

1 Introduction
Polyoxometalate (POM) clusters can be formed in acidic solution by condensation reactions of monomeric oxoanions of transition metals belonging mainly to groups V and VI [1]. In particular, a huge number of species featuring tungsten, molybdenum, vanadium and/or niobium atoms have been isolated. In this family of compounds, it has to be distinguished between the compounds comprising only one type of heavy atom, called isopolyoxometalates, and the compounds obtained in presence of a {XO4} additional group (X = PV, AsV, SiIV, GeIV…), called heteropolyoxometalates. To date, four families of compounds and their derivatives dominate the chemistry of POMs, namely Lindquist {M6O19}, Keggin {XM12O40}, Dawson {X2M18O62} and Anderson {M’M6O24} (where M’ is an octahedrally coordinated cation) type systems. For these species, the heavy atoms are generally in their highest oxidation state (WVI, MoVI, VV, NbV,…), but, in some cases, related reduced POMs can be chemically or electrochemically obtained, as shown, for example, by the characterization of the mixed-valence four electron reduced β-[H4PMo12O40]3− polyanion [2]. It has to be noticed that, beside these archetypal families of POMs, the chemistry of molybdenum mixed-valence POM clusters developed by Achim Müller and his collaborators has afforded spectacular, very high nuclearities compounds, such as a species which contains as many as 368 metal atoms [3]. Focusing on polyoxomolybdate systems where all the molybdenum centers are reduced in their V oxidation state, a large number of species have been isolated both in organic or aqueous media. In all cases, these polyanions contain the dinuclear dicationic [MoV2O4]2+ unit, where a metallic bond connects the two d1 centers, leading to a diamagnetic behavior of this fragment even at room temperature. The connection of [MoV2O4]2+ units by various organic ligands (oxalate, squarate, polycarboxylate, carbonate, ethanolate, methanolate, etc.) has afforded systems containing from one to six dinuclear fragments [4]. On the other hand, the ability of the related [MoV2O2S2]2+ oxothiofragment to form polyoxothiometalates has also been studied, affording cyclic systems with nuclearities ranging from 8 to 16 [5].
In our group, we have focused our investigations involving the [MoV2O4]2+ building unit on the polycondensation of this dinuclear fragment via pyrophosphate [O3POPO3]4− and methylenediphosphonate [O3P(CH2)PO3]4− ligands in the eventual presence of other coordinating groups (carboxylate, carbonate, sulfite…) in aqueous media. Our investigations have allowed us to isolate a wide variety of pure MoV cyclic POM clusters of various nuclearities. For example, the triangular cluster [(MoV2O4)3(O3P(CH2)PO3)3(CH3AsO3)]8− has been obtained in presence of methyl arsenate,[6] while ellipsoidal octanuclear clusters have been isolated when the synthetic process is performed in presence of sulfite or carbonate [7]. A larger complex, a dodecanuclear wheel which adopts a cyclohexane-like ring in a chair conformation has been obtained in absence of any additional coordinating groups. Nevertheless, the presence of ammonium cations in the reacting medium is crucial for the obtaining of this species, highlighting the importance of the nature of the counter-ions on the resulting POM [6]. In addition to these cyclic systems, bicyclic compounds have also been obtained. The isolated isostructural complexes [Na2{(MoV2O4)10(O3P(X)PO3)10(CH3COO)8}]24− (X = O, CH2) are made of two interconnected and nearly perpendicular octanuclear and dodecanuclear wheels, respectively [8]. Recently, we have shown that such diphosphonate MoV species are able to form and stabilize metal nanoparticules (NPs), which, from an electrocatalytical point of view, can combine the properties of metal NPs and POM complexes. Moreover, such nanosystems are obtained in a one-pot procedure, in water, at room temperature and without any additional surfactant. For example, by simply mixing the cyclohexane-like dodecanuclear compound mentioned above and PdII cations in aqueous solution, NPs with diameter of 9–14 nm containing a Pd0 core and capped by POMs are obtained [6]. Additionally, it has also been shown that Ag0 [9] and Pt0 [10] POM capped NPs can be obtained using this procedure. Beside the observation that this family of POMs allows the formation of NPs, it has also been stated that the sizes and shapes of the obtained nanosystems depend on the nature of the polynuclear MoV complex used. In order to synthesize NPs with the desired morphology, it is thus essential to elaborate a library of reduced POMs with different structural and redox characteristics.
