

1 Introduction
The arborescent dendritic structure is one of the most pervasive, prolific, and influential natural pattern that can be observed on earth, at all dimension length scales (from nm to km), at once in the inert, the virus and the living worlds [1,2]. Such a natural hyperbranched architecture has reached an unrivalled level of perfection and provides maximum interfaces for efficient contacts and interactions, as well as for optimum information collection, transport and distribution. The elaboration and the synthesis of such aesthetically-challenging architectures have been driven by the need to mimic the macroscopic natural branching networks and to convey their functions at the molecular level. Dendrimers and dendrons (or elementary dendritic units) may be considered as polymers with geometrically-restricted structures [2].
These compartmentalized and hierarchical artificial supermolecules possess a regular (monodisperse) and controlled three-dimensional branched topology, the result of sophisticated genealogical directed syntheses. Their construction is based on controlled and sequential reiterative synthetic elementary steps which are reproduced at each new generation (convergent or divergent sequential construction): at each new cycle, the molecule is incremented by an additional generation or shell [2]. The ultimate dendritic architecture can be finely tuned by the “intrinsic dendritic connectivity” comprising the multiplicity of the core (NC), the degree of branching (NB), and the number of junctions (J) and generations (G), and also, to some extent, by the chemical nature of the dendrimer, the type and density of the functional peripheral groups, and their coupling to the dendritic scaffold. This leads to hyperfunctionalized systems with a large number of peripheral groups (Z) per volume unit (high density), which in the specific case of end functionalisation increases geometrically as Z = NCNBG. This starburst growth is, however, not infinite, essentially for steric reasons, the so-called congestion effect as predicted by de Gennes in the early eighties [3], and, as a consequence, beyond a certain generation level, the branching, defined by J, is no longer regular, nor is the number of end-groups, Z: the creation of structural defects is accelerated and may occur at lower generation when the degree of branching (NB) is high, since J increases exponentially with NB and G according to J = NC[(NBG − 1)/(NB − 1)].
2 Liquid-crystalline dendrimers
Molecular engineering of liquid crystals is an important issue for controlling the self-assembling ability and the self-organizing processes of single moieties into periodically ordered meso- and nanostructures [4]. Moreover, ordered supramolecular assemblies can considerably enhance the functions of the single molecules [5]. Dendrimers, dendrons, dendronized and hyperbranched polymers have proved particularly to be versatile candidates as novel and original scaffoldings for the elaboration of new LC functional materials and research in this area has experienced an outstanding development during the last years, overlapping polymer chemistry and supramolecular chemistry [6]. In particular, the dendritic structure appeared as an interesting framework where mesomorphism can be modulated by very subtle modifications of the intrinsic dendritic connectivity. LC dendrimers are now representative of an important class of mesogens where new types of mesophases and original morphologies may be discovered [6].
The most commonly studied LC dendrimers are the so-called peripherally attached end-group supermolecules. Their overall structure generally consists of mesogenic or pro-mesogenic promoters attached to the termini of the branches of the flexible dendritic network; these units may be terminally (end-on, Fig. 1a) or laterally (side-on, Fig. 1b) attached, with such an orientation of attachment having dramatic consequences on the mesomorphic properties. For end-on systems, induction and stabilization of mesophases result from both the anisotropic interactions, winning over the tendency of the dendritic core to adopt a globular, isotropic conformation, and the microphase separation, due to the chemical and structural incompatibilities between the flexible dendritic core and the terminal groups. The decoupling between both moieties (e.g. spacer length, topology of attachment) also plays an important role, as this is the case for the side-on systems which mainly display a nematic phase [6]. These dendrimers likely adopt constrained conformational structures consequently to the occupation of specific regions by the different, incompatible, molecular parts (segmented molecular structures) in order to generate the most stable and favorable supramolecular organization. The type of mesogen, the supermolecular architecture, the dendritic chemical nature and connectivity (Fig. 1) therefore strongly determine the mesomorphic properties of the entire supermolecule, and all these structural parameters can be adjusted independently for an optimum control of the properties [6].
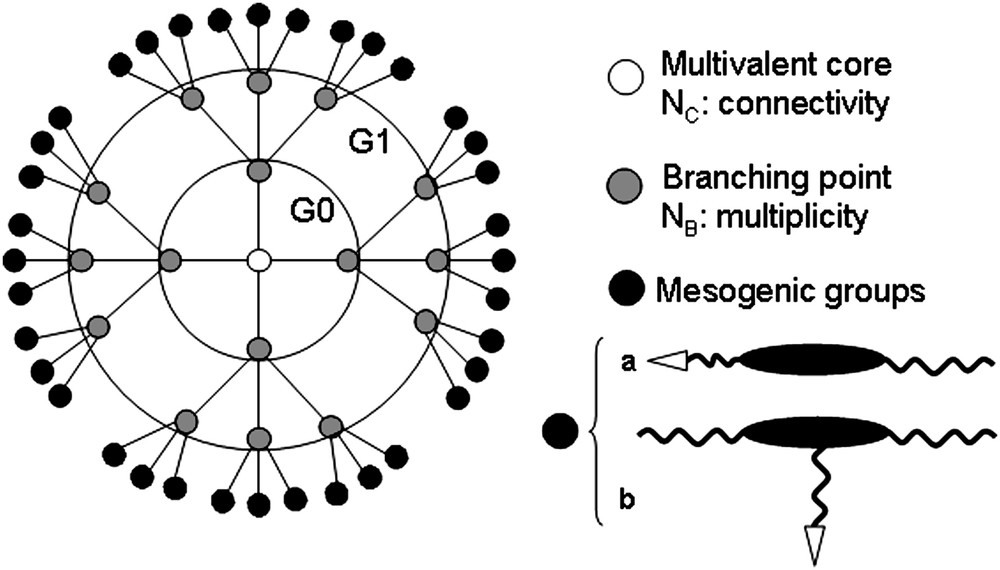
Schematic 2D representation of a side-chain LC dendrimer (NC = 4, NB = 3, G = 1). The mesogen is terminally (a) or laterally (b) connected.
3 Main-chain liquid-crystalline dendrimers
The insertion of rigid and linear segments within such scaffolds leads to another class of potentially interesting dendrimers, the so-called main-chain systems. Regarding the structure of these dendrimers, the junctions are no longer single and spherical atoms (C, N, P, Si, etc.) but consist of anisotropic molecular moieties instead. These units are linked together through long and flexible alkyl spacers, generating therefore the dendritic matrix. The differentiation between side-chain (peripherally-attached) and main-chain occurs at the first generation onwards; furthermore, for equivalent generations, the main-chain systems contain a larger number of functional units (Z∗ = Z + J) than side-chain LC dendrimers (Z). Therefore, an amplification of the properties is expected either by additive effects (sum of the properties of the individual elements of a single-component system) and/or by synergy (combination or association of the properties of different elements of a multi-component system) (Fig. 2). As far as self-organization into mesophases is concerned, the anisotropic groups present at every level of the dendritic hierarchy are responsible for some loss of conformational freedom: the dendrimers are forced to adopt more constrained molecular conformations than their side-chain homologues with their more flexible and deformable core, and the anisometric branches do not radiate isotropically, but, on the contrary, favor preferentially an anisotropic order at an early stage of the organizing process by a gain in the enthalpy of the system, in order to produce the most stable structure. These main-chain dendrimers may therefore represent an interesting alternative for the development of novel LC molecular materials.
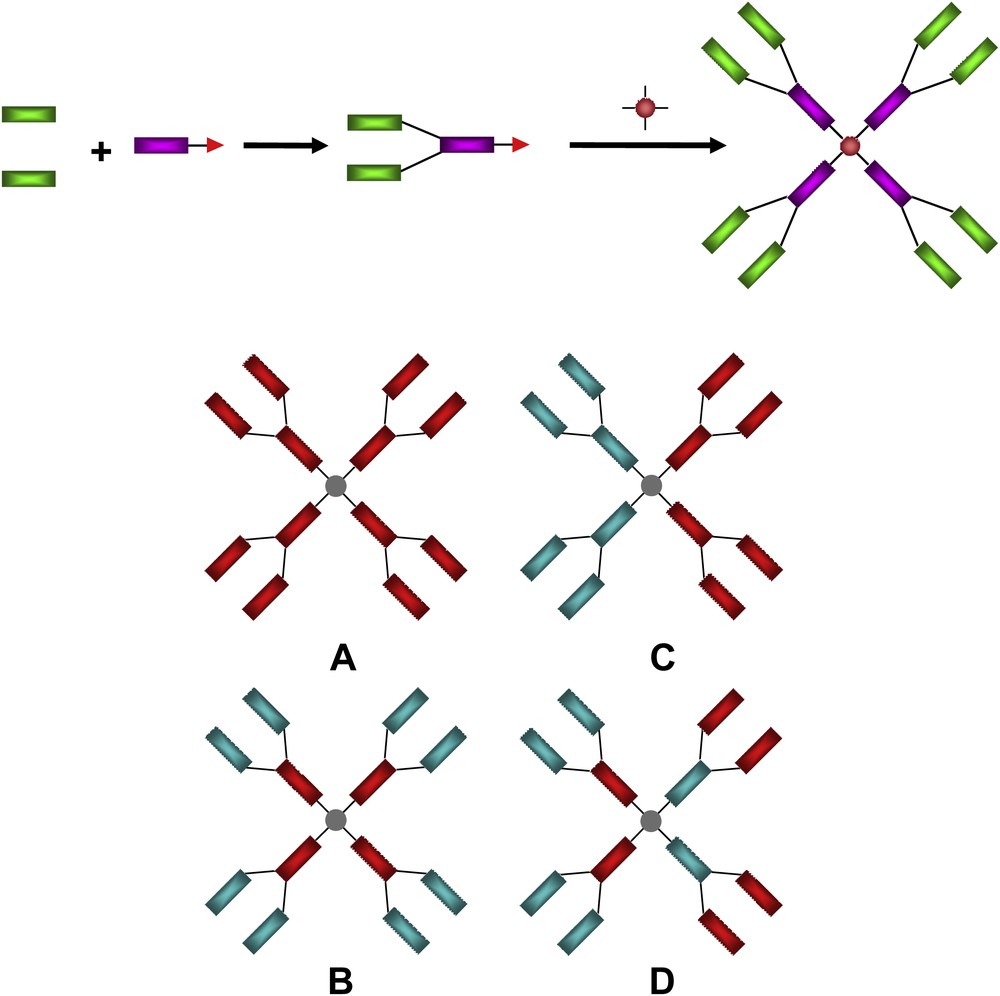
Schematic representation of the modular construction of a G1 octopus dendrimer (top), and a selection of some of the various possible molecular architectures that can be envisaged: symmetrical dendritic structures with homolithic (A), heterolithic alternated (B) and segmented (C, D) topologies (bottom).
3.1 Octopus LCs
Up to now two families of such systems have been identified, namely the willow-like and the octopus dendrimers. The so-called willow-like dendrons and corresponding dendrimers based on terphenylene monomeric AB2–type units, were first reported and showed mainly enantiotropic N and smectic phases, depending on some specific structural features [7]. As for the octopus dendrimers, they result from the coupling of segmented and symmetrical dendritic branches containing mesogenic moieties at each generational level (junction) onto a small tetra-podand core (Fig. 2). The modular construction elaborated for their synthesis (independent construction of individual blocks prior to their covalent connections according to a predefined framework) allowed a wide range of possible interesting structural combinations to be envisaged for the elaboration of original multicomponent supermolecules, including homolithic (Fig. 2, one type of building block, A) and heterolithic (Fig. 2, several different blocks, B–D) dendrimers with alternated and/or segmented supermolecular architectures.
In our first study, the anisotropic units selected were tolane- and stilbene-based moieties, because of both their excellent thermal stability and chemical versatility. Homolithic and alternated heterolithic octopus dendritic materials (Fig. 3, 1) bearing these units within the framework were synthesized by an efficient modular synthesis. It consisted of the preparation of the individual anisotropic segments by Sonogashira and Heck Pd-catalyzed cross-coupling reactions, respectively, for tolane- and stilbene-based systems [8], followed by their selective association into precursory dendritic branches, and their final connection to a central tetra-podand amino-core unit (derived from polypropyleneimine: PPI). Such poor mesogenic segments, despite respecting structural requirements for mesophase induction (adequate intrinsic anisotropy and rigidity), were chosen on purpose, in order to evaluate the “mesogenic amplification effect” by means of additivity and/or synergy between the individual components forming these novel structures, i.e. whether mesomorphism could be induced solely by the dendrimerization process. Thus, a number of homolithic (Fig. 3, 1, X = Y) and alternated heterolithic (Fig. 3, 1, X ≠ Y) octopus dendrimers were synthesized, further differing in the number of terminal chains (Fig. 3, 1, R1, R2, R3 = H or OC12H25) [9].
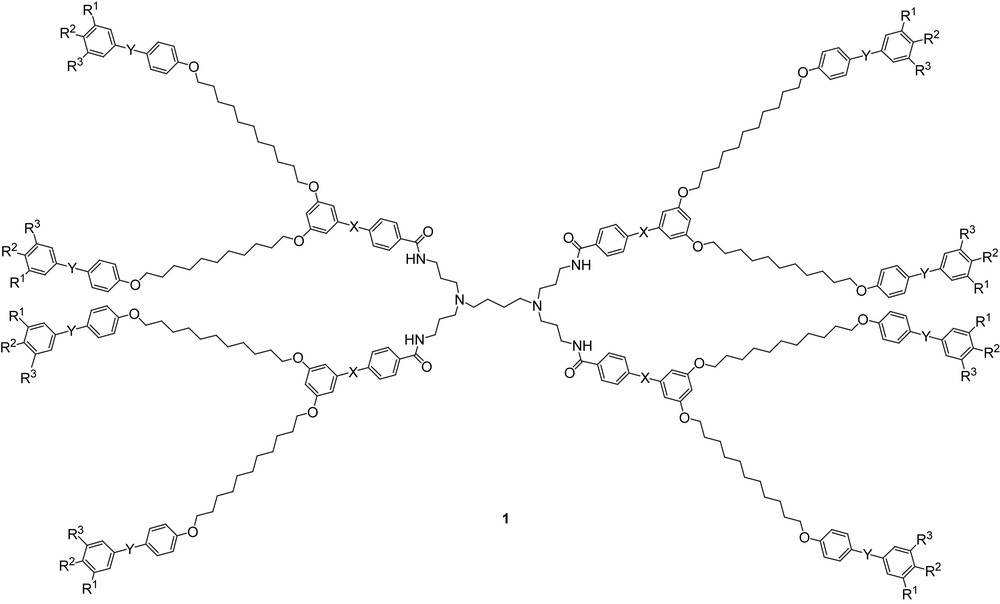
G1 octopus dendrimers (1: X, Y: =/≡; R1, R2, R3 = H/OC12H25).
