

1 Introduction
Ferro- and ferrimagnetic materials have been receiving a growing interest for both technological and theoretical reasons, especially in the context of magnetic recording. However, the properties of finely divided magnetic materials closely depend on the size of the particles and their state of dispersion and aggregation 〚1〛. It is therefore very important to carefully control the synthesis of particles and their surface state.
Various chemical techniques 〚2〛 for controlling the particle size consist in limiting the space available for the particle growth, by precipitating ions in microemulsions, vesicles or polymer solutions. However, such methods raise difficulties in getting out particles free from polymer, surfactant or ligands mostly inducing surface effects influencing the magnetic behaviour of the particles. Other techniques often used are gel impregnation 〚3〛 and sol–gel methods 〚4〛, involving aqueous metal ions and subsequent thermal treatment. In these conditions, well-defined materials regarding the crystal structure, the mean particle size, the size distribution, the morphology and the state of dispersion appear very difficult to obtain, because it is difficult to control the reactions.
Precipitation or coprecipitation of cations in aqueous solutions is an easy and cheap route of synthesis of metal oxide particles 〚5〛. Thus, spinel iron oxides (magnetite Fe3O4, maghemite γ-Fe2O3 and substituted magnetites, MFe2O4 with M = Fe, Co, Ni...) are easily formed by coprecipitating Fe3+ and M2+ ions. However, the physicochemistry of the systems have to be carefully controlled to adjust the characteristics of particles. Our studies showed that the particle size can actually be calibrated over one order of magnitude in the nanometer range, and that the aqueous dispersions of maghemite particles can be used to elaborate a variety of nanocomposites where the particle dispersion is controlled.
2 Synthesis and tailoring of iron oxide nanoparticles
Magnetite is easily obtained by coprecipitating aqueous Fe3+ and Fe2+ ions. At the stoichiometry Fe2+/Fe3+ = 0.5, crystallisation of spinel phase is quasi-immediate at room temperature. The mean particle size is monitored by the conditions of the medium: pH and ionic strength, I, imposed by a salt. Typical experimental conditions 〚6, 7〛 are: 2.5 ml of a solution containing a mixture of FeCl2 (1 mol l–1) and Fe(NO3)3 (2 mol l–1) were slowly, automatically injected into 25 ml of a solution of NaNO3 at a concentration in between 0.5 and 3 mol l–1. The pH of the medium was fixed and kept constant (ΔpH ≈ 0.1) by addition of NaOH, using an automatic potentiometric device. All solutions were carefully de-aerated with nitrogen, which was continuously bubbled during the precipitation. Ageing of the suspensions was achieved under argon atmosphere. Various samples were prepared in the range 8.5 ≤ pH ≤ 12 and 0.5 ≤ I ≤ 3 mol l–1).
Electron micrographs (Fig. 1) show spheroidal particles whatever the synthesis conditions, but with very different mean sizes. X-ray Diffraction (XRD) patterns (Fig. 2) are typical of the spinel phase. The line positions are the same for all samples, indicating no significant structural variation between the samples.

Transmission Electron Micrographs (TEM) of particles synthesised under various conditions: (a) pH = 9, I = 0.5 mol l–1 〚D = 12.5 nm; σ(D) = 2.5 nm〛; (b) pH = 12, I = 0.5 mol l–1 〚D = 6.5 nm; σ(D) = 1 nm〛; (c) pH = 12, I = 3 mol l–1 〚D = 1.6 nm; σ(D) = 0.2 nm〛 (T = 25 °C ; 8-day aged suspensions).