In this article, we present our results concerning the synthesis and the characterization of polynuclear MoV POMs obtained by condensation of the dinuclear [MoV2O4]2+ fragment with the 1-hydroxyethane 1,1-diphosphonate (or etidronate) ligand [O3P(C(CH3)(OH))PO3]4− (Fig. 1a). Our goal was to answer two questions: (a) do the potentially available MoV/etidronic POMs display the same topologies as those obtained by condensation of dinuclear [Mo2O4]2+ fragments with methylenediphosphonic ligands; and (b) can the hydroxo group grafted on the carbon center linking the two phosphorous atoms in [O3P(C(CH3)(OH))PO3]4− act as a ligand for the MoV cations? We thus report here the structural characterization of the tetranuclear compound (CN3H6)6[(MoV2O4)2(O3P(C(CH3)O)PO3)2]·12H2O (1), the dodecanuclear complex (NH4)18[(MoV2O4)6(OH)6(O3P(C(CH3)(OH))PO3)6]·35H2O (2) and the tetradecanuclear species (CN3H6)18Na3[(MoV2O4)7(O3P(C(CH3)O)PO3)7(CH3COO)7]·5CH3COONa 52H2O (3). Their stability in aqueous media has also been studied by 31P NMR.

(a) Representation of the [O3P(C(CH3)(OH))PO3]4− etidronate ligand; (b) Mixed ball-and-stick and polyhedral representation of the tetranuclear [(MoV2O4)2(O3P(C(CH3)O)PO3)2]4− polyanion in 1. The hydrogen atoms are omitted for clarity. Yellow octaedra, MoO6; green spheres, P; black spheres, C; red spheres, O.
2 Experimental section
2.1 Preparation of a [MoV2O4]2+ solution
Two hundred and ten microliters (4.29 mmol) of N2H4·H2O are added to a suspension of 2.30 g (16.0 mmol) of MoO3 in 80 mL of 4 M HCl. The mixture is then stirred at 80 °C for ca. 3 h. The resulting deep red solution is allowed to cool down to room temperature before further use.
2.2 Synthesis of (CN3H6)6[(MoV2O4)2(O3P(C(CH3)O)PO3)2]·12H2O (1)
One hundred and forty-one milligrams (0.63 mmol) of etidronic acid H2O3P(C(CH3)(OH))PO3H2·H2O are dissolved in 6.25 mL of a solution of [MoV2O4]2+ prepared as described above. A 8 M NaOH solution is added dropwise until reaching pH = 6.4, followed by the addition of 1 g (10.4 mmol) of guanidinium chloride. The resulting deep red solution is filtrated and left to evaporate slowly at room temperature. Red parallelepipedic crystals are collected by filtration after one day. Yield: 309 mg (67% based on Mo). Anal. calc. for C10H66O34N18P4Mo4 (found): C 8.06 (7.73), H 4.46 (3.83), N 16.92 (15.94), P 8.31 (8.38), Mo 25.75 (25.68). IR (KBr pellets): ν = 1449 (w), 1443 (w), 1369 (w), 1191 (m), 1140 (sh), 1127 (s), 1046 (s), 1000 (s), 965 (s), 951 (sh), 930 (m), 903 (m) cm−1.