We showed that all these dendrimers were mesomorphic and that the mesophases structures were solely dependent on the ratio number of terminal chains/end groups [9]. The dendrimers bearing one terminal aliphatic chain per peripheral unit (Fig. 3, 1, X: = /≡ and Y: ≡, R1 = R3 = H and R2 = OC12H25) exhibited smectic phases (homolithic tolane derivative: SmB 101 SmA 121 I; heterolithic derivative: SmB 109 SmA 132 I); in contrast, none of the precursory branches was mesomorphic. The mesomorphic behaviour was explained by the elongated (prolate) molecular conformation adopted by the dendrimers. Indeed, the lamellar periodicities were rather large (10–12 nm), consistent with a fully stretched molecular conformation, and with the mesogenic groups homogeneously distributed on both sides of the tetravalent core [9b]. The formation of the smectic phases resulted therefore from the parallel disposition of these giant rod-like supermolecules into layers. In this case, the structure of the smectic phases is quite unique and consists of a highly segregated, subleveled, molecular organization made of an internal sub-layer containing tilted rigid segments (segments of the first generation, randomly tilted to compensate the molecular area), flanked by outer slabs inside which the mesogenic groups are arranged perpendicular to the layer (Fig. 4); these various sublayers are separated by the aliphatic continuum. Molecular modeling supports this view of strongly segregated multilayer structures, with interfaces between the various molecular parts. Obviously, these interfaces are not so well defined due to thermal fluctuations. Nevertheless, let us point out that because of this peculiar structural feature, such layered mesophases cannot exactly be described as purely SmA or SmB phases, and were referred to as “supersmectic” phases.
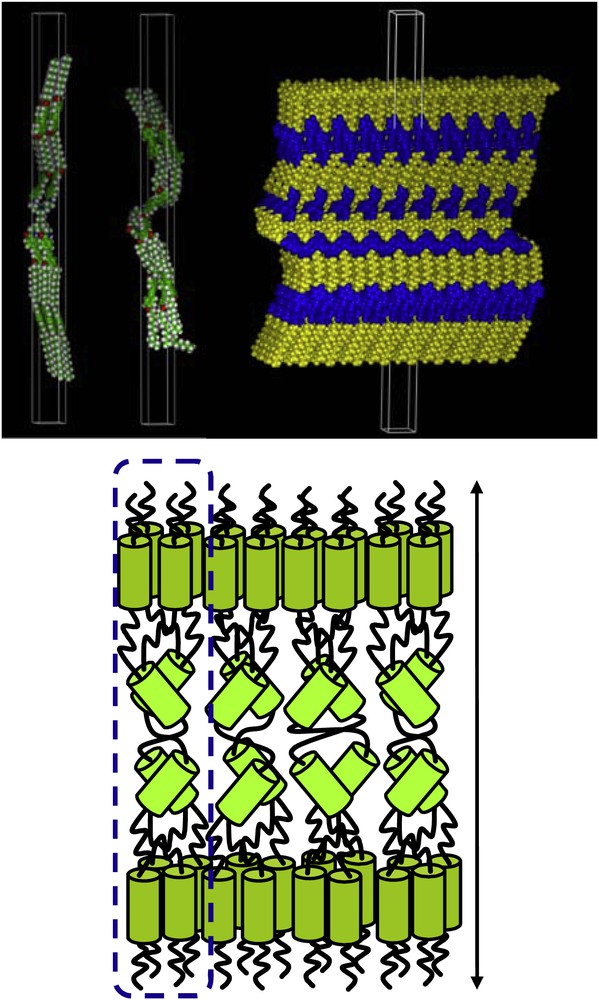
Modeling and schematic representation of the multilayer smectic phase structure formed by 1.
In contrast, the increase of the chain/end-group ratio prevents such a parallel disposition of the pro-mesogenic groups, and the dendrimers bearing two or three aliphatic chains per outer tolane or stilbene unit (Fig. 3, 1, X,Y: =/≡, R1 = R2 = H and R3 = OC12H25, R1 = R3 = H and R2 = OC12H25, or R1 = R2 = R3 = OC12H25) formed systematically a Colh phase [9]. The formation of these columnar superstructures is a consequence of the mismatch between the surface areas of the aromatic cores and the cross-sections of the aliphatic chains, resulting in the curvature of all the interfaces, as in polycatenar liquid crystals [10] and dendromesogens [11]. Consequently, the octopuses adopt a wedge-like conformation (or cone-like), authorized by the great flexibility of the G0-PPI core (zeroth generation polypropyleneimine), allowing the pro-mesogenic groups to be radially distributed with uniform interfaces at the different generational levels. With such a folded or fan conformation, the dendrimers self-assemble into (supra)molecular discs or columns which further self-organize into a hexagonal net (Fig. 5, top). Depending on the number of terminal chains per terminal mesogenic units (2 or 3), 3 or 2 folded dendrimers, respectively, can perfectly pave the hexagonal lattice, consistent with the hexagonal parameters (9–10 nm). Considering the diblock, alternated chemical nature of these dendrimers, a leek morphology for the columns is most likely (intracolumnar segregation) (Fig. 5). This model of a multi-segregated internal structure of the columns was supported by molecular dynamics on the homolithic stilbenoid system (Fig. 3, 1, X = Y: = ; R1 = R2 = OC12H25, R3 = H). It showed good segregation at the molecular level by means of interlocked concentric crowns of stilbenoid units belonging to the same generation, such crowns being further stabilized by intermolecular interactions. Each crown is separated by “inert” aliphatic films (Fig. 5).

Self-assembling and self-organization process into columns (top) and molecular dynamics snapshot of the hexagonal phase (bottom) for the octopus dendrimers 1. Inset: folded dendrimer into the wedge shape.
The mesophase stability was found to greatly depend on the disposition of the terminal chains attached onto the peripheral groups (positions 3,5-, 3,4- and 3,4,5-) and, for the heterolithic systems, on the precise localization of these various units (tolane/stilbene), and thus of their respective proportion, within the branched scaffolding (Fig. 6). Thus, the homolithic stilbene octopuses are all mesomorphic. Whilst systems with the 3,5- and 3,4,5-chain-substitution per terminal units exhibit a room temperature Colh phase, the derivative with the 3,4-chain substitution exhibits a much higher phase stability, and is crystalline up to ca 100 °C, before self-assembling into the Colh mesophase (Fig. 6, top). Interestingly, mesomorphism was induced in the latter compound while its corresponding precursory branch was deprived of mesophase, in contrast to the other branches (with the chain' substitution 3,5- and 3,4,5- both having a room temperature Colh phase). Note also the substantial decrease of the Colh phase lattice parameters in the case of the dendritic branches with respect to that of their parent octopuses (15–20 Å smaller), resulting in a lesser number of free branches (about 6 with respect to 8–10 i.e. 2–2.5 folded dendrimers) to sufficiently fill an elementary stratum of column 4.5 Å-thick [9a]. The proportion and localization of those groups within the dendritic skeleton have also dramatic effects on the mesophase stability; here only the systems with the 3,4-chain substitution were considered. A great enhancement of the Colh mesophase stability was clearly observed on increasing the number of stilbene units at the expense of the tolane ones (Fig. 6, bottom). This illustrates rather nicely the strength of the intra- and intermolecular interactions within the octopuses (and thus the different types of mesogenic units and their respective position within the dendritic frame), the higher the number of stilbene groups, the more stabilized the dendritic wedge conformation, and thus the column. Quasi similar observations were made with the non-mesomorphic corresponding acidic dendritic branches, for which the melting temperatures were found to increase from the pure tolane to the pure stilbene system. Interestingly, only one of these branches was mesomorphic, namely the heteroltithic one with two stilbene groups in the periphery (Fig. 3, 1, X: ≡, Y: =, R1 = R2 = OC12H25 and R3 = H).
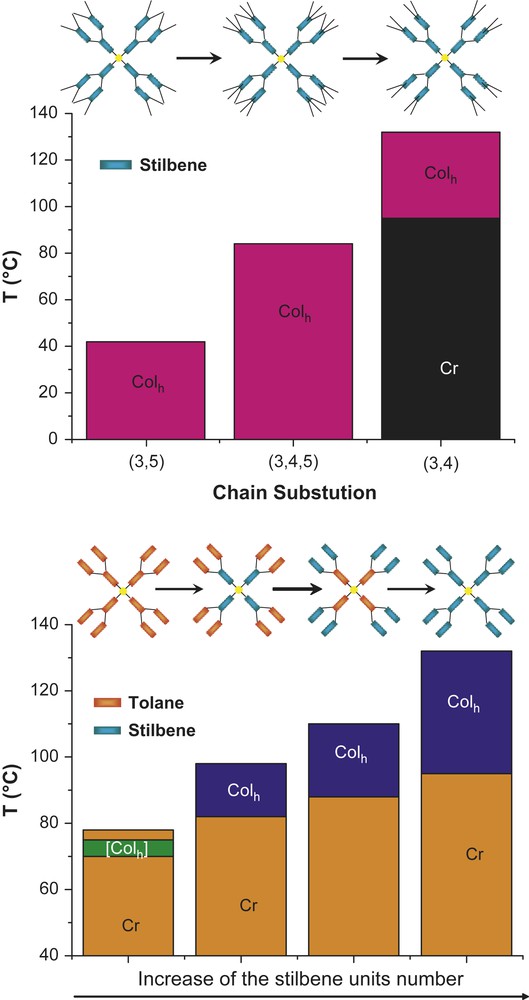
Phase diagrams showing the effect of the chain position (top, 1: R1, R2, R3 = OC12H25 or H) and the evolution of the mesophase stability from tolane-rich to stilbene-rich octopus dendrimers (bottom, 1: R1 = R2 = OC12H25, R3 = H).
In this study, new potentially challenging LC materials, possessing a segmented block structure have been synthesized, and it was shown that straightforward structural modifications of the dendritic scaffold or the change in the chains substitution pattern drastically modify the mesomorphism of these dendrimers. These variations permit to better control and tune the mesophase types and their stability, potentially interesting in the design of specific multi-component, polyfunctional materials for technological applications.
Several structurally related octopus dendrimers bearing longer oligo(phenylene–vinylene) terminal groups were also prepared in order to combine luminescent properties with self-organisation (Scheme 1) [12]. For the dendrimer shown in Scheme 1, the terminal OPV3 unit was prepared by two consecutive Heck coupling reactions, and the dendritic branches (4–6) and the final dendrimer (7) were prepared according to the same procedures used for dendrimers 1 [9]; the only difference was in the activation of the acid to react with the PPI-G0 core, which was achieved by the formation of the corresponding pentafluorophenol-derived ester (6). Both the acidic branch (5) and the dendrimer (7) exhibit a Colh phase (G 47 Colh 93 I and Cr 90 Colh 145 I, respectively), with a great enhancement of the phase stability with respect to the homologous homolithic stilbene octopus 1 (Cr -3 Colh 84 I) and corresponding acid (Colh 52 I) (Fig. 3, 1, X, Y: =; R1 = R2 = R3 = OC12H25) [9b]. This phase stability increase is clearly due to the elongation of the terminal chromophore, and the consequent increase of intermesogenic interactions. Three of these dendrimers self-assemble together into a supramolecular disc of 5.7 Å thick, and the paving of the hexagonal lattice (a = 100 Å) is total; as for the dendritic branches, 12 of them fulfil the geometrical requirement of the hexagonal lattice (a = 98 Å). Both the dendron and dendrimer exhibit interesting luminescent properties in solution [12].
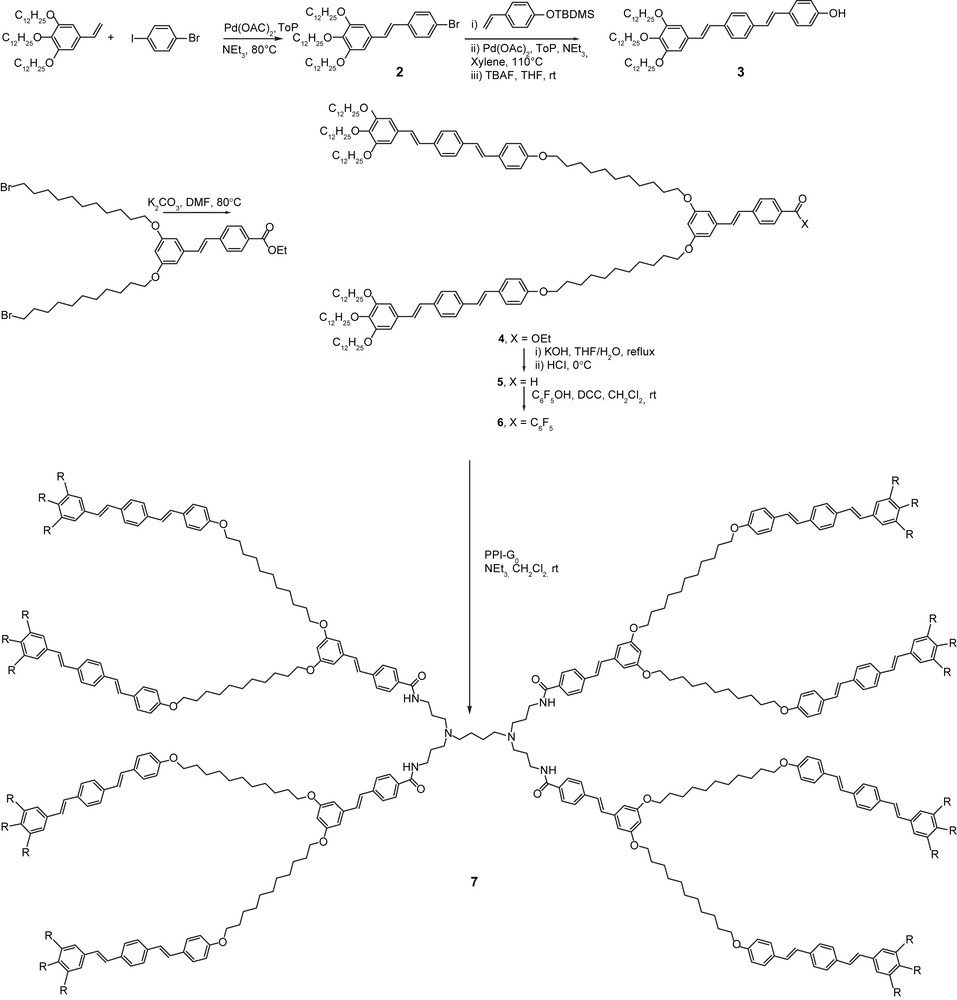
Preparation of the luminescent octopus 7 (R = OC12H25).