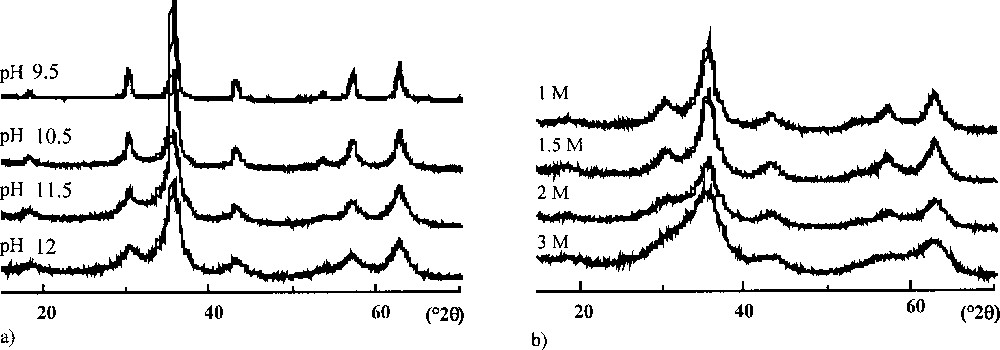
X-ray diffraction patterns (λCu Kα = 1.5406 Å) of particles synthesised (a) at different pHs with I = 1 mol l–1 and (b) at pH = 12 with different ionic strengths (T = 25 °C; 8-day aged suspensions).
Fig. 3 shows the influence of the precipitation conditions on the mean size of the particles after 8-day ageing. The mean size is strongly dependent on the acidity and the ionic strength of the precipitation medium, and clearly decreases with increasing pH and I.

Mean diameter of particles after 8-day ageing (a) as a function of pH (NaNO3 1 mol l–1) and (b) as a function of ionic strength (pH = 12) (T = 25 °C).
It is interesting to observe that such an influence of acidity on the particle size is relevant to thermodynamics rather than kinetics (nucleation and growth processes) 〚7, 8〛. The pH acts on protonation–deprotonation equilibriums of surface hydroxylated groups and, hence, on the electrostatic surface charge. The ionic strength carries out more or less electrostatic screening between charged groups and, then, at fixed pH, the electrical charge increases with I. Consequently, a change in the surface electrical charge due to pH leads to a change in the chemical composition of the interface, inducing a decrease of the interfacial tension, γ, as stated by Gibbs’s law, dγ = –Γi dμi, where Γi is the density of adsorbed species i with chemical potential μi. Finally, the surface contribution, dG = γ dA (A is the surface area of the system) to the free enthalpy of the formation of particles is lowered, allowing the increase in surface area of the system 〚9〛.
Maghemite γ-Fe2O3 does not form directly in solution by precipitation of ferric ions, but a small proportion of Fe2+ (x ≥ 0.1) induces the crystallisation of all the iron to spinel. Studies of the early precipitate revealed that all Fe2+ ions were incorporated into a Fe2+-ferrihydrite and gave rise to high electron mobility, as evidenced by Mössbauer spectroscopy 〚10, 11〛. This mixed valence material transforms with time into spinel by two simultaneous competing pathways: (i) solid state reaction with dehydration and spinel ordering at short range without particle size variation; (ii) dissolution of Fe2+–Fe3+ complexes from the surface, followed by crystallisation of spinel oxide (Ostwald ripening), this process taking place with considerable particle growth. The endproducts of the coprecipitation are single phase only for x ≥ 0.33, process (ii) seeming dominant during the evolution from the initial stage of precipitation, as noticeable growth of particles is observed.
The comparison with the cases where M2+ is different from Fe2+ emphasises the role of electron mobility between Fe2+ and Fe3+ ions in the crystallisation process. Mobility in the bulk presumably drives a local cubic close-packed ordering, made possible by the loose structure of the early material. This local ordering gradually extends to the whole fine particles (process (i)) and to the growing particles by process (ii). With the other divalent cations, intervalence transfers are generally negligible and a spinel ferrite forms only by process (ii) (dissolution–crystallisation) 〚11〛. Nevertheless, a control of particle size is possible on a wide range, because the solubility of the solid is strongly dependent on the pH of the medium 〚12〛 (Fig. 4).
