

1 Introduction
Control of magnetic coupling, in addition to the possibility of inducing a spin-state modification at the molecular scale, are among the most challenging themes in the field of molecular magnetism. Considering a large variety of magnetically interesting molecules [1], special attention has been paid in our lab to exchange coupled multinuclear transition metal paramagnetic complexes [2–4]. Systems presenting a lower nuclearity are of interest at least as versatile building blocks for cooperatively associated magnetic systems [5]. Due to the dependence of coupling pathways on the structure and symmetry of the organic ligands, dinuclear molecular systems offer interesting possibilities to tune metal-to-metal interactions by subtle structural changes within the organic periphery.
We earlier reported on an interesting dinuclear MnIII complex [Mn2(HL)4(μ-OCH3)2] (H2L is 2-salicyloylhydrazono-1,3-dithiolane) which was found to exhibit the largest J value (J = +19.7 cm−1) reported so far for a MnIII–MnIII interaction [4]. On this basis, we have been working on slight modifications of its organic peripheral backbone as well as on using other metal centers. The peculiarity of the [Mn2(HL)4(μ-OCH3)2] solid-state structure arises from an unsymmetrical arrangement of the ligands, a feature most likely responsible for its ferromagnetic ground state. Recent work in our lab clearly established that the latter ferromagnetic MnIII complex only exists in the solid state [6]. A similar observation was made for the analogous FeIII complex supported by the same H2L ligand) that features, as expected, an antiferromagnetic exchange coupling behavior [7].
In an effort to gain more insight into the structural factors controlling and influencing the formation and the stabilization of both mono- and/or dinuclear complexes in the crystalline state (with possible impact on their magnetic behavior), the original H2L ligand (named L1 in this work) was modified at specific positions (in this work the ligand 2-salicyloylhydrazono-1,3-dithiane – named L2 – was synthesized). Moreover, the chemistry of other transition metals has been investigated systematically. In the present article, we report on the synthesis and structural characterization of novel CoII and CoIII compounds supported by these two benzoic hydrazide ligands.
2 Results and discussion
The initial task of the present work dealt with the synthesis and characterization of benzoic hydrazide ligand derivatives – 2-salicyloylhydrazono-1,3-dithiane (L2) – which are related to the original ligand we used (2-salicyloylhydrazono-1,3-dithiolane), yet are slightly modified (the five membered cycle – dithiolane, ligand L1 – is replaced by a six member cycle – dithiane, ligand L2). Both ligands (L1 and L2) were prepared according to literature procedures [8–10]. During the present work, the crystal structure of L2 has been determined from single crystal X-ray diffraction. This result is briefly described hereafter.
2.1 Solid-state structures of the bidentate ligand L2
Crystal data and refinement details are gathered in Table 1 while selected bond lengths and angles are included in the figure caption (Fig. 1). The ligand L2 crystallizes (colorless single crystals) in the monoclinic P21/c space group. In the solid state, the L2 molecules are not planar (the angle between the phenol cycle and the average plan crossing the dithiane cycle is about 30° as clearly shown on the Fig. 1a), which contrasts with our previous observations in related ligands [6] (the ligand L1 for instance). This rotation starts from the C6C7 single bond (Fig. 1b), preventing the possible covering of both delocalized π electrons system (phenol group and hydrazine group) occurring in such molecule. This phenomenon could be a crucial parameter for metal complex stability as well as their physical properties (magnetic or photophysical for example). One classical strong hydrogen bond is detected between N1 and O1 with the specific positions of both hydrogens bonded on N1 and O1 clearly depicted on Fig. 1b (dashed line).
Crystal data and refinement details for L2 and complexes 1, 2 and 3·CH2Cl2.
| Compound | Ligand L2 | Complex 1 | Complex 2 | Complex 3·CH2Cl2 |
| Formula | C11H12N2O2S2 | C28H34CoN4O6S4 | C26H32CoN6O6S4 | C23H26Cl4CoN4O4S4 |
| Formula weight | 268.35 | 709.76 | 711.75 | 751.45 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Crystal color | Colorless | Orange | Orange | Yellow |
| Crystal size | 0.14 × 0.12 × 0.10 | 0.14 × 0.12 × 0.10 | 0.14 × 0.12 × 0.10 | 0.14 × 0.12 × 0.10 |
| Space group | P 21/c | P-1 | C 2/c | P 21/c |
| a (Å) | 12.4480(5) | 9.4170(2) | 17.0940(6) | 9.0089(2) |
| b (Å) | 8.3280(4) | 9.7000(4) | 8.6200(2) | 11.2411(4) |
| c (Å) | 12.0840(6) | 18.1100(7) | 22.4670(8) | 15.5647(6) |
| α (°) | 90.00 | 99.3100(16) | 90 | 90.00 |
| β (°) | 105.880(3) | 92.216(2) | 108.272(12) | 105.083(2) |
| γ (°) | 90.00 | 105.494(2) | 90 | 90.00 |
| V (A3) | 1204.90(10) | 1567.30(9) | 3143.6(3) | 1521.93(9) |
| Z | 4 | 2 | 4 | 2 |
| Density (g.cm−3) | 1.479 | 1.504 | 1.504 | 1.640 |
| mu (Mo Kα) (mm−1) | 0.432 | 0.862 | 0.861 | 1.227 |
| F(000) | 560 | 738 | 1476 | 766 |
| Temperature (K) | 173(2) | 173(2) | 173(2) | 173(2) |
| Theta (min – max) | 2.98–30.03 | 1.14–30.02 | 1.91–34.97 | 2.26–27.47 |
| Dataset [h, k, l] | −17/17, −11/10, −16/14 | −13/13, −13/13, −25/25 | −27/13, −13/11, −36/36 | −10/11, −14/13, −19/20 |
| Tot., Uniq. Data, R(int) | 8755, 3500, 0.0712 | 17410, 9070, 0.0521 | 13326, 6763, 0.0505 | 13746, 3495, 0.0393 |
| Observed data (> 2σ(I)) | 3022 | 6319 | 5322 | 2739 |
| Nreflections, Nparameters | 3500, 154 | 9070, 399 | 6763, 200 | 3495, 196 |
| R2, R1, wR2, wR1, Goof | 0.0715, 0.0606, 0.1492, 0.1441, 1.112 | 0.0814, 0.0472, 0.1344, 0.1169, 1.039 | 0.0633, 0.0449, 0.1559, 0.1386, 1.068 | 0.0639, 0.0451, 0.1407, 0.1241, 1.086 |
| Max. and Av. Shift/Error | 0.001, 0.000 | 0.002, 0.000 | 0.001, 0.000 | 0.000, 0.000 |
| Min, Max. Res. (e.Å−3) | −0.623, 0.755 | −1.193, 0.473 | −1.512, 1.524 | −0.616, 0.853 |