An octopus dendrimer based on a pentaerythritol core was prepared in order to test the effect of another type of core on the mesomorphic properties. Dendrimer 11 was prepared from an extended pentaerythritol core (10), as shown in Scheme 2. The coupling reaction between the dendron and this core was realized in dichloromethane using DMAP, CMC (1-cyclohexyl-3-(2-morpholinoethyl)carbodiimide) instead of DCC, and yielded the corresponding pure dendrimer although in rather low yield (25%). Unfortunately, mesophase formation was simply inhibited and a room temperature oily material was obtained instead. The absence of mesomorphism was credited to the relative rigidity of the tetrahedral pentaerythritol unit versus the deformable PPI one. The latter will force the branches to radiate isotropically to generate a protean structure unable to self-organize into a mesophase rather than the required wedge-like structure (the branch itself was mesomorphic, vide infra). The chemical nature of the core is therefore another important aspect in the design and the control of the properties of these octopus dendrimers. It can however be speculated that the grafting of a more anisometric monomeric units at the periphery of dendrimer 11, such as for example the OPV3 moiety instead of the stilbene one, would have likely promoted the formation and stabilization of a mesophase.
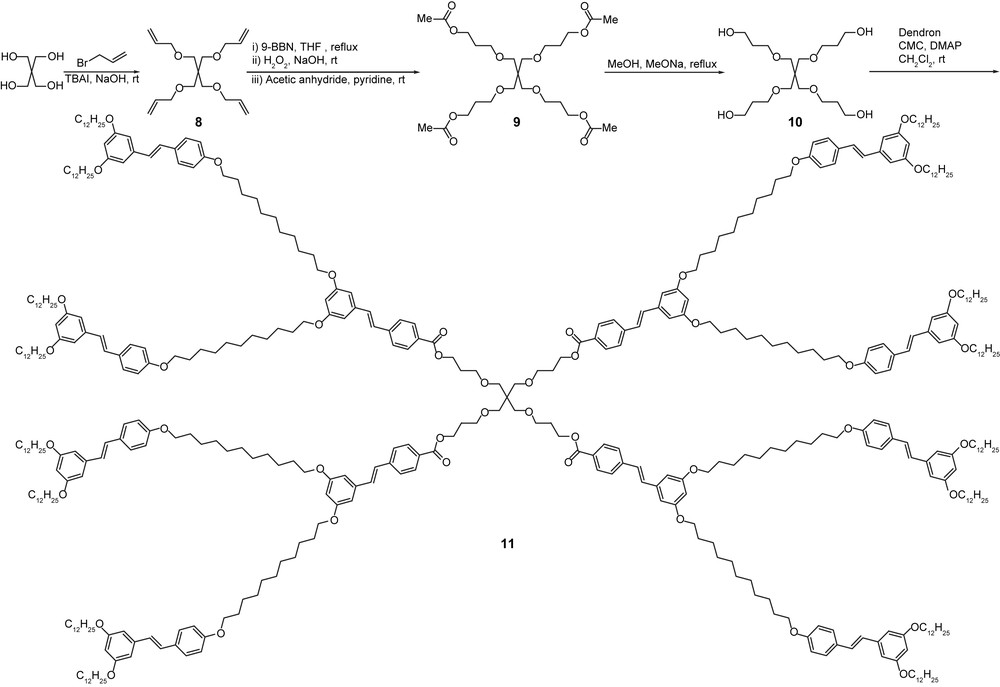
Octopus dendrimer with a pentaerythritol-based core.
There are other examples in the literature of pentaerythritol based-liquid crystals which support our observations that the pentaerythritol core is not such a judicious choice for designing liquid crystalline dendrimers due to restricted molecular topologies and limited molecular flexibility with respect to other G0 cores [6e]. However, providing that the peripheral units, preferentially grafted in the end-on configuration, are strong liquid-crystalline promoters (e.g. calamitic [13], discotic [14], or conical “DOBOB” [15] pro-mesogens), or that micro-segregation is enhanced by the insertion of amphipathic groups either between the core and the rigid units (e.g. silylated spacers [16]) or at the periphery (e.g. carbohydrate groups [17], fluorinated chains [18]), induction of stable mesophases (enantiotropic phases) is nevertheless observed. This core was considered as a potential source for the fabrication of non-symmetrical segmented dendrimers (Fig. 2, structures C and D) by regio-selective chemistry leading to a “selective multifunctionalization” of the pentaerythritol unit [16,19]. However, the synthesis of the symmetrical dendrimer was already found to be rather difficult itself, and this approach was later abandoned as totally inapplicable for the synthesis of Janus-like dendrimers.
3.2 Multipodal main-chain LCs
The effect of the core connectivity on the mesomorphism was also investigated [20]. Three main-chain dendrimers (with the same heterolithic branch, Fig. 7) were prepared from various amido-cores of zeroth generation and with different multiplicities: 1,4- diaminobutane (NC = 2), tris(2-aminoethyl) amine (NC = 3) and PAMAM-G0 (NC = 4); the latter was also prepared for the possibility to rigidify the core by additional hydrogen-bonds (amido groups) and enhance the mesomorphic temperature range as already observed with some side-chain LCDs [21]. They all exhibit a broad Colh phase (expected since there are two terminal chains per end-group) and the transition temperatures and mesophase stability were found to be influenced by the nature and the connectivity number of the core. In particular, the mesophase stability was greatly enhanced in the tetrapode 12 and the PAMAM derivative 14 (Fig. 7). Moreover, a stepwise increase of the hexagonal lattice parameter was correlated to the increase of the core connectivity (a = 81 → 86 → 95 Å for NC = 2 → 3 → 4, respectively). The mesophases are formed through the same process of self-organisation as described above. But since the overall number of peripheral chains per dendritic molecule is changed, and while the lattice parameter of the Colh phases increases concomitantly with the NC value, the number of dendrimers self-assembling within an elementary slice of column (same height) is therefore different; this number is deduced by the total number of radiating chains, varying from 36 to 48 throughout the series. Thus, within a slice of 4.6 Å, it was found that 4.5 molecular equivalents of 12, 3.3 of 13 and 3 of 14 (3 with the PPI-G0 core, Fig. 3, 1, X: =,Y: ≡, R1 = R2 = OC12H25 and R3 = H) were needed to completely fill the available volume.
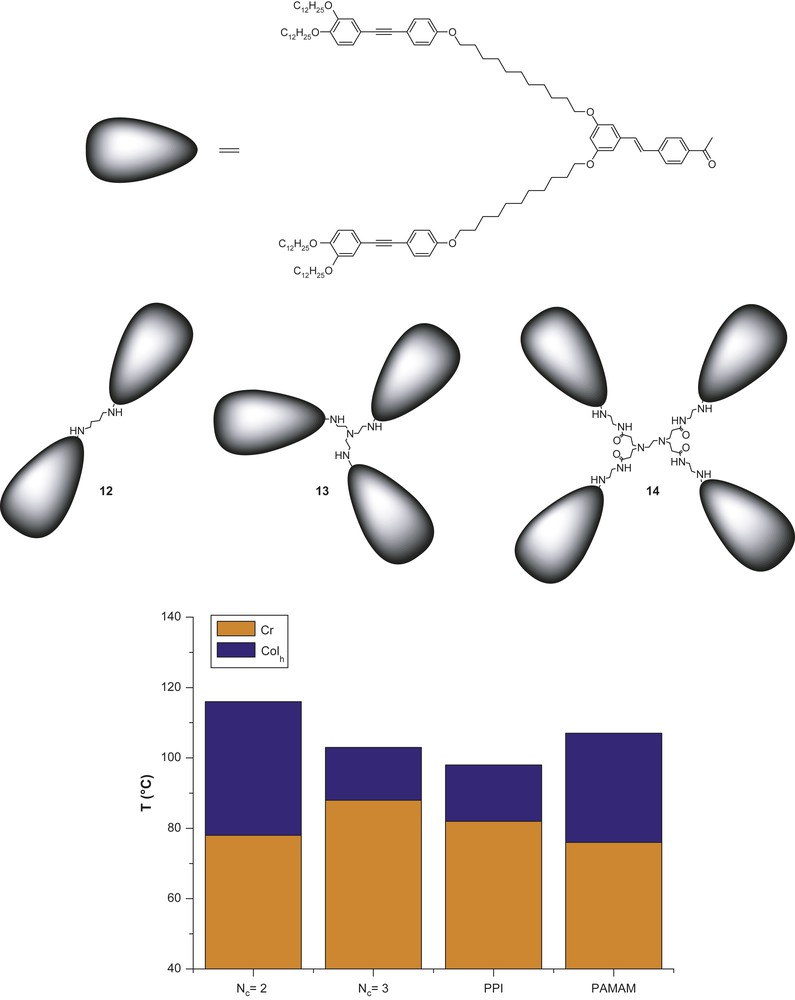
Evolution of the mesophase stability as a function of the core connectivity: NC = 2 (12) → 3 (13) → 4 (14); see Fig. 6 for the PPI derivative.
The next structural modification concerned the effect of the degree and toplogy of branching (NB) on the mesomorphic properties. A dendrimer with NB = 1 was prepared (Scheme 3, 18), whilst none with ternary branching (NB = 3) could be prepared, as due to strong steric constraints, the branches could not be grafted onto the core. Thus, following the same global methodology, 18 was prepared and obtained as a pure material. It exhibited a Colh phase (Cr 106 Colh 153 I). In this case, considering a similar mode of packing into columns (fan-shape conformation, self-assembling) about 6 tetrapodes (or molecular equivalents) associate to fill a slice of 4.5 Å.
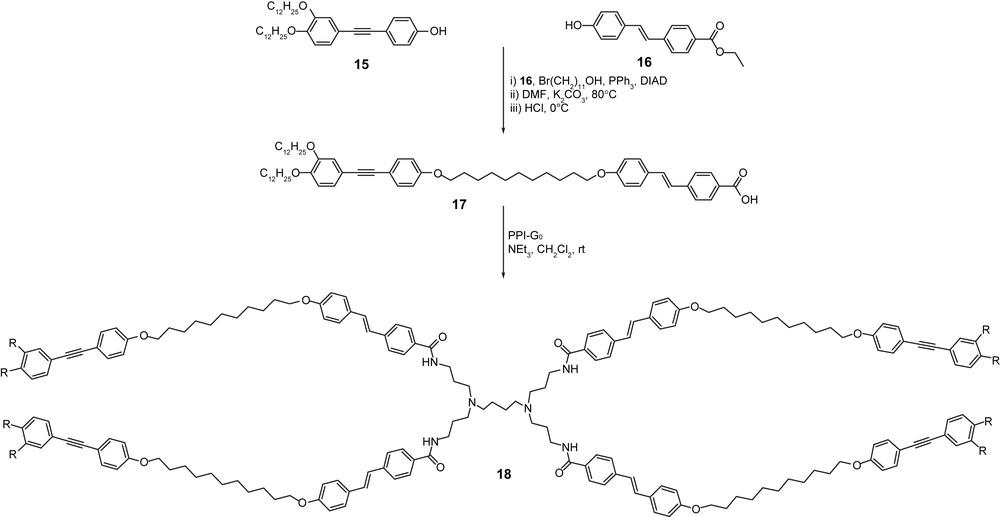
Synthesis of the tetrapodal LC 18 with NB = 1, (R = OC12H25).
3.3 Effect of generation
For a fully comprehensive study, several G0 dendrimers (tetramers) were synthesized, as well as the preparation of a second generation was attempted, in order to evaluate the impact of the generation on the mesomorphic properties. The various G0 systems (21–23, 27) were directly obtained from the coupling of the acidic anisotropic segment onto the PPI-G0 core, as shown in Scheme 4. A side-chain G1 was also prepared from one of the available acids (Scheme 4, 24) and PPI-G1.
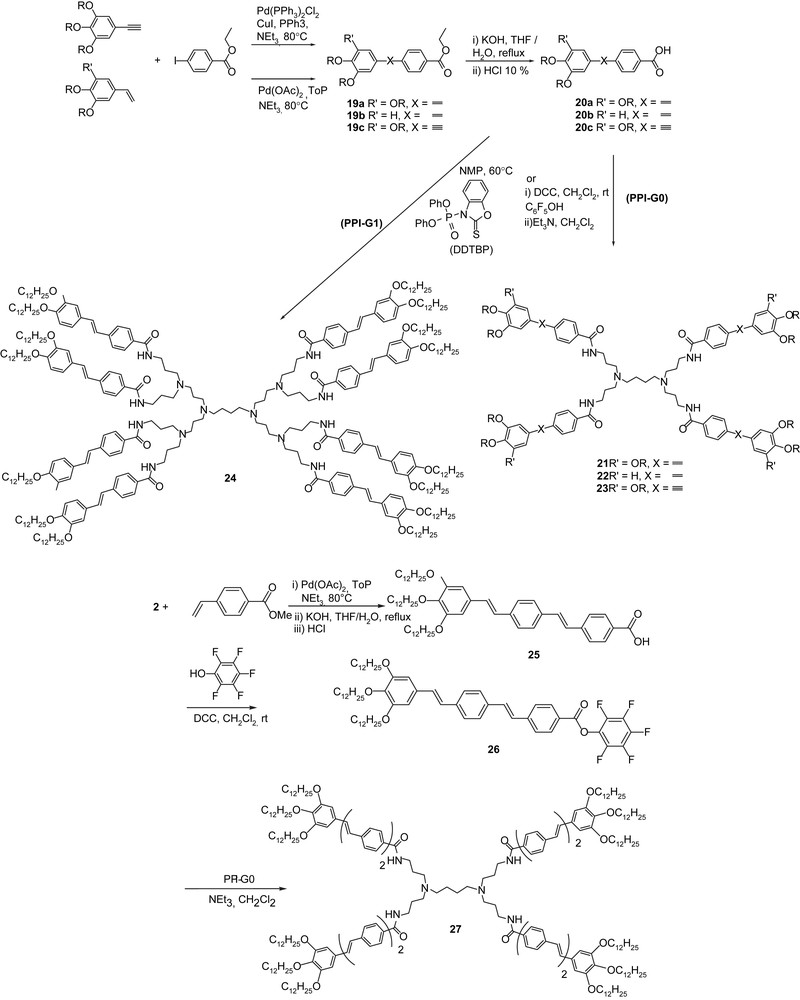
Synthesis of tetrapodal LCs (G0: 21, 22, 23, 27) and octopode G1 (24); R = OC12H25.