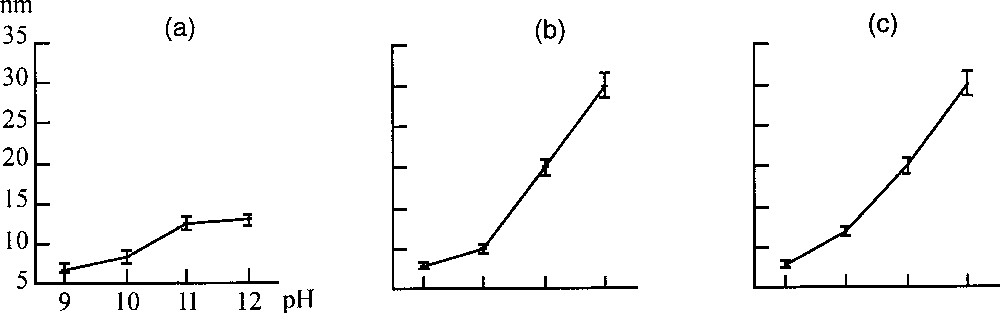
Mean diameter of particles of (a) CoFe2O4, (b) MnFe2O4, (c) NiFe2O4 after 24-h ageing at 100 °C of suspensions as a function of pH.
3 Oxidation of magnetite particles. Formation of aqueous sols of maghemite
Due to the high electron mobility in the bulk, the magnetite nanoparticles are very unstable against oxidation, leading, at term, to maghemite. Different mechanisms 〚13〛 involving ionic and/or electronic interfacial transfers are implicated, depending on the pH of the suspension. In basic medium, the oxidation of Fe2+ ions proceeds by oxygen reduction at the surface of particles (electron transfer only) and coordination of oxide ions, while in acidic medium and anaerobic conditions, surface Fe2+ ions are desorbed as hexaaquo complexes in solution (electron and ion transfer) according to:
Maghemite nanoparticles, bearing a high positive charge density (σ ≈ 0.3 C m–2 at pH 2, and low ionic strength, 10–2/5.10–2 mol l–1), can easily be dispersed in acidic water, forming cationic sols practically free from aggregation 〚14–16〛. On raising the pH, σ decreases, the repulsions between particles weaken, the degree of agglomeration increases. Aggregation remains limited provided that pH < PZC. The aggregate configuration, dependent on the pH, ranges from small chain-like clusters of a few particles to branchy chains of more than ca 50 particles. In every case, the structure is loose and the particles are not in close contact. At pH ∼ PZC, σ ∼ 0, a floc forms. At pH > PZC, the particle re-disperse, but aggregation is generally more significant than at pH < PZC, because the surface is less solvated.
4 Dispersion of maghemite nanoparticles in polymers and silica glasses. Nanocomposites
The particles synthesised and dispersed in aqueous medium can be trapped in different solid matrices keeping their dispersion state.
By adding a hydrosoluble polymer in the sol and drying of the mixture, solid samples were obtained. The oxide/polymer mass ratio determines the average distance between the objects existing in the sol, isolated particles or aggregates. This technique was used to prepare series of composites made up of maghemite particles with the same size distribution and different aggregation/dispersion states in polyvinylic alcohol, typically isolated particles with volume concentration ranging from ∼1 to 20%, clusters of varying length and flocs, in order to study the magnetic properties of noninteracting particles and the interparticle interaction effects 〚1, 15, 17–19〛. Such materials allowed us to give the first demonstrative experimental verification of the Néel–Brown model for superparamagnetic relaxation 〚17〛
Composites made up of well-dispersed maghemite particles in an epoxy resin were obtained by polymerisation of the resin inside an organosol of particles 〚20〛. The transfer of particles from aqueous medium to an organic solvent (sulfolane, propylene carbonate, hexamethylphosphorus-triamide) needs the adsorption of a coupling agent such as a phosphonic acid (phenylphosphonic acid, PPA). The head of the molecule, the phosphate group, strongly adsorbs onto particles, as evidenced by infrared and Mössbauer spectroscopies. The room temperature Mössbauer spectra of uncoated and coated particles are shown in Fig. 5. The quadrupole doublet observed for the coated particles demonstrates the formation of a paramagnetic surface complex, due to the strong complexing ability of PPA for iron 〚21〛.