2.3 Synthesis of (NH4)18[(MoV2O4)6(OH)6(O3P(C(CH3)(OH))PO3)6]·35H2O (2)
One hundred and forty-one milligrams (0.63 mmol) of etidronic acid H2O3P(C(CH3)(OH))PO3H2·H2O are dissolved in 6.25 mL of a solution of [MoV2O4]2+ prepared as described above. A 33% NH3 solution is added dropwise until reaching pH = 7.4. The resulting deep red solution is filtrated and left to evaporate slowly at room temperature. Red parallelepipedic crystals are collected by filtration after three days. Yield: 110 mg (28% based on Mo). Anal. calc. for C12H172O107N18P12Mo12 (found): C 3.79 (3.78), H 4.56 (4.25), N 6.63 (6.88), P 9.77 (9.65), Mo 30.26 (29.61). IR (KBr pellets): ν = 1398 (s), 1144 (s), 1131 (sh), 1082 (m), 1052 (s), 1007 (s), 961 (w), 920 (m), 903 (m) cm−1.
2.4 Synthesis of (CN3H6)18Na3[(MoV2O4)7(O3P(C(CH3)(OH))PO3)7(CH3COO)7]·5CH3COONa 52H2O (3)
In 10 mL of 4 M CH3COONa/CH3COOH buffer (pH = 4.9) are dissolved 242 mg (1.0 mmol) of Na2MoO4·2H2O, 112 mg (0.5 mmol) of etidronic acid H2O3P(C(CH3)(OH))PO3H2·H2O, 500 mg (5.2 mmol) of guanidinium chloride and 50 μL (1.0 mmol) of hydrazine hydrate. The resulting suspension is stirred for 15 min at room temperature before filtration. The red filtrate is left to evaporate slowly at room temperature. Small red cubic crystals are collected by filtration after one day. Yield: 240 mg (55% based on Mo). Anal. calc. for C56H294N54O153P14Mo14Na8 (found): C 10.96 (11.01), H 4.83 (4.07), N 12.33 (12.37), P 7.07 (7.05), Mo 21.90 (22.45) Na 3.00 (2.91). IR (KBr pellets): ν = 1558 (m), 1539 (m), 1434 (m), 1162 (m), 1136 (m), 1109 (w), 1081 (w), 1037 (s), 1002 (s), 966 (sh), 923 (w) cm−1.
2.5 X-ray structure determination of 1, 2 and 3
Intensity data collections were carried out with a Siemens SMART three-circle diffractometer for 1 and 2 and with a Bruker Nonius X8 APEX 2 diffractometer for 3, each equipped with a CCD bidimensional detector using the monochromatized wavelength λ(Mo Kα) = 0.71073 Å. The absorption corrections were based on multiple and symmetry-equivalent reflections in the data set using the SADABS program [11] based on the method of Blessing [12]. The structures were solved by direct methods and refined by full-matrix least-squares using the SHELX-TL package [13]. In the structures of 2, NH4+ and H2O could not be distinguished according to the observed electron densities; therefore all the positions were labelled O and assigned the oxygen atomic diffusion factor. As 3 rapidly looses water at room temperature, a single crystal of this compound was glued in Paraton-N oil and the data set collected at 100 K. However, despite many efforts, it was not possible to obtain an entirely satisfying data set for 3 probably because of its great instability. It was thus not possible to locate the guanidinium counter-ions. The number of guanidinium cations indicated in the formula of compound 3 has then been determined considering the elemental analysis results. As generally observed for POM compounds, in all three structures there is a discrepancy between the formulae determined by elemental analysis and the formulae deduced from the crystallographic atom list because of the difficulty in locating all the disordered water molecules and counter-ions [8]. The data sets for 2 and 3, which contain large voids occupied by solvent molecules were corrected with the program SQUEEZE, a part of the PLATON package of crystallographic software used to calculate the solvent disorder area and remove its contribution to the overall intensity data [14]. Crystallographic data are given in Table 1. Selected bond lengths are given in Table 2.