The four G0 dendrimers (Table 1) exhibit a hexagonal columnar phase characterized by a series of three or four sharp and intense diffraction peaks in the small-angle region, in the ratio 1, √3, √4, √7 which is typical for a hexagonal two-dimensional arrangement of columns. The X-ray diffraction patterns contain also a diffuse signal at 4.5–4.6 Å, which is attributed to the liquid-like ordering of the molten terminal aliphatic chains (Table 1). The hexagonal cell parameters are almost the same for 21 and 23 bearing tricatenar sub-units, and do not vary significantly as a function of temperature within the stability temperature range of the mesophase. The stilbene-containing dendrimer also exhibit more stable phases than the tolane system. The decrease of the number of chains coincides with an increase of the phase stability but with a narrowing of the temperature range (21 versus 22). The phase stability is enhanced upon generation increase (22 versus 24) or by elongation of the rigid unit (21 versus 27). In a first approximation, the three G0 dendrimers 21–23 likely adopt a folded conformation, with an angle at the summit of the fan-shape of nearly 120° for 21 and 23 and ca 90° for 22, which then self-assemble to form columns (see Fig. 8 for a typical columnar optical texture). Using a straightforward geometrical approach, which takes into account the relationships between the planar angle α, that represents the projection of the tapered dendrons solid angle, ω, ω/2 = α, and N, the number of dendrimers covering the columnar cross-section area, through the equation α = 2π/N [11,22], it was found that three (21, 23) and four (22) dendrimers occupy an “elementary” slice of column with a thickness close to 8–9 Å. The presence of the numerous terminal chains (36 and 32 chains, respectively) forces the rigid sub-units to tilt out of the plane in order to accommodate a tight packing of the chains. For 27, bearing longer arms, in its fan-like shape, the cone angle is a smaller than for 21, closer to 90°. Considering a similar thickness (h = 8 Å), about 4.5 molecular equivalents are required to pave the hexagonal lattice, in agreement with the increase of the lattice dimensions, much larger than that of 21. The columnar mesophase is formed by the self-assembling of folded dendrimers (3–4.5) into a supramolecular disk or column; this model of these zeroth generation (G0) dendrimers is similar to that already described for mesomorphic side-chain dendrimers [21].
Mesomorphic properties of main-chain liquid crystalline dendrimers
| Compounds | Mesomorphisma | dexp/Åb | Ic | [hk/hkl]d | dtheo/Åb,e | Parametersb,e | h/Å,f N |
| 18 | Cr 106 (14) | 83.4 | VS (sh) | 10 | 83.6 | T = 120 °C | h ∼ 4.5, |
| Colh 153 (7) I | 48.5 | M (sh) | 11 | 48.2 | a = 96.5 Å | N ∼ 6 | |
| 41.9 | M (sh) | 20 | 41.8 | S = 8070 Å2 | |||
| 31.4 | S (sh) | 21 | 31.6 | Colh-p6 mm | |||
| 4.5 | VS (br) | hch | |||||
| 21 | CrX 80 (–) | 41.6 | VS (sh) | 10 | 42.1 | T = 120 °C | h ∼ 8, |
| Colh 119 (70) I | 24.0 | S (sh) | 11 | 24.2 | a = 49 Å | N ∼ 3 | |
| 20.85 | S (sh) | 20 | 21.05 | S = 2080 Å2 | |||
| 16.3 | M (sh) | 21 | 15.9 | Colh-p6 mm | |||
| 4.5 | VS (br) | hch | |||||
| 22 | CrX 165 (–) | 40.3 | VS (sh) | 10 | 40.2 | T = 140 °C | h ∼ 9.0, |
| Colh 175 (12) I | 23.2 | S (sh) | 11 | 23.2 | a = 46.4 Å | N ∼ 4 | |
| 20.05 | S (sh) | 20 | 20.1 | S = 1870 Å2 | |||
| 15.2 | S (sh) | 21 | 15.2 | Colh-p6 mm | |||
| 4.5 | VS (br) | hch | |||||
| 23 | Cr 44 (–) | 43.5 | VS (sh) | 10 | 43.0 | T = 60 °C | h ∼ 8, |
| Colh 104 (14) I | 24.6 | S (sh) | 11 | 24.8 | a = 49.65 Å | N ∼ 3 | |
| 21.4 | S (sh) | 20 | 21.5 | S = 2135 Å2 | |||
| 4.5 | VS (br) | hch | Colh-p6 mm | ||||
| 24 | Cr 149 (148) | 48.9 | VS (sh) | 10 | 49.1 | T = 190 °C | h ∼ 9, |
| Colh 204 (6.9) I | 28.5 | S (sh) | 11 | 28.4 | a = 56.7 Å | N ∼ 3 | |
| 24.6 | S (sh) | 20 | 24.6 | S = 2790 Å2 | |||
| 4.5 | VS (br) | hch | Colh-p6 mm | ||||
| 25 | CrX 76 (67) | 51.0 | VS (sh) | 10 | 51.0 | T = 150 °C | h ∼ 4.5, |
| Colh 184 (–) I | 30.2 | S (sh) | 11 | 29.45 | a = 58.9 Å | N ∼ 8–9 | |
| 24.5 | S (sh) | 20 | 25.5 | S = 3000 Å2 | |||
| 19.55 | S (sh) | 21 | 19.25 | Colh-p6 mm | |||
| 4.5 | VS (br) | hch | |||||
| 27 | CrX1 74 (14) CrX2 110 | 55.6 | VS (sh) | 10 | 55.65 | T = 200 °C | h ∼ 8, |
| (18) Cub 190 (0.3) Colh | 32.3 | W (sh) | 11 | 32.1 | a = 64.0 Å | N ∼ 4.5 | |
| 232 (–) I | 27.7 | W (sh) | 20 | 27.8 | S = 3560 Å2 | ||
| 4.5 | VS (br) | hch | Colh-p6 mm | ||||
| G 49 (4.7) Cub 185 | |||||||
| (0.6) Colh 232 (–) I | 71.44 | VS (sh) | 200/220 | 71.35 | T = 170 °C | ||
| 63.8 | W (sh) | 210/310 | 63.8 | a = 142.7 Å (P) | |||
| 58.15 | W (sh) | 211/222 | 58.25 | a = 201.75 Å (I) | |||
| 4.5 | VS (br) | hch | a = 285.3 Å (F) |
a Abbreviations: Colh = hexagonal columnar phase; Cub = cubic phase; Cr: crystalline phase; CrX: solid phase; G: glassy phase; I: isotropic liquid. Values in brackets correspond to enthalpy of transition (ΔH) in kJ mol−1; in some cases, this value could not be measured (–).
b dexp and dtheo are the experimentally measured and theoretical diffraction spacings. The distances are given in angstroms.
c Intensity of the reflections: VS: very strong, S: strong, M: medium, W: weak, VW: very weak; br and sh stand for broad and sharp reflections, respectively.
d [hk/hkl] are the Miller indices of the reflections; hch stands for the diffuse scattering corresponding to the molten alkyl chains.
e dtheo and the mesophases parameters a, S, are deduced from the following mathematical expressions. For the Colh phase, the lattice parameter a = 2[Σhkdhk(h2 + k2 + hk)1/2]/√3Nhk where Nhk is the number of hk reflections, and the lattice area (i.e. columnar cross-section) S = a231/2/2. For the Cub phase, the lattice parameter a = [Σhkdhk(h2 + k2 + l2)1/2]/Nhk.
f N is the number of molecules (or molecular equivalents). For the columnar phases, it is defined as N = h.S/Vmol; Vmol is the molecular volume; h is the intracolumnar repeating distance, deduced directly from the measured molecular volume and the columnar cross-section according to h = NVmol/S.

Typical columnar optical texture of 23.
A good agreement is also found with the relation dendron–dendrimer between the precursory, mesomorphic acid 25 and the corresponding dendrimer 27, where, considering the same thickness value for the columnar slice, almost four times more of the former are needed for a good filling of the cylinder. Nevertheless, one striking result is the transition from a hexagonal columnar phase to a cubic mesophase when the length of the peripheral group increases (21 → 27). Once more, the observation of such mesophases is not surprising when considering the strong curvature at the interface between the dendritic cores and the peripheral groups.
Thus, a clear stabilization, or even induction, of the mesophases is observed upon dendrimerisation, i.e. from the acid to the corresponding dendrimer. None of the precursory acids of 21–23 were mesomorphic, while their connection to PPI-G0 produced large temperature ranges Colh phases; between 25 and 27, not only the phase stability is enhanced, the apparition of a new phase was also observed. The same observations can be made for the G1 systems, i.e. between 5 and 7 and between the precursory acids of 1 and dendrimers 1 [9]. The effect of generation is not as positive as expected: except for the non-mesomorphic stilbene acid precursors, the increase of generation is concomitant with a decrease of the mesophase stability (reduction of the mesophase temperature ranges, substantial decrease of the isotropic transition temperatures e.g. 25 → 5, 21, 22 → 1, 27 → 7).
Below the Colh phase, 27 exhibits a cubic phase as well. The small-angle X-ray diffraction pattern exhibits three sharp, small-angle reflections, for which the reciprocal d-spacings were in the ratios √4:√5:√6, pointing to a cubic supramolecular organization [(h2 + k2 + l2)1/2 = a/dhkl] and an additional, very intense, but diffuse, halo in the wide-angle region at about 4.4–4.6 Å confirming the liquid nature of the mesophase. Cubic mesophases are ordered three-dimensional supramolecular edifices, with a multi-continuous [23–25], or a discrete micellar structure [26], depending on the molecular self-organisation within the cubic cell [11,22,27]. Bi- and tri-continuous cubic structures consist of two or three regular, interwoven, infinite, interconnected rod networks (referred to as Im-3m (P) and Im-3m (I) structures, respectively [24]); the columnar rods are formed by the rigid part of the molecules, and the networks are separated by lipophilic films (their associated minimal surfaces). The difference between the bicontinuous and the micellar structures lies in the periodic discontinuities along the rods in the x, y, z directions (bicontinuous model) that appear at mid-height of the cubic-cell edges; the junctions between three connecting rods then correspond to the gravity centres of the micelles (micellar model). It is therefore not trivial to choose one model preferentially to the other. For the present system, the micellar model is unlikely due to the too high number of molecules per cubic cell (about 470 for P, 1300 for I and 3700 for F), which should be further distributed equitably within the micelles. As far as the bicontinuous structure is concerned, the molecules need to be able to stack to form the columns, with the required molecular conformation as that found in the columnar phase. Thus, on the basis of their molecular conformations and packing considerations, the micellar model does not appear appropriate in the present situation and a multi-continuous model is more probable.
The determination of the cubic symmetry is generally not trivial, but the possibilities can be considerably reduced. All non-centro-symmetric groups (groups with the 23, 432 and -43m Laue classes) can be disregarded, a consequence of the Friedel's law [28,29], thus leaving only 17 cubic space groups to consider out of initially 36 of them. However, with three reflections only, the correct determination is unlikely. Indeed, this sequence of ratios is compatible with a face centred cubic network (Fm-3, Fm-3m and Fm-3c), primitive Bravais lattice (Pm-3, Pa-3, Pm-3m and Pm-3n) and with a body-centered cubic lattice (Im-3, Im-3m). The F-type can be a priori disregarded since a number of absent reflections is not explained by the conditions on the reflections. However, this is difficult to discriminate between a body-centered and primitive space group. The distribution of the reflections' intensities often helps distinguishing both phases: on the basis of other studies [30], the intensity of the reflections decreases on increasing the diffraction angle, in agreement with a body-centered symmetry.
An aggregation into the highest symmetry is generally admitted and accordingly, the Im-3m (No. 229) [29] space group can be retained. For the body-centered cubic lattice, the considered sequence in this case is √8:√10:√12 and the reflections can be indexed as (220), (310) and (222). The multicontinuous models of the Im-3m phase could also render account of the self-organization. In the network model of the Im-3m (P) bicontinuous cubic phase, two networks are interpenetrated and separated by lipophilic fragments, whereas in the triple network (Im-3m (I) structure), there are three interpenetrating continuous networks (Fig. 9) [24].
