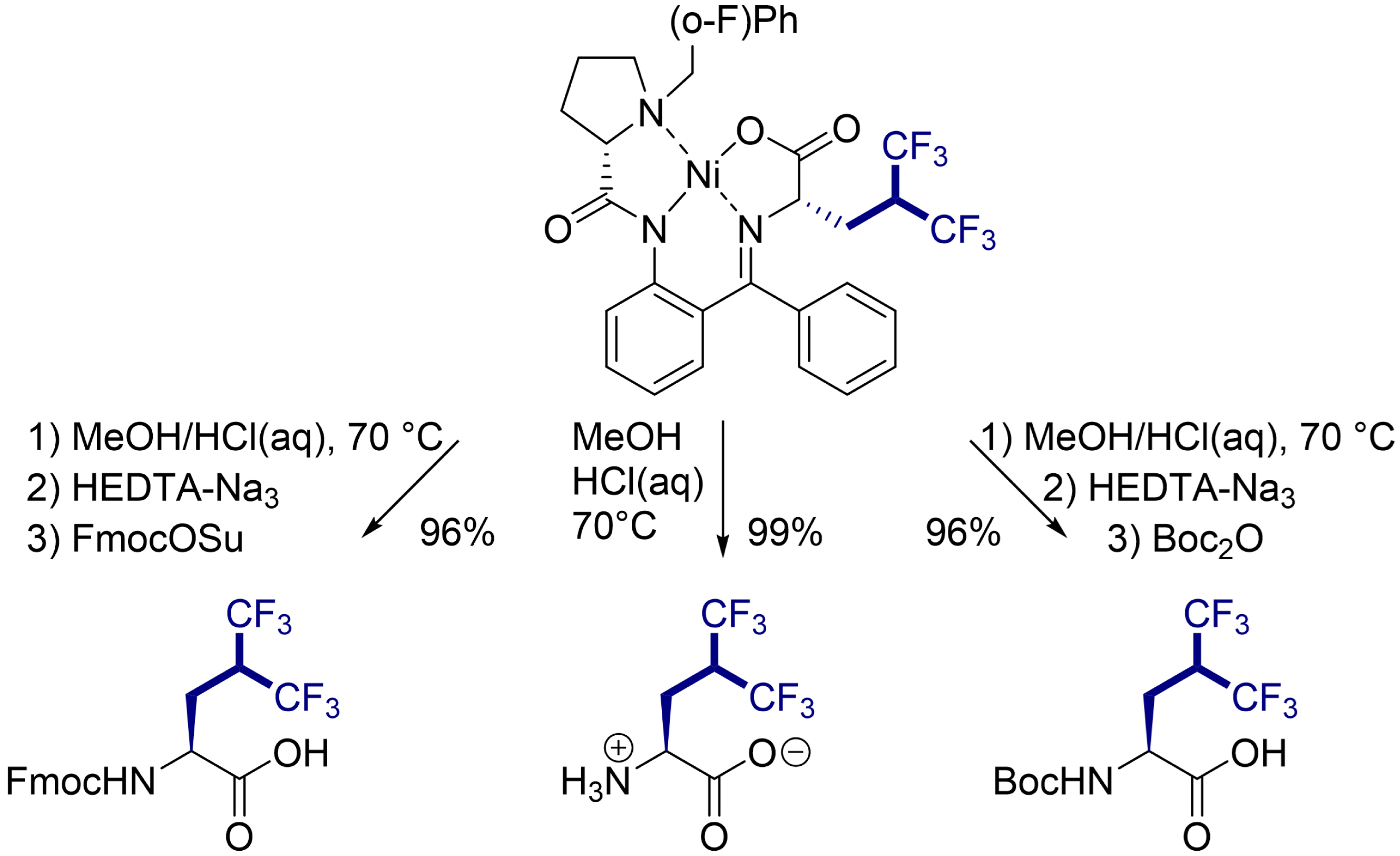
2.3. Mechanistic aspects
In situ NMR experiments were performed and allowed the identification of several intermediates. First, the brominated electrophile underwent an elimination of HBr under basic conditions, leading to hexafluoroisobutene after a few minutes (Scheme 4). Hexafluoroisobutene, with its electron-poor double bond, is highly electrophilic and reacts with the deprotonated substrate via an SN2′ mechanism, leading to the pentafluoroalkene intermediate.