Crystallographic data and details of diffraction experiments for 1–3.
| 1 | 2 | 3 | |
| Empirical formula | C10H66O34N18P4Mo4 | C12H172O107N18P12Mo12 | C56H294N54O153P14Mo14Na8 |
| Formula weight (g) | 1490.39 | 3804.53 | 6133.94 |
| Crystal system | Triclinic | Monoclinic | Monoclinic |
| Space group | P-1 | C2/c | C2/m |
| a/Å | 12.362(1) | 31.040(5) | 24.868(4) |
| b/Å | 13.085(1) | 17.130(2) | 33.141(6) |
| c/Å | 15.595(1) | 23.651(3) | 28.910(5) |
| α/deg | 89.170(2) | 90 | 90 |
| β/deg | 89.418(2) | 101.017(4) | 101.15(1) |
| γ /deg | 89.430(2) | 90 | 90 |
| V/Å3 | 2522.0(3) | 12344(3) | 23376(7) |
| Z | 2 | 4 | 4 |
| T/K | 293 | 293 | 100 |
| ρcalc/g cm−3 | 1.943 | 1.986 | 1.483 |
| μ/mm−1 | 1.212 | 1.451 | 0.917 |
| Data/parameters | 14282/622 | 17856/606 | 15455/681 |
| Rint | 0.0328 | 0.0711 | 0.1070 |
| GOF | 1.075 | 1.043 | 0.940 |
| R (>2σ(I)) | R1 = 0.0566 | R1 = 0.0789 | R1 = 0.0696 |
| wR2 = 0.1435 | wR2 = 0.1990 | wR2 = 0.1884 |
Selected bond lengths (Å) for 1–3.
| 1 | 2 | 3 | |
| MoVO | 1.677(4)–1.687(4) | 1.679(6)–1.706(6) | 1.630(4)–1.704(5) |
| [1.684] | [1.691] | [1.667] | |
| MoVOba | 1.941(3)–2.076(3) | 1.918(6)–2.107(6) | 1.838(5)–2.151(4) |
| [2.014] | [2.011] | [2.016] | |
| MoVOtb | 2.329(3)–2.477(3) | 2.209(5)–2.240(5) | 2.221(5)–2.332(4) |
| [2.394] | [2.222] | [2.282] | |
| MoVMoV | 2.5755(6)–2.5801(6) | 2.5752(11)–2.5852(9) | 2.5482(9)–2.5708(10) |
| [2.578] | [2.580] | [2.561] | |
| PO | 1.489(4)–1.577(4) | 1.498(6)–1.519(6) | 1.434(6)–1.522(7) |
| [1.504] | [1.508] | [1.449] | |
| POMo | 1.547(4)–1.577(4) | 1.513(6)–1.550(6) | 1.475(4)–1.564(4) |
| [1.562] | [1.531] | [1.521] | |
| PC | 1.828(5)–1.852(5) | 1.813(9)–1.848(10) | 1.736(10)–1.941(7) |
| [1.838] | [1.825] | [1.809] | |
| POM size | 7 × 12 | 16 × 18 | 23 × 23 |
a MoO bond with bridging oxygen atoms.
b MoO bond trans to the MoO bond.
2.6 NMR spectroscopy
The 31P NMR spectra were recorded on a Brüker AC-300 spectrometer operating at 121.5 MHz in 5-mm tubes with 1H decoupling.
2.7 Elemental analysis
The elemental analysis of complexes 1, 2 and 3 were performed by the Service central d’analyse élémentaire, CNRS, 69390 Vernaison, France.
2.8 FTIR spectroscopic analysis
The IR spectra of complexes 1, 2 and 3 were recorded on an IRFT Magna 550 Nicolet spectrophotometer using the technique of pressed KBr pellets.