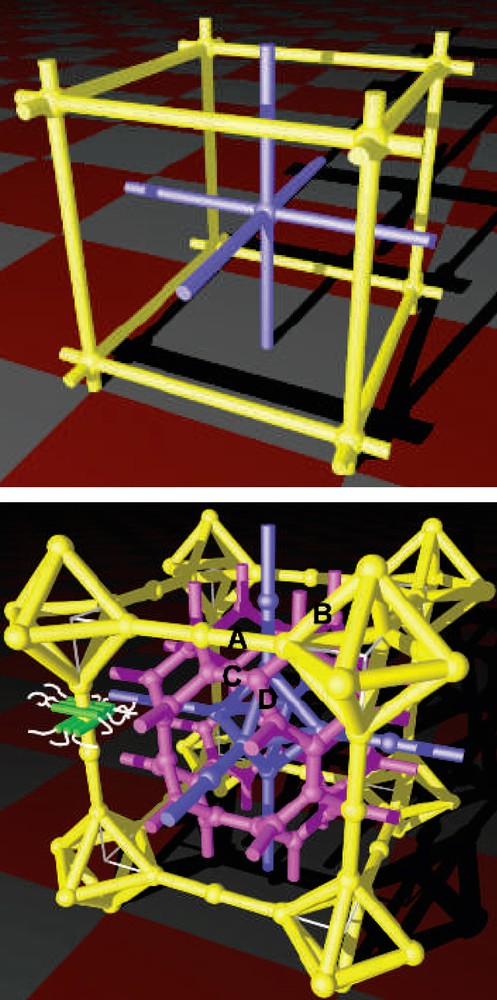
(Top) Network model of the Im-3m (P) bicontinuous cubic phase (the two interpenetrating networks are coloured blue and yellow). Model of the cubic unit cell with two layers made of alkyl chains separated by three core regions. (Bottom) Triple network representation of the Im-3m (I) structure (the three interpenetrating continuous networks are coloured purple, pink and yellow; the purple and yellow networks are identical but mutually displaced by (x/2,y/2,z/2)). Model of the cubic unit cell with three layers made of alkyl chains separated by four core regions (images taken from Ref. [24]). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
As for the G1 dendrimer, 24, a Colh phase was also observed. The thermal stability of the mesophase increases slightly when increasing the generation (22 → 24). Such a larger stability could be related to a better nano-segregation due to the larger core size, in agreement with previous results [21]. A molecular dynamic study of 24 (side-chain dendrimer) was performed in order to optimize the geometry of organization. Thus, three molecules were assembled in a hexagonal cell with a thickness of 9 Å, corresponding to the thickness of one folded molecule. The hexagonal parameter, a, is not imposed so that no constraint exists during the optimization process. It has to be noted that this simulation has been studied at a temperature lower than the stability domain of the mesophase, in order to avoid any eccentric distortion. The result of this molecular dynamic study at 80 °C is presented in Fig. 10a. The arrangement of the molecules is not random, and it is possible to distinguish three main zones. The central part is constituted by all the dendritic cores, while the rigid molecular moieties are building an internal corona; finally the disorganized aliphatic chains are located at the periphery. It is worth to mention that the hexagonal cell parameter determined after such a simulation is in good agreement with that found experimentally by X-ray diffraction. The organization of the columns in a two-dimensional hexagonal lattice is easily obtained through interdigitation of the aliphatic chains (Fig. 10b).
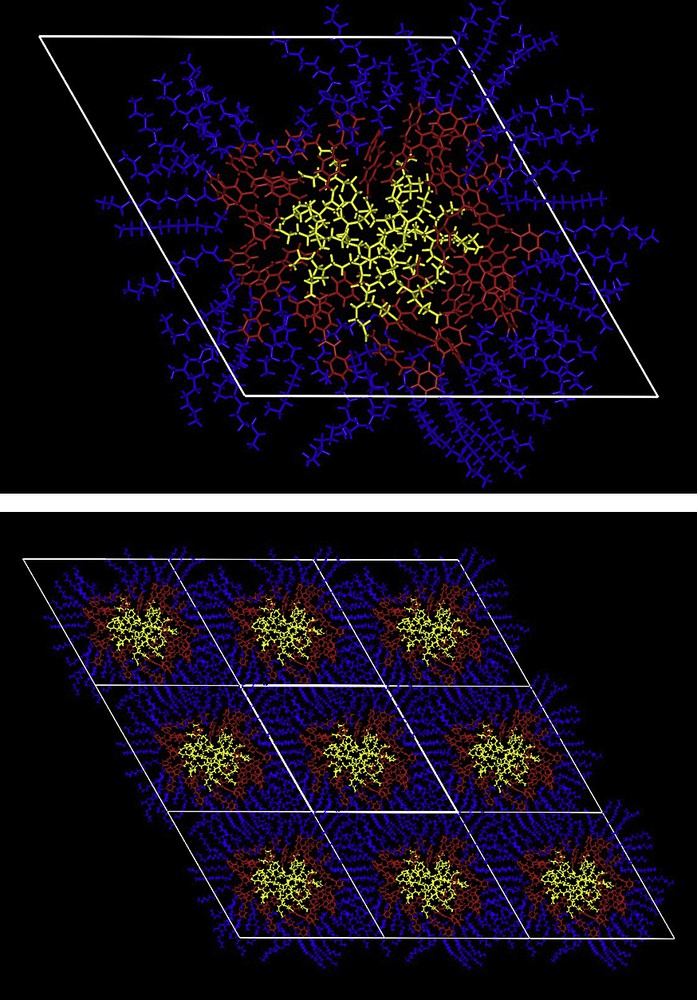
Top: Optimisation of the geometry of organization obtained by molecular dynamics at 80 °C with three dendrimers (24) located in a hexagonal cell. Bottom: Visualization of the 2D hexagonal organisation.
At this stage, it became obvious to synthesize main-chain dendrimers of the second generation, with the expectation to enlarge further the molecular variety as well as the mesophases morphology. A straight convergent synthesis was used for the preparation of the G2 dendrons 34 (Scheme 5). Connection of the readily available trialkoxytolanol derivative (28) with the core 29 by standard etherification led to the first precursory branch (30). The iodide group was converted into the acetylenic function (32) using the Sonogashira coupling, followed by deprotection of TMS by TBAF. A second Sonogashira coupling between 32 and iodophenol led to the desired functional branch 33. The final dendron was obtained by etherification between the precursory branching stilbene (vide supra) and 33. All the steps proceeded in good yields. However, despite various attempts, it was not possible to couple the acidic dendron onto the PPI-G0 core. Unfortunately, both the dendrons 34a and 34b were devoid of mesomorphism (33 was not liquid crystalline either), probably due to a too great flexibility and entropy winning over the enthalpic intramolecular interactions. It can, however, be speculated that mesomorphism could be induced in G2 dendrons, providing the use of longer anisotropic units, such as OPV3 basic units; this work is still in progress.
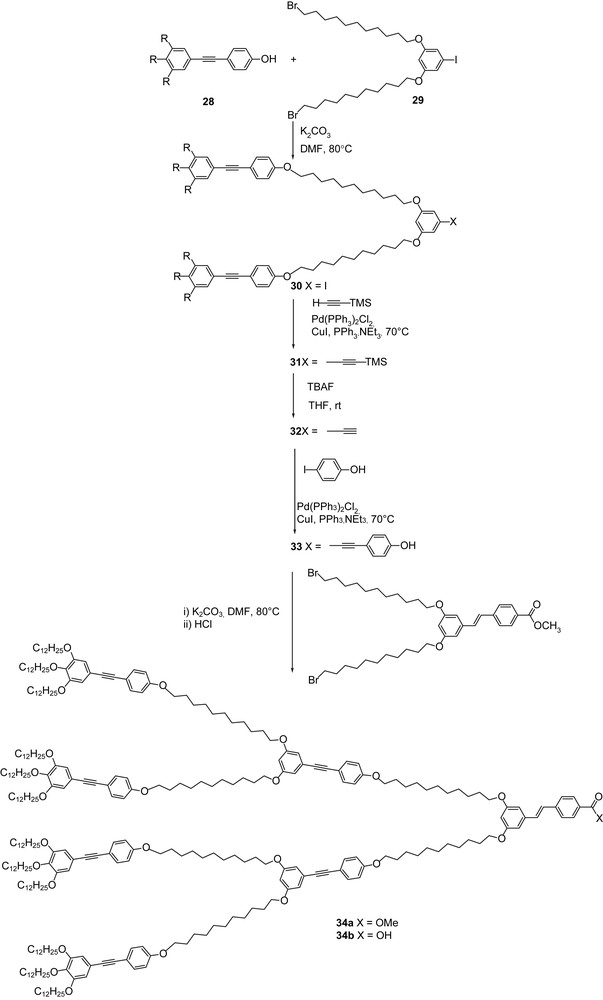
Convergent synthesis of the G2 dendrons 34 (R = OC12H25).
4 Conclusions
Thus, the above examples show that liquid-crystalline phases can be produced with high molecular weight monodisperse main-chain dendrimers and dendrons. Lamellar, columnar, cubic phases with unusual morphologies were obtained depending upon the dendritic chemical nature and connectivity and the type of mesogenic groups. For the dendrimers described in the present paper, the morphology of the mesophase is determined by the number of alkyl chains grafted on the peripheral mesogenic group, i.e. the change in the number of terminal chains per end group modifies the relationships between the hard parts and the soft parts, and consequently the molecules adopt either a parallel (prolate) or flat (oblate) fan conformation. The variation of the core connectivity or the degree of branching scarcely influences these mesomorphic properties, only affecting the phase stability and transition temperatures. However, those properties are strongly affected by the generation (G0 and G1 exhibit mesophases, while is absent in G2) or by the length of the terminal pendant group (cubic phase at G0). Mesophase stability is systematically enhanced upon dendrimerisation (acidic branch → dendrimer) but the opposite is true with generation (G0 → G1 → G2) due to a reinforcement of the micro-segregation. The formation of the smectic lamellar phases is the result of the parallel disposition of the mesogenic groups on both sides of the focal tetravalent core, the dendrimer adopting the shape of a giant elongated multipede, and then organizing into layers. In contrast, the grafting of additional terminal chains at the periphery prevents such a parallel disposition of the pro-mesogenic groups, which are forced into a radial arrangement around the central moiety: the dendrimer can adopt a fan or conical shape and self-arrange into supra-molecular columns or possibly in cubic phases, respectively.
This control of the mesomorphic dendritic structure by an appropriate molecular design makes possible the development of new liquid-crystalline materials containing active molecular units (metals [31], clusters [32], ferrocene [33], fullerenes [34], particles [35]) with specific physical properties (magnetic, optical, electronic) which may find uses in nano- and bio-technology. The high sensitivity of LC dendrimers with the surrounding environment (properties versus molecular structure) could be, in principle, beneficial to access some kinds of molecular sensors i.e. to use such supermolecules as tools to test how properties in general may be altered or modulated upon delicate external stimuli. The fabrication of self-assembling organic–inorganic nanohybrids by combining the magnetic, electronic and/or optical properties of metallic clusters, oxides and nanoparticles with the soft nature of organic dendrons could represent an interesting perspective in this area. In this case, the nanoparticle would play the role of a hard and multivalent core.
5 Experimental part
The detailed synthetic procedures of several of the dendrimers and precursors have already been published elsewhere [9,12,20], and only the synthesis of the new materials is described below. 9-BBN (9-Borabicyclo[3.3.1]nonane), DCC (N,N′-dicyclohexylcarbodiimide), DDTBP (diphenyl(2,3-dihydro-2-thioxo-3-benzoxazolyl)phosponate), DIAD (diisopropyl azodicarboxylate), DMAP (4-dimethylaminopyridine), NMP (N-methyl-2-pyrrolidone), TBAF (tetrabutylammonium fluoride), TBAI (tetrabutylammonium iodide), TBDMS (tert-butyldimethylsilane), ToP (tri(o-tolyl)phosphine).
5.1 Preparation of the octopus 7 (Scheme 1)
Compound 2: A solution of 3,4,5-tridodecyloxystyrene (5.6 g, 8.52 mmol) [9a], Pd(OAc)2 (70 mg, 0.3 mmol), ToP (374 mg, 1.23 mmol) and 4-bromoiodobenzene (2.9 g, 10.2 mmol) in NEt3 (50 mL) was stirred at 60 °C under argon (2 h) and at 80 °C (2 d). A yellow precipitate was observed during this time. The mixture was cooled to RT, diluted (CH2Cl2) and filtrated on celite. The crude product was extracted (CH2Cl2), washed (H2O), dried (MgSO4). Purification by column chromatography (SiO2, 1:4 CH2Cl2/cyclohexane) and crystallization in acetone gave a slightly yellow solid (5.35 g, 77%). 1H NMR (CDCl3): δ 7.46 (m, 2H), 7.36 (m, 2H), 6.95 (dd, 2H, J = 16 Hz), 6.7 (s, 2H), 4.0 (m, 6H), 1.8 (m, 6H), 1.3 (m, 54H), 0.89 (m, 9H).
Compound 3: Prepared in three steps: i) To a suspension of Ph3PCH3Br (42 g, 116 mmol) in THF (100 mL) under argon was added in a small portion t-BuOK (13.1 g, 116 mmol). The resulting yellow mixture was stirred at RT (30 min). The solution of the TBDMS-protected aldehyde (23 g, 97 mmol) in dry THF (30 mL) was then added dropwise [12]. The mixture was stirred at RT (2 h), and then was quenched by adding 30 mL of a saturated aqueous solution of NH4Cl. The crude product was extracted (CH2Cl2), washed (H2O), and dried (MgSO4). Purification by chromatography (SiO2, 3:7 CH2Cl2/cyclohexane) provided the TBDMS-protected styrene as a colourless oil (19.4 g, 85%). 1H NMR (CDCl3): δ 7.29 (d, 2H, 3J = 9 Hz), 6.8 (d, 2H, 3J = 9 Hz), 6.66 (dd, 1H, J = 11 Hz, J = 17 Hz), 5.61 (dd, 1H, J = 0.9 Hz, J = 17 Hz), 5.13 (dd, 1H, J = 0.9 Hz, J = 11 Hz), 0.99 (s, 9H), 0.26 (s, 6H). ii) As for 2: 2 (4.82 g, 5.94 mmol), Pd(OAc)2 (54 mg, 0.24 mmol), ToP (217 mg, 0.7 mmol), NEt3 (60 mL), 60 °C (2 h). The protected styrene (1.67 g, 7.12 mmol) in xylene (60 mL) was then added, and the reaction mixture was heated to 110 °C (2 d). The crude was diluted (CH2Cl2), filtrated (celite), extracted (CH2Cl2), washed (H2O), and dried (MgSO4). The yellow solid protected OPV3 derivative (4.6 g, 80%) was obtained by two successive chromatography columns (SiO2, 1:4 CH2Cl2/cyclohexane). 1H NMR (CDCl3): δ 7.47 (s, 4H), 7.4 (d, 2H, 3J = 8.55 Hz), 7.02 (m, 4H), 6.84 (d, 2H, 3J = 8.55 Hz), 6.72 (s, 2H), 4.0 (m, 6H), 1.8 (m, 6H), 1.27 (m, 54H), 1.01 (s, 9H), 0.89 (t, 9H, 3J = 6.9 Hz), 0.22 (s, 6H). iii) To a solution of this protected alcohol (2.3 g, 2.37 mmol) in THF (20 mL) was added dropwise TBAF (3.6 mL, 3.56 mmol). The yellow mixture was stirred at RT overnight. Purification by column chromatography (SiO2, CH2Cl2) yielded a fluorescent yellow solid (1.63 g, 80%). 1H NMR (CDCl3): δ 7.47 (s, 4H), 7.42 (d, 2H, 3J = 8.77 Hz), 7.03 (AB, 2H, 3J = 16.22 Hz), 6.99 (m, 2H), 6.84 (d, 2H, 3J = 8.55 Hz), 6.72 (s, 2H), 4.87 (s, 1H), 4 (m, 6H), 1.8 (m, 6H), 1.27 (m, 54H), 0.89 (t, 9H, 3J = 6.9 Hz).