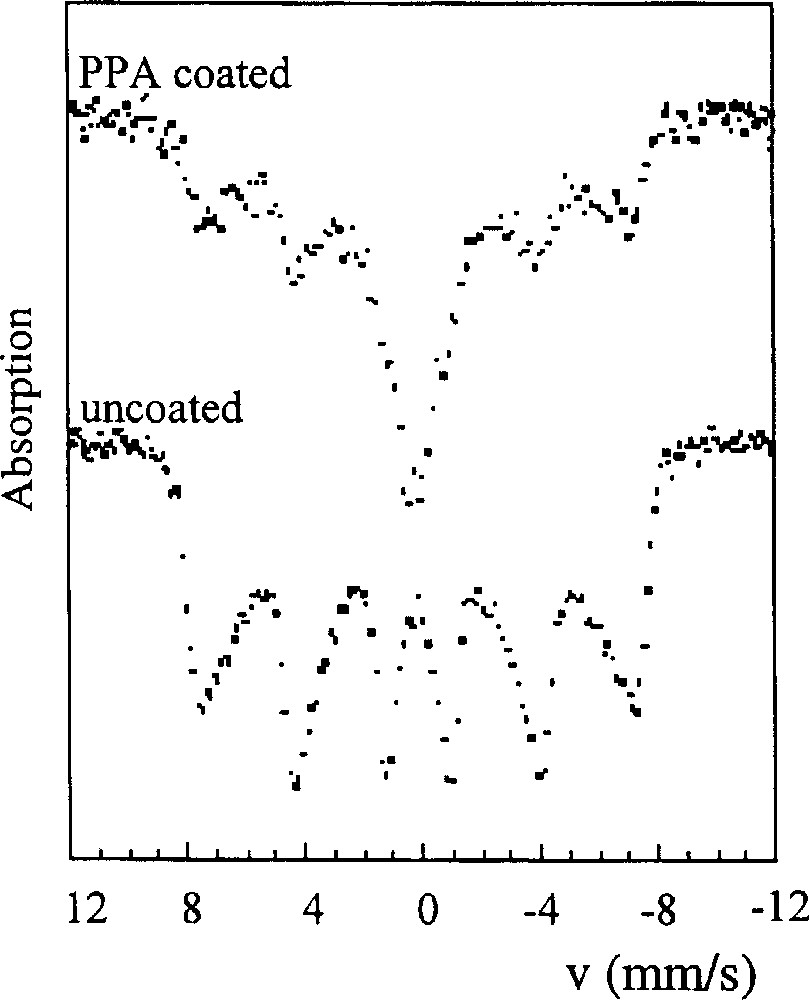
Mössbauer spectra (at room temperature) of uncoated and PPA coated maghemite particles (9.0 nm).
The hydrophobic tail (phenyl group) of the coupling agent allows the dispersion in the organic solvent and prevents aggregation of the particles forming a kinetically stable organosol, as indicated by light scattering. The components of CIBA–GEIGY epoxy resin (araldite LY556, hardener HY917 and accelerator DY070) introduced in the sol form a homogeneous hybrid composite material after heating at 60 °C for 24 h. The materials were examined as thin slices by transmission electron microscopy (Fig. 6).
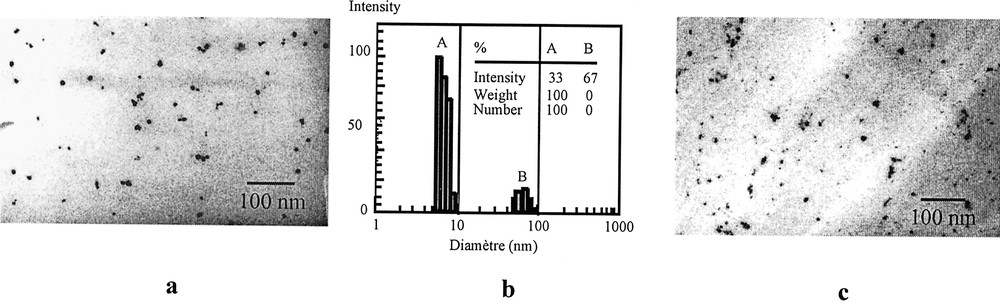
Transmission electron micrographs (a) and quasi-elastic light scattering observation (b) of maghemite particle (4.7 nm) dispersed in hexamethylphosphorus triamide; micrographs of magnetic composites (4.7 nm maghemite particle-epoxy resin, thin slice) (c).