3 Results and discussion
Compound 1 has been obtained by adding solid etidronic acid to a solution of [MoV2O4(H2O)6]2+. The pH of the mixture was raised to 6.4 with sodium hydroxyde, followed by an addition of guanidinium chloride. After slow evaporation of the filtrate, red crystals of 1 were obtained. A single crystal X-ray diffraction study revealed that 1 is made of a [(MoV2O4)2(O3P(C(CH3)O)PO3)2]6− tetranuclear polyanion (Fig. 1b) with six guanidinium counter-ions. Two crystallographically non-equivalent [(MoV2O4)2(O3P(C(CH3)O)PO3)2]6− subunits have been found in the unit cell. Nevertheless, these two subunits are highly similar and will be considered as identical for the forthcoming discussion. Each [(MoV2O4)2(O3P(C(CH3)O)PO3)2]6− unit contains a centrosymetric, rhomboid {Mo4O16} core with localized metal–metal bonds. Such topology is recurrent in the MoV chemistry [15]. The two crystallographically equivalent etidronate groups constituting each [(MoV2O4)2(O3P(C(CH3)O)PO3)2]6− fragment in 1 act as tetradentate ligands, respectively (see Scheme 1a). For each [O3P(C(CH3)O)PO3]5− ligand, one {PO3} group is monocoordinated to a MoV center, while the other phosphonato group is coordinated via two oxygen atoms to an adjacent metallic center. Additionally, the hydroxo group bound to the carbon atom connecting the PV centers is found to be deprotonated, and acts as a μ2O ligand. The characterization of 1 then allows one to answer the questions asked in the introduction section: the hydroxo group of an etidronate ligand can be deprotonated and connect MoV centers even in slightly acidic medium, and then the reactivity of this ligand can be different from the reactivity of the methylenediphosphonate ligand. Moreover, it should be mentioned that, despite the large number of MoV / methylenediphosphonate ligands characterized to date, no complex possessing a {MoV4O16} core had been observed using this ligand, confirming that the MoV chemistry involving the methylenediphosphonate ligand cannot be transposed to that involving the etidronate ligand. The stability of 1 in aqueous medium has been studied. The 31P NMR spectrum in water is represented in Fig. 2. It exhibits a doublet of doublets at 32.28, 32.10, 22.12 and 21.93 ppm (J = 22.1 Hz). Such a spectrum is in agreement with the presence of two independent and coupled phosphorous atoms, indicating that the structure observed in the solid state for the anion constituting 1 is maintained in solution.
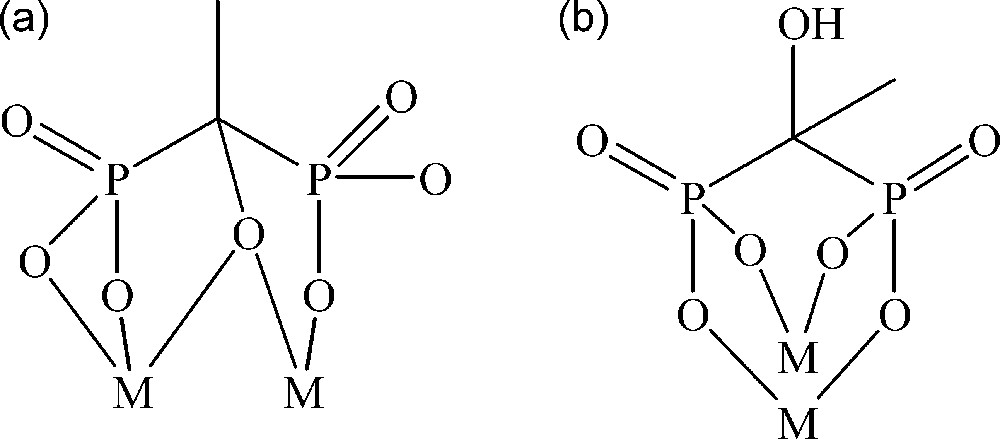
Representation of the coordination modes of the etidronate ligand in (a) complex 1 and (b) complexes 2 and 3.