Compound 4: To a solution of 3 (1 g, 1.17 mmol) and internal stilbene block (0.36 g, 0.48 mmol) [9a] in DMF (20 mL) was added slowly K2CO3 (1.6 g, 11.7 mmol). The mixture was stirred (80 °C, 2 d). The solvent was then removed under vacuum and the crude product extracted (CH2Cl2, 50 mL), washed (50 mL saturated solution of NH4Cl, and 50 mL of H20), dried (MgSO4), and purified by column chromatography (SiO2, 3:7 CH2Cl2/cyclohexane) to afford a yellow solid (1.03 g, 93%). 1H NMR (CDCl3): δ 8.02 (d, 2H, 3J = 8.85 Hz), 7.55 (d, 2H, 3J = 8.55 Hz), 7.47 (s, 10H), 7.44 (d, 4H, 3J = 8.99 Hz), 7.0 (m, 10H), 6.89 (d, 4H, 3J = 8.55 Hz), 6.72 (s, 4H), 6.68 (d, 2H, 3J = 2.19 Hz), 6.43 (t, 1H), 4.0 (m, 20H), 3.92 (s, 3H), 1.79 (m, 20H), 1.37 (m, 136H), 0.89 (t, 18H, 3J = 6.68 Hz).
Compound 5: To 4 (1.03 g, 0.45 mmol) in THF (20 mL) was added an aqueous solution of KOH (1.01 g, 18.04 mmol). The reaction mixture was heated at reflux (2 d). The acid was then precipitated in an aqueous solution of HCl (10%), cooled (0 °C), filtrated, washed (H2O), and dried (MgSO4). Crystallization in acetone afforded a yellow solid (0.963 g, 95%). 1H NMR (CDCl3): δ 8.08 (d, 2H, 3J = 8.55 Hz), 7.58 (d, 2H, 3J = 8.55 Hz), 7.47 (s, 8H), 7.44 (d, 4H, 3J = 8.99 Hz), 7.04 (m, 10H), 6.89 (d, 4H, 3J = 8.7 Hz), 6.72 (s, 4H), 6.69 (d, 2H, 4J = 2.19 Hz), 6.44 (m, 1H), 4.0 (m, 20H), 1.79 (m, 20H), 1.37 (m, 136H), 0.89 (t, 18H, J = 6.9 Hz). 13C NMR (CDCl3): δ 160.5, 158.9, 153.3, 142.6, 138.5, 138.3, 136.9, 136.3, 132.6, 131.8, 130.6, 129.9, 128.5, 128.1, 127.9, 127.7, 127.3, 126.6, 126.5, 126.4, 126, 114.7, 105.4, 105.2, 73.5, 69.2, 68.1, 68, 31.93, 31.91, 30.3, 29.74, 29.72, 29.7, 29.6, 29.53, 29.5, 29.4, 29.35, 29.25, 26.1, 26, 22.7, 14.1. Elemental analysis calculated for C153H232O12: C, 81.19, H, 10.33; found: C, 80.97, H, 10.77.
Compound 6: To a solution of 5 (1.5 g, 0.66 mmol) and pentafluorophenol (0.146 g, 0.79 mmol) in CH2Cl2 (10 mL) stabilized by amylene, was added under argon DCC (0.164 g, 0.79 mmol). The white solid precipitated suddenly. The reaction mixture was stirred at RT overnight. The dicyclohexylurea which is the secondary product of the reaction was then eliminated by filtration. The solvent was removed under reduced pressure. Purification by column chromatography (SiO2, 3/7 CH2Cl2/cyclohexane) afforded a yellow solid (1.39 g, 86%). 1H NMR (CDCl3): δ 8.18 (d, 2H, J = 8.33 Hz), 7.66 (d, 2H, J = 8.55 Hz), 7.47 (s, 8H), 7.45 (d, 4H, J = 8.98 Hz), 7.08 (m, 10H), 6.89 (d, 4H, J = 8.77 Hz), 6.72 (s, 4H), 6.7 (d, 2H, J = 2.2 Hz), 6.46 (m, 1H), 4.0 (m, 20H), 1.79 (m, 20H), 1.37 (m, 136H), 0.89 (t, 18H, J = 6.9 Hz). 13C NMR (CDCl3): 162.29, 160.56, 158.88, 153.27, 143.59, 138.30, 138.20, 136.85, 136.29, 132.62, 132.55, 131.17, 129.92, 128.50, 128.06, 127.65, 127.31, 126.68, 126.62, 126.49, 126.01, 125.36, 114.68, 105.49, 105.15, 101.76, 73.50, 69.15, 68.11, 68.03, 31.9, 30.33, 29.73, 29.51, 29.47, 29.24, 26.10, 22.73, 14.06.
Compound 7: To a solution of 6 (1.34 g, 0.55 mmol) in CH2Cl2 (5 mL) stabilized by amylene, PPI-G0 (39.7 g, 0.13 mmol) in CH2Cl2 (5 mL) was added slowly; a few drops of NEt3 were also added. After stirring (3 d), the crude product was taken up in CH2Cl2 and precipitated in cyclohexane. The precipitate was eliminated by filtration and the filtrate was concentrated in vacuum. Purification by column chromatography (Al2O3, with CH2Cl2 and then with THF) afforded a waxy yellow solid (270 mg, 23%). 1H NMR (CDCl3): δ 7.76 (d, 8H, 3J = 8.11 Hz), 7.46 (m, 40H), 7.42 (d, 16H, 3J = 8.99 Hz), 7.0 (m, 40H), 6.87 (d, 16H, 3J = 8.55 Hz), 6.71 (s, 16H), 6.61 (d, 8H), 6.39 (m, 4H), 4.0 (m, 80H), 3.48 (m, 8H), 2.48 (m, 12H), 1.77 (80H), 1.45 (m, 556H), 0.88 (m, 72H). 13C NMR (CDCl3): δ 167.12, 160.50, 158.90, 153.28, 140.15, 138.62, 138.36, 136.89, 136.31, 133.34, 132.59, 130.69, 129.92, 128.53, 128.09, 127.66, 127.47, 127.32, 126.64, 126.51, 126.01, 114.69, 105.22, 101.40, 73.52, 69.18, 68.05, 52.35, 43.44, 39.11, 31.91, 30.84, 30.36, 30.16, 29.75, 29.70, 29.66, 29.56, 29.47, 29.44, 29.38, 29.35, 29.31, 26.88, 26.14, 26.07, 22.68, 14.09. Elemental analysis calculated for C628H960N6O44: C, 81.12, H, 10.41, N, 0.9; found: C, 82.09, H, 10.62, N, 0.76. MS-MALDI-TOF (m/z): found 9298.1 g mol−1 [M + H]+, calculated 9298.36 g.mol−1. SEC: Mw = 12.633, Mn = 11.271, IP: 1.12.
5.2 Preparation of the octopus based on the pentaerythritol core, 11 (Scheme 2)
Compound 8: To pentaerythritol (9.2 g, 67 mmol) in 33% NaOH solution (150 mL), is added TBAI (7.24 g, 19 mmol), and the mixture is heated under stirring at 40 °C. Then, allyl bromide is slowly added (43.8 g, 0.36 mol), and the reaction is kept stirring at RT (2 d). The product is extracted (toluene), washed (H2O), dried (MgSO4), filtered and the solvent evaporated. The crude product was purified by chromatography (SiO2, CH2Cl2) yielded 7 g of a yellowish oily liquid (35%). 1H NMR (CDCl3): δ 5.89 (m, 4H), 5.21 (m, 8H), 3.96 (m, 8H), 3.47 (s, 8H).
Compound 9: Under argon, 8 (1 g, 3.4 mmol) in THF (200 mL) and 9-BBN (60 mL, 30 mmol) are heated at reflux (1 h), under stirring. The excess of hydride is neutralized by small portion of H2O. The solution is cooled down to 0 °C, 30 mL of NaOH 3 M added dropwise, and then 30 mL of a 30% of H2O2 solution. Stirring is maintained at RT (4 h), and then the solution saturated with K2CO3. The product is extracted (THF), dried (MgSO4), filtered, and the solvent evaporated. Then, the residue is solubilised in 25 mL of pyridine, and 25 mL of acetic anhydride added, and stirred at RT (8 h). Purification by chromatography (SiO2, 4:6 EtOAC/petroleum ether) afforded 1.35 g of a colorless oil (75%). 1H NMR (CDCl3): δ 4.13 (t, 3J = 7 Hz, 8H), 3.44 (t, 3J = 7 Hz, 8H), 3.35 (s, 8H), 2.05 (s, 12H), 1.86 (q, 3J = 7 Hz, 8H).
Compound 10: To a vigorously stirred solution of 9 (1.35 g, 2.5 mmol) in MeOH (60 mL), a piece of sodium is added, and the mixture kept at RT under vigorous stirring (12 h). The solution is then neutralized (DOWEX, W50 H+). After filtration and evaporation 0.9 g (100%) of a yellowish oil is obtained. 1H NMR (CD3OD): δ 3.72 (t, 3J = 7 Hz, 8H), 3.56 (t, 3J = 7 Hz, 8H), 3.45 (s, 8H), 1.83 (q, 3J = 7 Hz, 8H).
Compound 11: Under argon, 10 (0.012 g, 0.03 mmol) and CMC (0.138 g, 0.33 mmol) are dissolved in CH2Cl2 stabilized by amylene (5 mL). The suspension is then stirred at RT (4 d), and the appropriate dendritic branch [9a] (0.46 g, 0.27 mmol) and a catalytic quantity of DMAP are introduced. The product is then extracted (CH2Cl2), washed (H2O), dried (MgSO4), filtered and the solvent evaporated. Purification by chromatography (SiO2, 7:3 CH2Cl2/petroleum ether) yielded 50 mg of a viscous white solid (25%). 1H NMR (CD2Cl2): δ 7.95 (d, 3J = 9 Hz, 8H), 7.50 (d, 3J = 9 Hz, 8H), 7.32 (d, 3J = 9 Hz, 16H), 7.00 (m, 40H), 6.61 (d, 4J = 2 Hz, 24 H), 6.33 (t, 4J = 2 Hz, 12H), 4.33 (t, 3J = 7 Hz, 8H), 3.95 (m, 64H), 3.51 (t, 3J = 7 Hz, 8H), 3.40 (s, 8H), 1.95 (q, 3J = 7 Hz, 8H), 1.78 (m, 64H), 1.27 (m, 400H), 0.89 (m, 48H). 13C NMR (CDCl3): δ 160.5, 158.9, 139.6, 129.9, 129.8, 129.3, 129.1, 128.6, 127.7, 126.6, 126.3, 114.7, 104.9, 68.1, 31.9, 29.7, 29.6, 29.4, 29.3, 26.1, 22.7, 14.1. Elemental analysis calulated for C469H716O44: C, 79.80, H, 10.22; found: C, 79.61, H, 10.31. SEC: IP = 1.06. MS-MALDI-TOF: calculated, 7058.82 g mol−1; found, 7060.06 g mol−1.
5.3 Preparation of tetrapode 18 (Scheme 3)
Compound 16: Prepared in two steps. i) By a Heck coupling as 2: TBDMS-protected styrene (7.75 g, 0.033 mol, see 3), Pd(OAc)2 (0.3 g, 0.001 mol), ToP (0.6 g, 0.002 mol), ethyl-4-iodobenzoate (9.13 g, 0.033 mol). Purification by column chromatography (SiO2, 3/7 CH2Cl2/petroleum ethers) afforded a yellow solid (12 g, 65%). 1H RMN (CDCl3): δ 8.02 (d, 3J = 8 Hz, 2H), 7.78 (d, 3J = 9 Hz, 2H), 7.53 (d, 3J = 8 Hz, 2H), 7.17 (d, 3J = 16 Hz, 1H), 6.98 (d, 3J = 16 Hz, 1H), 6.93 (d, 3J = 9 Hz, 2H), 4.39 (q, 3J = 7 Hz, 2H), 1.42 (t, 3J = 7 Hz, 3H), 1.0 (s, 9H), 0.26 (s, 6H). ii) as in 3iii: protected alcohol (12 g, 0.031 mol), THF (100 mL), TBAF (8.2 g, 0.031 mol), stirring (RT, 1 h) and filtration on silica gel. Purification by crystallization in petroleum ether yielded a white solid (5.1 g, 60%). 1H NMR (CDCl3): δ 8.02 (d, 3J = 8 Hz, 2H), 7.53 (d, 3J = 8 Hz, 2H), 7.44 (d, 3J = 9 Hz, 2H), 7.17 (d, 3J = 16 Hz, 1H), 6.98 (d, 3J = 16 Hz, 1H), 6.86 (d, 3J = 9 Hz, 2H), 4.39 (q, 3J = 7 Hz, 2H), 1.42 (t, 3J = 7 Hz, 3H).
Compound 17: Prepared in three steps. i) (E)-Ethyl 4-(4-(11-bromoundecyloxy)styryl) benzoate was prepared from 16 (5 g, 0.018 mol), bromoundecanol (6.18 g, 0.024 mol), PPh3 (5.31 g, 0.023 mol) and DIAD (4.75 g, 0.023 mol) according to the Mitsonobu procedure. Purification by chromatography (SiO2, 7:3 CH2Cl2/petroleum ethers), and crystallisation in petroleum ether yielded 7.5 g of a white solid (95%). 1H NMR (CDCl3): δ 8.02 (d, 3J = 8 Hz, 2H), 7.53 (d, 3J = 8 Hz, 2H), 7.46 (d, 3J = 9 Hz, 2H), 7.17 (d, 3J = 16 Hz, 1H), 6.98 (d, 3J = 16 Hz, 1H), 6.91 (d, 3J = 9 Hz, 2H), 4.39 (q, 3J = 7 Hz, 2H), 3.99 (t, 3J = 7 Hz, 2H), 3.41 (t, 3J = 7 Hz, 2H), 1.87 (m, 4H), 1.31 (m, 14H). ii) 15 [9b] (1.41 g, 2.51 mmol) was reacted with the alkyl bromide just prepared (1 g, 2.28 mmol) according to the Williamson etherification procedure. Purification by chromatography (SiO2, 7:3 CH2Cl2/petroleum ethers) yielded 1.6 g of the ester branch as a white solid is obtained (76%). 1H NMR (CDCl3): δ 7.98 (d, 3J = 9 Hz, 2H), 7.53 (d, 3J = 9 Hz, 2H), 7.45 (m, 4H), 6,92 (m, 9H), 4.39 (q, 3J = 7 Hz, 2H), 3.99 (m, 8H), 1.82 (m, 8H), 1.27 (m, 53H), 0.89 (m, 6H). iii) As in 5: Crystallisation from petroleum ether (1.4 g, white solid, 100%). 1H NMR (CDCl3): δ 7.94 (d, 3J = 9 Hz, 2H), 7.53 (d, 3J = 9 Hz, 2H), 7.45 (m, 4H), 6.93 (m, 9H), 3.97 (m, 8H), 1.82 (m, 8H), 1.27 (m, 50H), 0.89 (m, 6H). MS-FAB+: calculated, 955.42 g mol−1; found, 954.85 g mol−1.