Proposed general mechanism.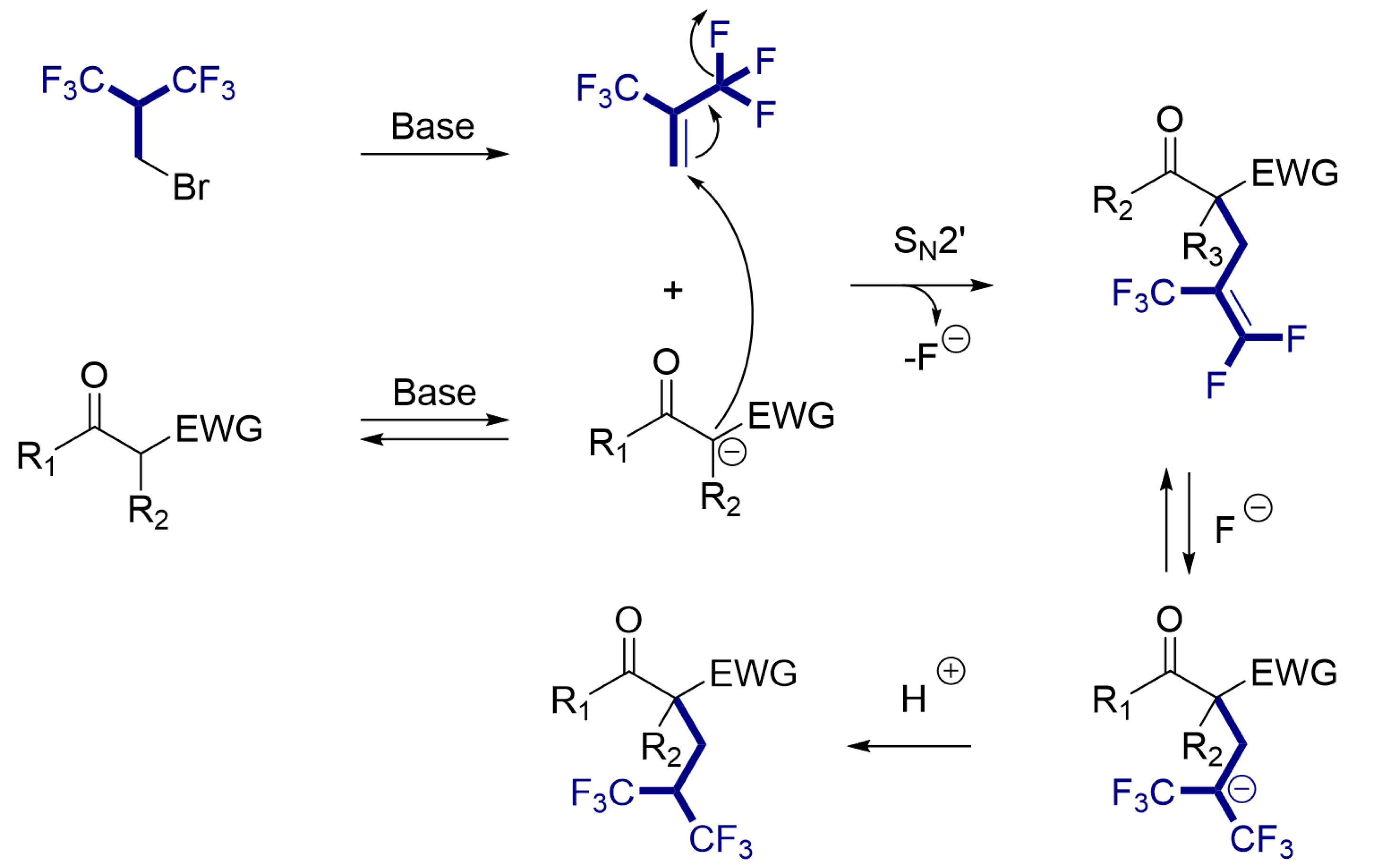
Using an excess of fluoride anions (TBAF), we have shown that hydrofluorination of the newly formed double bond is favored, yielding the saturated hexafluorinated chain. With alkali metal bases, the fluoride released in the medium forms inorganic fluoride salts that are not nucleophilic enough to react with the fluorinated alkene. In contrast, the organic base DBU generates a DBU∙HF salt that preserves the nucleophilicity of the fluoride ions released in situ and acts as a fluorinating agent, as demonstrated by our in situ NMR experiments (Figure 3A). In this reaction, the DBU∙HF salt thus enabled the successful hydrofluorination of the pentafluorinated intermediate without the need for an external source of nucleophilic fluoride ions (Figure 3B). In a more general context, it is well known that α-(trifluoromethyl)vinyl groups generally react with nucleophiles via an SN2′ mechanism, leading to β-fluoride elimination [55, 56, 57], which is observed in α-CF3-carbanion chemistry [58]. In our study, we showed that this reaction can be successfully reversed in the presence of nucleophilic fluoride ions, restoring the saturated chain containing two CF3 groups.