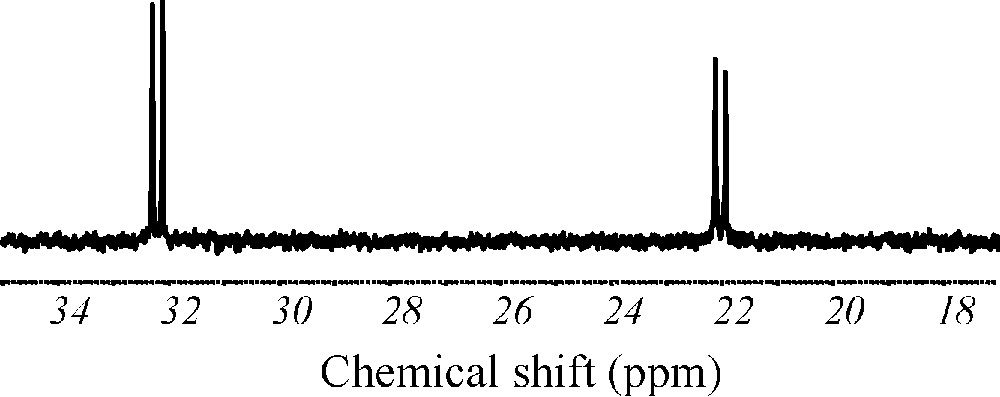
31P NMR spectrum of a ca. 0.02 mol.L−1 solution of (CN3H6)6[(MoV2O4)2(O3P(C(CH3)O)PO3)2]·12H2O (1) in unbuffered H2O/D2O (9:1).
Compound 2 has been synthesized using a similar procedure to that described for compound 1, but in this case the pH of the solution was adjusted to 7.3 with ammonia and no additional cationic species was added. Its structure (Fig. 3) is similar to that previously described for the [(MoV2O4)6(OH)6(O3P(CH2)PO3)6]18− methylenediphosphonato complex that we recently reported [6]. It consists in six connected {(MoV2O4)(O3P(C(CH3)(OH))PO3} units which form a cyclohexane-like ring in a chair conformation and where the tetracoordinated etidronate ligands (see Scheme 1b) are at the vertices and the edge-sharing metallic octahedra are at the edges, leading to a wheel with a pseudo S6 symmetry. Bond valence summations indicate that six MoV bridging oxygen atoms are protonate [16]. Additionally, contrary to compound 1, in 2 the hydroxo groups of the etidronate ligands are not deprotonated, explaining why the analogous methylenediphosphonate complex can be obtained. As previously found for this last compound, 31P NMR studies indicate that 2 degrades rapidly in pure water into unknown products. Moreover, we have not been able to find an aqueous medium (1 M LiCl, 1 M NaCl…) where compound 2 can be stabilized.

Mixed ball-and-stick and polyhedral representations of the dodecanuclear [(MoV2O4)6(OH)6(O3P(C(CH3)O)PO3)6]18− polyanion in 2. The hydrogen atoms are omitted for clarity. Yellow octaedra, MoO6; green spheres, P; black spheres, C; red spheres, O.