Compound 18: This compound was prepared from 17 (0.3 g, 0.34 mmol), DDTBP (0.14 g, 0.37 mmol) and PPI-G0 (0.022 g, 0.07 mmol) as in 7. Purification by column chromatography (Al2O3 with CH2Cl2 and then THF) afforded a yellow solid (160 mg, 60%). 1H NMR (CDCl3): δ 7.72 (d, 3J = 9 Hz, 8H), 7.38 (m, 28H), 7.04 (m, 12H), 6.85 (m, 24H), 3.95 (m, 32H), 3.50 (m, 8H), 2.50 (m, 8H), 2.40 (m, 4H), 1.81 (m, 44H), 1.27 (m, 200H), 0.89 (m, 24H). 13C NMR (CDCl3): δ 148.7, 132.8, 128.0, 126.1, 114.7, 114.5, 31.9, 29.7, 29.6, 29.5, 29.4, 29.3, 29.2, 26.0, 22.7, 14.1. Elemental analysis calculated for C272H392N6O20: C, 80.35, H, 9.72, N, 2.07; found: C, 80.54, H, 10.01, N, 1.85. MS-MALDI-TOF: found, 4066.84 g mol−1; calculated, 4066.13 g mol−1.
5.4 Prepration of dendrimers G0, 21, 22, 23, 27, (Scheme 4)
Compound 19a: As for 2: under argon 3,4,5-tridodecyloxystyrene (3 g, 6.35 mmol) [9a], Pd(OAc)2 (0.13 g, 0.57 mmol), ToP (1.05 g, 3.46 mmol), 4-ethyl iodobenzoate (0.96 mL, 5.77 mmol), NEt3 (50 mL), stirring (2 h, 60 °C and for 24 h at 80 °C). The crude product was extracted (CH2Cl2), washed (H2O), and dried (MgSO4). Purification by column chromatography (SiO2, 4:6 CH2Cl2/petroleum ethers) yielded a white solid (2.4 g, 60%). 1H NMR (CDCl3): δ 8.02 (d, 3J = 8 Hz, 2H), 7.54 (d, 3J = 8 Hz, 2H), 7.05 (m, 5H), 4.39 (q, 3J = 7 Hz, 2H), 4.04 (m, 4H), 1.85 (m, 4H), 1.37 (m, 39H), 0.89 (m, 6H).
Compound 19b: As in 2: 3,4-didodecyloxystyrene [9a] (7 g, 10.7 mmol), Pd(OAc)2 (87 mg, 0.04 eq.), ToP (354 mg, 0.12 eq.), 4-ethyl iodobenzoate (2.27 g, 9.7 mmol), NEt3 (50 mL), stirring (2 h at 60 °C under argon and for 2 d at 80 °C). The mixture was cooled to RT, diluted (CH2Cl2) and filtrated on celite, and the crude extracted (CH2Cl2), washed (H2O), and dried (MgSO4). Purification by column chromatography (SiO2, 4:6 CH2Cl2/cyclohexane) yielded a white solid (6.25 g, 78%). 1H NMR (CDCl3): δ 8.03 (d, 2H, 3J = 8.33 Hz), 7.54 (d, 2H, 3J = 8.33 Hz), 7.06 (AB, 2H, 3J = 16.22 Hz), 6.74 (s, 2H), 4.4 (t, 2H, 3J = 7 Hz), 4 (m, 6H), 1.8 (m, 6H), 1.36 (m, 57H), 0.88 (t, 9H, 3J = 6.6 Hz).
Compound 19c: Prepared in three steps from 3,4,5-tridodecyloxybenzaldehyde [9a]: i) CBr4 (25.2 g, 0.076 mol), PPh3 (19.96 g, 0.076 mol), and Zn (4.96 g, 0.076 mol) are dissolved in CH2Cl2 (200 mL) and stirred at RT (1 d). Then, 3,4,5-tridodecyloxybenzaldehyde (10 g, 0.015 mol) in CH2Cl2 (100 mL) is added and the mixture left stirred (1 d). Filtration over silica, purification by column chromatography (SiO2, 5:5 CH2Cl2/petroleum ethers) and crystallisation in petroleum ethers yielded 11 g of a white solid (90%). 1H NMR (CDCl3): δ 7.39 (s, 1H), 6.56 (s, 2H), 3.95 (t, 3J = 6 Hz, 6H), 3.0 (s, 1H), 1.77 (m, 6H), 1.27 (m, 54H), 0.89 (m, 9H). ii) Under argon, a solution of the dibromo derivative just prepared (15 g, 0.018 mol) in THF (50 mL) is cooled down to −78 °C, and LDA (10 g, 0.09 mol) added dropwise. The temperature is slowly raised to RT, and the mixture stirred (3 h); the reaction mixture is then quenched by addition of a saturated solution of NH4Cl, diluted (hexane), washed (H2O), and dried (MgSO4). Purification by column chromatography (SiO2, 3:7 CH2Cl2/petroleum ethers) yielded 9.5 g of a white solid (80%). 1H NMR(CDCl3): δ 6.69 (s, 2H), 3.95 (t, 3J = 6 Hz, 6H), 3.0 (s, 1H), 1.77 (m, 6H), 1.27 (m, 54H), 0.89 (m, 9H). iii) A solution of ethyl-4-iodo-benzoate (1.23 g, 0.44 mmol), Pd(PPh3)2Cl2 (0.16 g, 0.22 mmol), CuI (0.034 g, 0.18 mmol) and PPh3 (0.023 g, 0.09 mmol) in 50 mL of NEt3 was stirred under argon (15 min) and the solution of 3,4,5-tridodecyloxyphenylacetylenene (3 g, 4.67 mmol) in 50 mL of NEt3 was added. The mixture was heated at 70 °C (2 d). Purification by column chromatography (SiO2, 4:6 CH2Cl2/petroleum ethers), and crystallization in methanol yielded a white solid (3.2 g, 90%). 1H NMR (CDCl3): δ 8.03 (d, 3J = 9 Hz, 2H), 7.57 (d, 3J = 9 Hz, 2H), 6.76 (s, 2H), 4.39 (q, 3J = 7 Hz, 2H), 3.99 (t, 3J = 7 Hz, 6H), 1.78 (m, 6H), 1.28 (m, 57H), 0.89 (m, 9H).
Compound 20a: As 5: 19a (2.2 g, 3.54 mmol), THF (30 mL), KOH (8 g, 0.14 mol), H2O (10 mL), reflux (2 d), and then HCl (10%, 300 mL) at 0 °C, filtrated, solubilized (CHCl3), washed (H2O), dried (MgSO4). Purified by crystallization in THF/petroleum ethers afforded 2 g of a white solid (100%). 1H NMR (CDCl3): δ 8.08 (d, 3J = 8 Hz, 2H), 7.58 (d, 3J = 8 Hz, 2H), 7.05 (m, 5H), 4.04 (m, 4H), 1.85 (m, 4H), 1.27 (m, 36H), 0.89 (m, 6H).
Compound 20b: As 5: 19b (6.25 g, 7.8 mmol), THF (20 ml), KOH (17.44 g, 312 mmol, 40 eq.), reflux (2 d). Precipitation in HCl (10%), cooled to 0 °C. Crystallization in acetone (5.9 g, 98%, white solid). 1H NMR (CDCl3): δ 8.09 (d, 2H, 3J = 8.55 Hz), 7.59 (d, 2H, 3J = 8.55 Hz), 7.08 (AB, 2H, 3J = 16.22 Hz), 6.74 (s, 2H), 4 (m, 6H), 1.8 (m, 6H), 1.36 (m, 54H), 0.88 (t, 9H, 3J = 6.6 Hz). 13C NMR (CDCl3): 171.44, 153.34, 142.81, 138.96, 131.93, 131.78, 130.64, 127.68, 126.37, 126.19, 105.58, 73.54, 69.22, 31.88, 30.31, 29.71, 29.66, 29.62, 29.57, 29.39, 29.34, 29.33, 26.08, 22.65, 14.06. Elemental analysis calculated for C51H84O5: C, 78.81, H, 10.89; found: C, 79.48, H, 11.05.
Compound 20c: As 5: 19c (3 g, 3.79 mmol). Purification by crystallization in THF/petroleum ethers (2.3 g, 80%, white solid). 1H NMR (CDCl3): δ 8.10 (d, 3J = 9 Hz, 2H), 7.61 (d, 3J = 9 Hz, 2H), 6.77 (s, 2H), 3.99 (t, 3J = 7 Hz, 6H), 1.80 (m, 6H), 1.29 (m, 54H), 0.89 (t, 9H). 13C NMR (CDCl3): δ 171.1, 153.0, 139.6, 131.4, 130.1, 129.1, 128.3, 116.9, 110.3, 93.4, 87.3, 73.5, 69.2, 31.9, 30.3, 29.7, 29.6, 29.5, 29.4, 29.3, 26.0, 14.1. Elemental analysis calculated for C51H82O5: C, 79.02, H, 10.66; found: C, 78.90, H, 10.68. MS-FAB+: found, 774.g mol−1; calculated, 775.21 g mol−1.
Compound 21: As in 18: 20a (0.5 g, 0.84 mmol), DDTBP (0.36 g, 0.93 mmol), Et3N (0.2 mL), NMP (5 mL), 60 °C (3 h), then PPI-G0 (0.056 g, 0.17 mmol), NMP(1 mL). The mixture was stirred at 60 °C (2 d) and then precipitated in an aqueous solution of NaHCO3 (1%, 100 mL). The solid was recovered by filtration, washed (H2O), purified by column chromatography (Al2O3 with CH2Cl2 and then THF). Crystallization in THF/petroleum ether yielded a white solid (160 mg, 35%). 1H NMR (CDCl3): δ 7.76 (d, 3J = 8 Hz, 8H), 7.44 (d, 3J = 8 Hz, 8H), 7.3 (m, 4H), 6.93 (m, 20H), 4.01 (m, 16H), 3.50 (m, 8H), 2.53 (m, 12H), 1.61 (m, 172H), 0.89 (m, 24H). 13C NMR (CDCl3): δ 177.5, 171.1, 167.2, 149.3, 148.8, 140.6, 132.9, 130.5, 129.8, 127.4, 126.1, 125.6, 120.5, 113.5, 111.6, 69.3, 69.1, 31.9, 29.7, 29.6, 29.5, 29.4, 29.3, 26.1, 22.7, 14.1. Elemental analysis calculated for C172H272N6O12: C, 78.98, H, 10.48; found: C, 78.53, H, 10.42. MS-FAB+: found, 2617.2 g mol−1, calculated, 2616.08 g mol−1.
Compound 22: As in 18: 20b (1 g, 0.13 mmol), PPI-G0 (0.085 g, 0.27 mmol), DDTBP (0.55 g, 1.4 mmol). Purification by column chromatography (Al2O3 with CH2Cl2 and then THF) afforded a white solid (298 mg, 33%). 1H NMR (CDCl3): δ 7.78 (d, 8H, 3J = 8.11 Hz), 7.49 (d, 8H, 3J = 8.33 Hz), 7 (AB, 8H, 3J = 16 Hz), 6.69 (s, 8H), 4.0 (m, 24H), 3.5 (m, 8H), 2.47 (m, 12H), 1.7 (m, 24H), 1.38 (m, 216H), 0.88 (t, 36H, 3J = 6.8 Hz). 13C NMR (CDCl3): δ 167.12, 153.32, 151.48, 140.41, 138.73, 135.74, 133.06, 131.93, 130.83, 128.20, 127.42, 126.39, 126.25, 125.47, 105.37, 73.50, 69.17, 53.95, 52.39, 39.14, 34.18, 31.89, 30.35, 30.28, 29.73, 29.68, 29.64, 29.44, 29.36, 29.33, 26.81, 26.12, 25.88, 24.92, 22.67, 21.14, 14.07. Elemental analysis calculated for C220H368N6O16: C, 78.8, H, 11.06, N, 2.51; found: C, 78.09, H, 11.75, N, 2.14. MS-MALDI-TOF (m/z): found, 3348.63 g mol−1 [M + H]+, calculated, 3353.31 g mol−1.
Compound 23: As in 18: 20c (0.5 g, 0.65 mmol), PPI-G0 (0.043 g, 0.14 mmol) and DDTBP (0.27 g, 0.72 mmol). Purification by column chromatography (Al2O3 with CH2Cl2 and then THF), and crystallization in THF–petroleum ethers afforded a white solid (140 mg, 30%) as. 1H NMR (CDCl3): δ 7.79 (d, 3J = 8 Hz, 8H), 7.53 (d, 3J = 8 Hz, 8H), 7.35 (m, 4H), 6.73 (s, 8H), 3.96 (m, 24H), 3.50 (m, 8H), 2.50 (m, 8H), 2.38 (m, 4H), 1.77 (m, 36H), 1.27 (m, 216H), 0.89 (m, 36H). 13C NMR (CDCl3): δ 166.8, 153.0, 139.4, 133.7, 131.5, 127.0, 126.5, 117.0, 110.2, 92.2, 87.3, 73.5, 69.1, 31.9, 30.3, 29.7, 29.6, 29.4, 29.3, 26.1, 22.6, 14.1. Elemental analysis calculated for C220H360N6O16: C, 78.99, H, 10.85, N, 2.51; found: C, 78.02, H, 10.77, N, 2.52. MS-FAB+: found, 3346.46 g mol−1; calculated, 3345.31 g mol−1. CES: Mn = 3650; Mw = 3800; IP = 1.03.