Silica composites were prepared by polymerising silicic acid or alkoxysilanes inside the aqueous sol 〚22, 23〛. Hydrolysis and condensation of the precursors take place in situ and the reactions yield a gel that leads, after drying at room temperature, to a transparent monolithic glass. Such a procedure leads to homogeneous solid composites (Fig. 7), containing up to 40 wt% Fe2O3. In both cases, infrared, near-infrared and Mössbauer spectroscopies indicate no detectable Fe–O–Si bond. In fact, the ‘dispersability’ of particles into the silica matrices results from solvation of silanol groups of the matrix by associated-water layers surrounding the particles, without other chemical surface interactions. The silica matrix acts as an antisintering agent, which stabilises the maghemite particles against the thermal transformation into hematite. Whereas with powdered uncoated particles, the γ → α-Fe2O3 transformation starts below 300 °C and is generally complete at 400 to 500 °C, in composites with sufficiently low particle concentration, no transformation occurs as long as the matrix prevents the migration and coalescence of particles that is, up to around 1000 °C, the temperature at which the glass softens or starts crystallising 〚24〛.
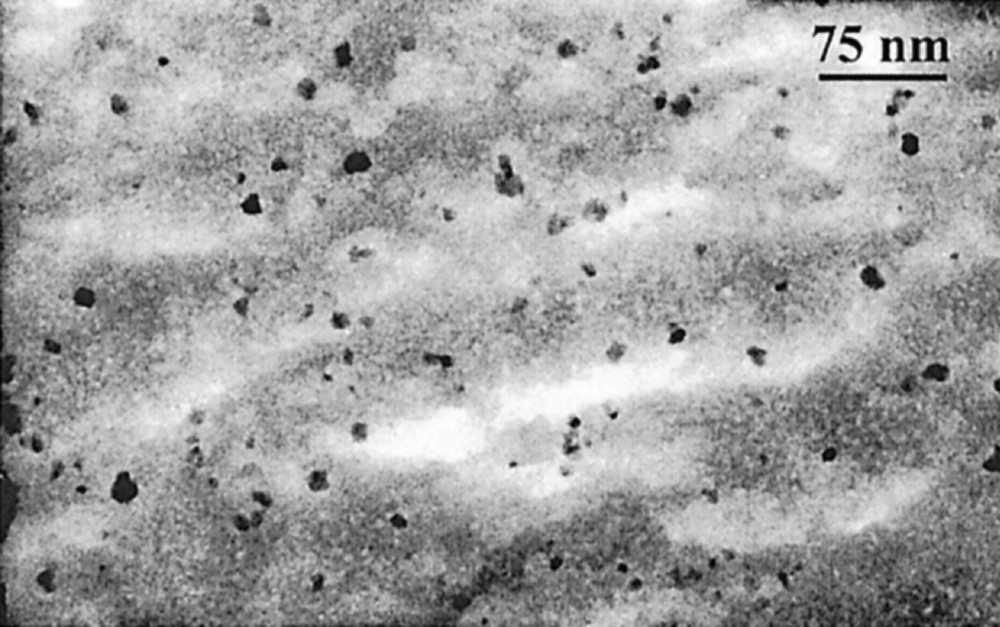
TEM micrograph of a maghemite–silica composite (18 wt% Fe2O3).
5 Conclusion
The aqueous chemistry of spinel iron oxide nanoparticles offers broad possibilities for tailoring materials for a wide range of utilisation. The careful control of the size and degree of dispersion of the particles in composite materials can allow one to reveal unexpected phenomena.
Acknowledgements
We are deeply grateful to our magnetician colleagues, M. Noguès, LMOV, University of Versailles, France, D. Fiorani, IC-Mat, CNR, Monterotondo Staz., Italy, F. Lucari and F. D’Orazio, Università L’Aquila, Italy, for appreciating our materials and to M. Lavergne, UPMC, Paris, France, for electron microscopy observations.