(A) In situ NMR experiments showing the hydrofluorination reaction of the pentafluoroalkene with DBU∙HF. (B) Proposed mechanism of the hexafluoroisobutylation reaction when DBU is employed.
3. Incorporation of Hfl into peptides and peptidomimetics
The incorporation of Hfl into peptides has previously been achieved using solid-phase peptide synthesis (SPPS). The SPPS allows the rapid preparation of peptides up to 30–40 residues in length, either manually or using automated peptide synthesizers. However, despite its similarity to canonical leucine, the integration of Hfl into peptides required optimization of synthetic procedures. The Boc strategy has been the most commonly used approach to obtain Hfl-containing sequences, particularly by the Kumar group [25, 31, 32, 59, 60] and the Marsh group [24, 28, 33, 61]. This method uses the in situ neutralization/N,N,N,N-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate protocol described by Schnölzer et al. [62] and 4-methylbenzhydrylamine resin. Coupling of Boc-protected Hfl sometimes required a prolonged coupling step (>2 h) [60]. The Boc strategy is robust but requires HF treatment at the end of the elongation process to release the peptide from the resin, which is a limitation for research laboratories that are not equipped to safely handle anhydrous HF. In some cases, the Boc strategy has been chosen over the more practical Fmoc strategy because of the ineffective synthesis of Hfl-containing peptides with the latter method [28]. MALDI-TOF analyses revealed that the coupling yields for the two residues following Hfl incorporation were very low. However, the reason for this remains unclear. Nevertheless, the incorporation of Hfl into other peptide sequences has been successfully achieved using the Fmoc strategy on Wang, Rink amide, and Fmoc-PAL-PEG-PS resins [22, 30, 34, 38] with a reduced amount of coupling reagents relative to the molar amount of Fmoc-Hfl-OH. Racemization of the Hfl residue was also described in one case during peptide synthesis, resulting in two diastereoisomers with identical masses (L/D ratio: 10/4) [22]. Finally, the Schepartz group reported β-peptides containing β-Hfl to investigate their supramolecular properties in the foldamer field [63]. The fluorinated oligomers were synthesized using Fmoc chemistry on Wang resin with PyAOP, HOAt, and DIEA for the coupling steps. Exploration of alternative coupling agents and optimization of conditions for deprotection of the amine protecting group to further improve the synthesis of Hfl-containing peptides and generalize their potential use are areas where our group is currently focused. In addition, diversifying the types of hexafluorinated monomers that can be used in foldamer chemistry is certainly a promising direction for future research.
4. Incorporation of Hfl into proteins
There are several approaches to synthesize proteins containing non-canonical amino acids, such as those using genetic code expansion or SPPS combined with native chemical ligations [64, 65, 66]. Some of these strategies have been used to incorporate fluorinated amino acids [12]. In the case of leucine analogues, several fluorinated derivatives have been successfully incorporated into large proteins using E. coli strains auxotrophic for leucine [67, 68, 69, 70]; for example, the introduction of (S)-5,5,5-trifluoroleucine into several proteins has been achieved using this method. However, the incorporation of Hfl still remains challenging with this approach and requires engineered cells overexpressing Leu-tRNA synthetase to overcome the unfavorable formation of the activated ester between Hfl and its corresponding tRNA [27]. Importantly, this approach does not result in complete incorporation of the fluorinated amino acid due to the presence of the canonical amino acid in the cells, resulting in a mixture of modified proteins. On the other hand, cell-free protein expression offers a powerful alternative to in-cell methods [71, 72, 73, 74] and allows the incorporation of non-natural amino acids using the translation machinery [75, 76, 77]. This method allows the synthesis of proteins that are difficult to produce in cell culture, including those that are toxic or prone to aggregation. In collaboration with the Loquet group at CBMN and IECB, we showed that the introduction of Hfl into amyloid fibrils can be successfully achieved [78]. The three Leu residues of the HET-s (218–289) prion were completely replaced by Hfl within this 72-residue protein. Interestingly, the fluorinated HET-s was well assembled and remained pathological. Solid-state 19F NMR reveals five out of six relatively narrow CF3 signals (the sixth is certainly overlapped by the others) reflecting the different local environment of the CF3 groups within the folded protein and a well-ordered conformation of the Hfl side chains (Figure 4). This method is thus well suited for the incorporation of highly fluorinated residues and nicely complements existing approaches for using this fluorinated amino acid to study protein structure and function.