Compound 25: In two steps: i) as in 2, 2 (7 g, 10.7 mmol), Pd(OAc)2 (30 mg, 0.04 eq.), ToP (121 mg, 0.12 eq.), NEt3 (30 mL), stirring (2 h at 60 °C), methylbenzoate styrene [12] (2.68 g, 3.3 mmol), xylene (30 mL), heating (110 °C, 2 d). The mixture was diluted (CH2Cl2), filtrated (celite), extracted (CH2Cl2), washed (H2O), and dried (MgSO4). Purification by column chromatography (SiO2 3:7 CH2Cl2/cyclohexane) yielded a yellow solid (2.11 g, 72%). 1H NMR (CDCl3): δ 8.04 (d, 2H, 3J = 8.33 Hz), 7.58 (d, 2H, 3J = 8.55 Hz), 7.52 (m, 4H), 7.18 (AB, 2H, 3J = 16.44 Hz), 7.00 (AB, 2H, 3J = 16.22 Hz), 6.73 (s, 2H), 4.01 (m, 6H), 3.93 (s, 3H), 1.8 (m, 6H), 1.36 (m, 54H), 0.88 (t, 9H, 3J = 6.7 Hz). ii) as 5: ester (2.17 g, 2.5 mmol), THF (20 ml), KOH (5.6 g, 100 mmol), reflux (2 d). Precipitation in HCl (10%) at 0 °C, filtrated, solubilized (CHCl3), washed (H2O), dried (MgSO4). Crystallization in acetone yielded a yellow solid (1.85 g, 87%). 1H NMR (CDCl3): δ 8.10 (d, 2H, 3J = 8.55 Hz), 7.61 (d, 2H, 3J = 8.55 Hz), 7.53 (m, 4H), 7.20 (AB, 2H, 3J = 16.23 Hz), 7.02 (d, 2H, 3J = 16 Hz), 6.73 (s, 2H), 4.0 (m, 6H), 1.8 (m, 6H), 1.36 (m, 54H), 0.88 (t, 9H, 3J = 6.6 Hz). 13C NMR (CDCl3): δ 171.25, 153.30, 142.71, 138.52, 137.53, 135.73, 132.37, 131.25, 130.67, 129.29, 127.85, 127.18, 127.12, 127.06, 126.73, 126.32, 105.31, 73.53, 69.19, 31.89, 30.31, 29.72, 29.67, 29.63, 29.43, 29.43, 29.39, 29.35, 29.32, 26.09, 22.65, 14.07. Elemental analysis calculated for C59H90O5: C, 80.59, H, 10.32; found: C, 80.69, H, 10.22.
Compound 26: As in 6: 25 (1.5 g, 1.8 mmol), pentafluorophenol (0.41 g, 2.2 mmol), CH2Cl2 stabilized by amylene (25 mL), DCC (0.378 g, 1.8 mmol). Purification by column chromatography (SiO2, 4:6 CH2Cl2/cyclohexane), afforded a yellow solid (1.71 g, 93%). 1H NMR (CDCl3): δ 8.19 (d, 2H, 3J = 8.55 Hz), 7.67 (d, 2H, 3J = 8.55 Hz), 7.54 (m, 4H), 7.27 (AB, 2H, 3J = 16.44 Hz), 7.03 (AB, 2H, 3J = 16.23 Hz), 6.74 (s, 2H), 4.04 (t, 4H, 3J = 6.57 Hz), 3.98 (t, 2H, 3J = 6.58 Hz), 1.8 (m, 6H), 1.38 (m, 54H), 0.89 (t, 9H, 3J = ,6.7 Hz).
Compound 27: As in 18: 26 (1.67 g, 1.64 mmol), CH2Cl2 stabilized by amylene (5 mL), PPI-G0 (123 mg, 0.39 mmol), CH2Cl2 (5 mL) and drops of NEt3. Stirring (RT, 3 d). The crude product was taken up in CH2Cl2 and precipitated in cyclohexane. Purification by column chromatography (Al2O3 with CH2Cl2 and then THF) afforded a waxy yellow solid (1.36 mg, 90%). 1H NMR (CDCl3): δ 7.77 (d, 8H, 3J = 8.33 Hz), 7.48 (d, 8H, 3J = 8.33 Hz), 7.44 (s, 16H), 7.08 (AB, 8H, 3J = 16.23 Hz), 6.96 (AB, 8H, 3J = 16.23 Hz), 6.69 (s, 8H), 4.01 (t, 16H, 3J = 6.57 Hz), 3.96 (t, 8H, 3J = 6.57 Hz), 3.5 (m, 8H), 2.47 (m, 12H), 1.7 (m, 24H), 1.38 (m, 216H), 0.88 (t, 36H, 3J = 6.8 Hz). 13C NMR (CDCl3): δ 167.15, 153.29, 140.28, 138.51, 137.26, 135.83, 133.27, 132.33, 130.10, 129.17, 127.45, 127.16, 127.05, 126.69, 126.38, 105.27, 73.49, 69.17, 53.36, 52.38, 39.13, 31.88, 30.34, 29.73, 29.69, 29.63, 29.44, 29.33, 26.13, 22.65, 14.05. Elemental analysis calculated for C252H392N6O16: C, 80.46, H, 10.05, N, 2.23; found: C, 80.61, H, 10.33, N, 2.00. MS-MALDI-TOF (m/z): found, 3762.36 g mol−1 [M + H]+, calculated 3761.84 g mol−1.
5.5 Prepration of dendrimer G1, 24 (Scheme 4)
Compound 24: As in 18: 20b (5 g, 2.53 mmol), DDTBP (1.07 g, 2.79 mmol), PPI-G1 (0.192 g, 0.26 mmol). Purification by column chromatography (Al2O3 eluting with CH2Cl2 and then THF), crystallization in petroleum ethers afforded a yellow solid (250 mg, 20%). 1H NMR (CDCl3): δ 7.79 (d, 3J = 9 Hz, 16H), 7.70 (m, 8H), 7.41 (d, 3J = 9 Hz, 16H), 6.94 (m, 24H), 6.74 (d, 3J = 9 Hz, 8H), 3.97 (m, 32H), 3.49 (m, 16H), 2.40 (m, 36H), 1.80 (m, 60H), 1.27 (m, 288H), 0.89 (m, 48H). 13C NMR (CDCl3): δ 167.3, 149.6, 149.2, 140.5, 132.8, 130.4, 129.8, 127.6, 126.1, 125.3, 120.5, 113.4, 111.5, 69.3, 69.1, 52.1, 38.9, 31.9, 30.3, 29.7, 29.6, 29.5, 29.4, 26.9, 26.1, 22.7, 14.1. Elemental analysis calculated for C352H560N14O24: C, 78.70, H, 10.51, N, 3.65; found: C, 78.01, H, 10.60, N, 3.63. MS-MALDI-TOF: found, 5373.44 g mol−1, calculated, 5372.40 g mol−1.
5.6 Prepration of dendrons G2, 34 (Scheme 5)
Compound 28: as 19ciii: 3,4,5-tridodecylophenylacetylenene (5 g, 7.78 mmol), 4-iodo-phenol (1.56 g, 7.07 mmol), Pd(PPh3)2Cl2 (0.25 g, 3.53 mmol), CuI (0.054 g, 2.83 mmol) and PPh3 (0.037 g, 1.41 mmol) in of NEt3 (100 mL). Purification by column chromatography (SiO2, CH2Cl2), yielded a white solid (4.5 g, 85%). 1H NMR (CDCl3): δ 7.41 (d, 3J = 8 Hz, 2H), 6.80 (d, 3J = 8 Hz, 2H), 3.97 (m, 6H), 1.78 (m, 6H), 1.28 (m, 54H), 0.89 (m, 9H).
Compound 29: In three steps: i) To a suspension of 3,5-dimethoxyaniline (10 g, 0.065 mol) in 20 mL of an aqueous solution of HCl (15%) was added dropwise NaNO2 (5.23 g, 0.076 mol) solubilized in 40 mL of H2O; the temperature was kept below 10 °C during the addition. The aqueous solution of KI (11 g, 0.066 mol) in 40 mL of H2O was then added with precaution. The mixture was stirred at RT overnight. The product was then extracted (ether), washed (saturated aqueous solution of NaHSO3), dried (MgSO4), filtrated and evaporated to dryness. The purification by column chromatography (SiO2, 3:7 CH2Cl2/petroleum ethers) afforded a white solid (8.2 g, 50%). 1H NMR (CDCl3): δ 6.86 (d, 4J = 2 Hz, 2H), 6.41 (t, 4J = 2 Hz, 1H), 3.77 (s, 6H). ii) The dihydroxy was prepared by demethylation of the dimethoxy (5 g, 0.032 mol) using BBr3. Purification by filtration over silica (THF), and crystallisation in petroleum ethers yielded 3 g of a white solid (70%). 1H NMR (CDCl3): δ 6.80 (d, 4J = 2 Hz, 2H), 6.32 (t, 4J = 2 Hz, 1H), 4.89 (s, 2H). iii) To a solution of 3,5-dihydroxy-iodobenzene (3 g, 5.58 mmol), bromoundecanol (9.57 g, 0.038 mol) and PPh3 (13.33 g, 0.05 mol) in 30 mL of THF, was added dropwise DIAD (10.27 g, 0.05 mol) at 0 °C. The mixture was stirred at RT overnight. The crude product was purified by column chromatography (SiO2, 2:8 CH2Cl2/petroleum ethers) to yield a white solid (3 g, 75%). 1H NMR (CDCl3): δ 6.86 (d, 4J = 2 Hz, 2H), 6.41 (t, 4J = 2 Hz, 1H), 3.96 (t, 3J = 7 Hz, 4H), 3.41 (t, 3J = 7 Hz, 4H), 1.87 (m, 8H), 1.27 (m, 28H).
Compound 30: 28 (3,65 g, 4,97 mmol), 29 (1.58 g, 2.26 mmol) as in 4. Purification by column chromatography (SiO2, 3:7 CH2Cl2/petroleum ethers) afforded 3 g (50%) of a yellow oil. 1H NMR (CDCl3): δ 7.45 (d, 3J = 9 Hz, 4H), 6.84 (m, 6H), 6.71 (s, 4H), 6.39 (t, 4J = 2 Hz, 1H), 3.97 (m, 20H), 1.78 (m, 20H), 1.27 (m, 136), 0.89 (m, 18H).
Compound 31: 30 (3 g, 1.47 mmol), CuI (0.011 g, 0.06 mmol), PPh3 (0.08 g, 0.03 mmol), trimethylsilylacetylene (0,176 g, 1,79 mmol) as in 19c. Purification by column chromatography (SiO2, 1:1 CH2Cl2/petroleum ethers) afforded a yellow oil (2.5 g, 85%). 1H NMR (CDCl3): δ 7.45 (d, 3J = 9 Hz, 4H), 6.84 (d, 3J = 9 Hz, 4H), 6.71 (s, 4H), 6.63 (d, 4J = 2 Hz, 2H), 6.39 (t, 4J = 2 Hz, 1H), 3.97 (m, 20H), 1.78 (m, 20H), 1.27 (m, 136), 0.89 (m, 18H), 0.26 (s, 9H).
Compound 32: 31 (2.5 g, 1.25 mmol), TBAF (0.12 g, 0.44 mmol) as in 3iii. Purification by column chromatography (SiO2, 1:1 CH2Cl2/petroleum ethers) afforded a white solid (2.3 g, 100%). 1H NMR (CDCl3): δ 7.45 (d, 3J = 9 Hz, 4H), 6.85 (d, 3J = 9 Hz, 4H), 6.71 (s, 4H), 6.65 (d, 4J = 2 Hz, 2H), 6.43 (t, 4J = 2 Hz, 1H), 3.96 (m, 20H), 3.05 (s, 1H), 1.80 (m, 20H), 1.27 (m, 136H), 0.89 (m, 18H).
Compound 33: 32 (2.5 g, 1.29 mmol), Pd(PPh3)2Cl2 (0.044 g, 0.06 mmol), CuI (0.009 g, 0.05 mmol), PPh3 (0.006 g, 0.02 mmol), 4-iodophenol (0.27 g, 1.25 mmol) as in 19c. Purification by column chromatography (SiO2, CH2Cl2) afforded a viscous white solid (2 g, 80%). 1H RMN (CDCl3): δ 7.42 (m, 6H), 6.85 (d, 3J = 9 Hz, 4H), 6.78 (d, 3J = 9 Hz, 2H), 6.71 (s, 4H), 6.65 (d, 4J = 2 Hz, 2H), 6.44 (m, 1H), 5.04 (s, 1H), 3.96 (m, 20H), 1.80 (m, 20H), 1.27 (m, 136H), 0.89 (m, 18H).
Compound 34a: 33 (0.5 g, 0.25 mmol), the internal branch (0.08 g, 0.11 mmol) [9] as in 4. Purification by column chromatography (SiO2, 3:7 CH2Cl2/petroleum ethers) afforded a yellow oil (0.25 g, 50%). 1H NMR (CDCl3): δ 8.02 (d, 3J = 8 Hz, 2H), 7.55 (d, 3J = 8 Hz, 2H), 7.43 (d, 3J = 9 Hz, 12H), 7.11 (m, 2H), 6.85 (d, 3J = 9 Hz, 12H), 6.69 (m, 14H), 6.43 (t, 4J = 2 Hz, 3H), 3.96 (m, 51H), 1.78 (m, 48H), 1.27 (m, 300H), 0.89 (m, 36H).
Compound 34b: 34a (0.24 g, 0.05 mmol) as in 5: Triturating in petroleum ether afforded a yellow oil (0.24 g, 100%). 1H NMR (CDCl3): δ 8.04 (d, 3J = 8 Hz, 2H), 7.59 (d, 3J = 8 Hz, 2H), 7.43 (d, 3J = 9 Hz, 12H), 7.13 (m, 2H), 6.85 (d, 3J = 9 Hz, 12H), 6.69 (m, 14H), 6.43 (t, 4J = 2 Hz, 3H), 3.96 (m, 48H), 1.78 (m, 48H), 1.27 (m, 300H), 0.89 (m, 36H).
Acknowledgement
All the authors thank the Thai government (SB), the Ministry of Research (LG), the ANR DENDRIMAT (SB and BD), the CNRS and University Louis Pasteur (Strasbourg) for support and funding. We also thank Dr Cyril Bourgogne (IPCMS) for modelling the supramolecular arrangements.