Compound 3 was prepared by mixing sodium molybdate, etidronic acid, guanidinium chloride and hydrazine in 4 M CH3COONa/CH3COOH buffer (pH = 4.9). After slow evaporation of the filtrate for one day, crystals of 3 were obtained. In 3, seven {(MoV2O4)(O3P(C(CH3)(OH))PO3)(CH3COO)} units (Fig. 4a) are interconnected, forming a tetradecanuclear wheel (Fig. 4b) with an internal diameter of ca. 12 Å. In each dinuclear building unit, the oxygen atoms of the μ1,1 acetato ligand are located in axial position of the distorted {MoVO6} octahedra, in trans position of the terminal Mo = O oxo groups. As in complex 2, the hydroxo groups of the etidronate ligands are not deprotonated. It follows that in each {(MoV2O4)(O3P(C(CH3)(OH))PO3)(CH3COO)} building unit, the [O3P(C(CH3)(OH))PO3]4− groups are doubly coordinated to the [MoV2O4]2+ fragment (see Scheme 1b), the oxygen atoms of the etidronate ligand connected to the MoV center occupying two equatorial positions. The connection of seven building units affords an almost planar cyclic system. From a crystallographic point of view, a mirror plane passing through the phosphorous atoms of an etidronic ligand and the carbon atoms of an acetate ligand (see Fig. 4b) is observed. The presence of this symmetry element induces a crystallographic disorder of the methyl and the hydroxo group of the etidronate ligand located on the mirror plane, these groups occupying two crystallographic sites with an occupancy factor of 0.5, respectively. It then follows that 3 exhibits only a Cs symmetry. The solution behaviour of this compound has also been studied. As for compound 2, 31P NMR studies revealed that 3 degrades into unknown products in aqueous media.

(a) Mixed ball-and-stick and polyhedral representation of the {(MoV2O4)(O3P(C(CH3)(OH))PO3)(CH3COO)} building units in 3; (b) the tetradecanuclear [(MoV2O4)7(O3P(C(CH3)O)PO3)7(CH3COO)7]21− polyanion in 3, with the representation of the σv mirror plane. The double arrow indicates the two methyl and hydroxo groups disordered in 3. The hydrogen atoms are omitted for clarity. Yellow octaedra, MoO6; green spheres, P; black spheres, C; red spheres, O.
4 Conclusion
We have shown here that it is possible to isolate MoV/etidronate cyclic systems of various shapes and nuclearities. Moreover, depending on the synthetic conditions, it appears that the hydroxo group of the [O3P(C(CH3)(OH))PO3]4− can either be deprotonated and then connect MoV centers (compound 1) or remain protonated (compound 2 and 3). The experimental factors governing the possible deprotonation of this ligand in presence of MoV cations have not been clearly elucidated, but it can be remarked that the deprotonation processes cannot be attributed exclusively to the pH of the reacting medium. Indeed, 1 has been isolated at pH = 6.4 while 2 was obtained at pH = 7.4. The role of the counter-ions on the formation of the final product then seems crucial, as previously observed for the family of methylenediphosphonate POMs [8]. Obviously, when deprotonation occurs, systems that cannot be obtained using the methylenediphosphonate ligands are isolated, but it can be noticed that to date, surprisingly, no methylenediphosphonato MoV complex analogous to complex 3 has been isolated. Finally, to the best of our knowledge, complex 3 represents the homometallic MoV polyoxometalate cyclic system with the highest nuclearity reported to date. The next step is now to study the ability of these systems to form and stabilize metal nanoparticules. Additionally, the synthesis of analogous POM compounds containing functionnalized diphosphonate ligand [O3P(C(OH)R)PO3]4− where R is an alkyl group bearing amino, phenolato, thiophene, etc. functional groups will be published soon. Finally, the reactivity of these diphosphonate ligands with MoVI cations must be studied [17].
5 Supplementary materials
The supplementary material has been sent to the Cambridge Crystallographic Structural Database, 12 Union Road, Cambridge, CB2 1EZ, UK, as supplementary material CCDC Nos. 734898, 734899 and 734900, for 1, 2 and 3, respectively, and can be obtained by contacting the CCDC, (fax: +44-1223-336033; e-mail: deposit @ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk).
6 Table of content
The reactivity of the [MoV2O4]2+ dinuclear unit with the [O3P(C(CH3)(OH))PO3]4− etidronate ligand has been investigated for the first time. Three complexes of nuclearities ranging from four to 14 have been isolated and characterized by IR spectroscopy, elemental analysis and single crystal X-Ray diffraction studies. Their stability in aqueous solution has been studied by 31P NMR.