19F NMR at 60 kHz magic-angle spinning solid-state experiments of cell-free synthesized HET-s containing three Hfl residues.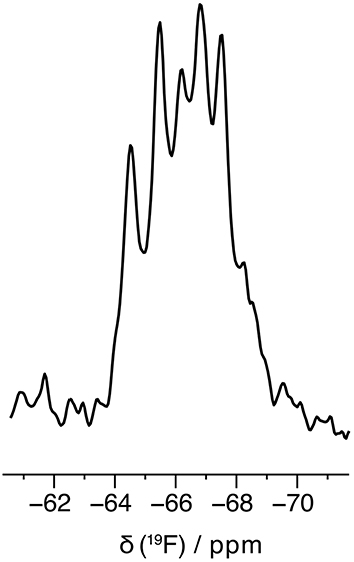
5. Conclusion and outlook
In summary, our work has led to several key developments with promising prospects.
- We have developed a general method for incorporating the hexafluoroisobutyl group into carbonylated compounds. This process allows the introduction of a fluorinated chain in a single step through a tandem mechanism using a non-gaseous fluorinated electrophile. In particular, this process overcomes the β-fluoride elimination (SN2′ mechanism) thanks to an in situ fluorination reaction when using an excess of fluoride sources or DBU as a strong organic base, allowing the formation of the hexafluorinated chain. The SN2′ mechanism can thus be reversed. Future work could explore the application of tandem SN2′/hydrofluorination reactions to other α-(trifluoromethyl)vinyl groups. This method now makes it possible to introduce this fluorinated group and investigate its potential interest in drug discovery programs. In addition, this fluoroalkylation reaction was successfully applied to the enantiopure synthesis of Hfl, which can now be produced on a large scale using a chiral Ni complex. Furthermore, although fluoride elimination was not desirable for synthesizing the fluorinated chain, the central acidic proton of highly fluorinated—though not perfluorinated—groups, next to the CF3, could be used to promote fluoride ion elimination. This, in turn, could potentially trigger the degradation of the side chain, which may be of interest in the search for degradable fluorinated motifs [79].
- We have demonstrated the potential of DBU∙HF as a fluorinating agent. The nucleophilicity of the fluoride ions in this salt has yet to be determined and compared to other fluoride sources, as well as its stability at high temperatures. This salt could potentially be (i) less acidic than other widely used nucleophilic fluoride sources such as Et3N∙3HF and (ii) less prone to Hofmann elimination compared to TBAF [80]. DBU∙HF could thus complement the existing arsenal of fluorinating agents and find a wide range of applications in fluorine chemistry.
- The introduction of the Hfl into peptides has been previously achieved by standard SPPS methods, including Boc and Fmoc strategies. However, problems such as inefficient coupling steps and residue racemization have been reported. There is potential for further improvements in the synthesis of peptides containing this important fluorinated residue, which would enhance its application in peptide science and foldamer chemistry. Although there are some studies on bioactive peptides containing Hfl [32, 33, 34, 35, 36, 37, 38], in-depth studies on the rational design of peptide ligands to accurately incorporate Hfl and exploit the full potential of this highly fluorinated group to enhance (bio)molecular recognition properties are still needed.
- Finally, we have shown that cell-free protein expression is not only effective but particularly well suited for introducing Hfl into large proteins—in our study, amyloid fibrils—that can be difficult to synthesize using other approaches, such as SPPS or using auxotrophic bacterial strains. This method opens new avenues for studying protein structure and function with this highly fluorinated residue.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article and have declared no affiliation other than their research organizations.
Funding
This work has been supported by grants from Agence Nationale de la Recherche (ANR, ANR-20-CE06-0008), Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation (MESRI), and Région Nouvelle Aquitaine. They are gratefully acknowledged.
Acknowledgments
The authors thank the IECB Biophysical and Structural Chemistry Platform (BPCS), CNRS UAR 3033, INSERM US001, and University of Bordeaux, especially Estelle Morvan for NMR facilities and Dr. Brice Kauffmann for X-ray diffraction analyses. They thank Dr. Antoine Loquet and his team for the fruitful collaboration on cell-free synthesis of Hfl-containing amyloids.