a and b: Two perpendicular ORTEP views of the ligand L2 with full labeling scheme. The ellipsoids enclose 50% of the electronic density. Selected distances (Å): C8-N2: 1.295(3); N2-N1: 1.393(3); N1-C7: 1.359(3); C7-O2: 1.221(3).
2.2 Synthesis and structural characterization of CoII complexes
We then decided to explore the chemistry of CoII complexes with the supported ligands L1 and L2. Such complexes are paramagnetic compounds and thus potentially molecular magnets considering the polynuclear species. Moreover, comparison could be made with the previous MnII and FeII complexes previously obtained during our systematic work [7,8]. The synthesis and crystal structure of these complexes is described here.
Considering the new complexes synthesized in this present work, commercial crystalline CoCl2 was selected as main metal source. Direct reactions were successively carried out with L1 and L2 in different solvents (THF, DMF and Methanol).
The general way to obtain the CoII complexes is described in Scheme 1. Three new compounds were obtained and characterized (see experimental section and Table 1) following this procedure: complex 1 [CoII(L1)2(THF)2], complex 2 [CoII(L1)2(DMF)2] and complex 3 [CoII(L2)2Cl2]. Their crystal structures were established from single crystal X-ray diffraction.

General procedure for the synthesis of the CoII complexes. Solvents are THF, DMF or CH2Cl2. (n = 1 or 2 for L1 and L2 respectively).
Remark: the charge (0 or −1) of the ligands has been clearly identified by single crystal X-ray diffraction, i.e., presence (charge 0) or absence (charge −1) of the hydrogen atom on N1 (Fig. 1b) as well as significant variation of dC---O(»1.245 Å for charge 0 and »1.280 Å for charge −1) and significant variation of dC---N(»1.345 Å for charge 0 and »1.325 Å for charge −1). These phenomenons have been confirmed for more than 12 other complexes synthesized and structurally characterized in our lab. These results will be published shortly. These observations indicate that the charge −1 for deprotonated ligands is delocalized between O2 and N2 – Fig. 1b.
The orange complex 1 [CoII(L1)2(THF)2] (Fig. 2) crystallizes in the triclinic centrosymmetric space group P-1. The asymmetric unit is constituted by two almost identical molecules of 1. Only one of them is described here (see also the corresponding cif file for further information). The crystal structure clearly shows that the CoII ion is surrounded by two L1 (with the charge −1) ligands in trans situation. The Fourier map differences (crystal structure refinement with Shelxl97) clearly shows the absence of hydrogen atoms on N1 (or N1a, Fig. 2), confirming the charge (1 for the ligand). The octahedral sphere is completed by two THF molecules, indicating the relative lability of the chloride atoms in such CoII system. This lability is not observed in the case of FeII species [7,11]. All distances and angles (Fig. 2 caption) are coherent with such CoII complex. Quite similar species were observed previously with MnII ion (complex 4b in [8]).
ORTEP view of one molecule of the asymmetric unit of complex 1 with partial labeling scheme. The ellipsoids enclose 50% of the electronic density. Selected distances (Å): Co1-O2: 1.992(1); Co1-N2: 2.133(1); Co1-O3: 2.179(1); N2-N1: 1.412(2); O2-C7: 1.277(2). Symmetry codes for equivalent positions: a: −x + 1, −y + 1, −z + 1.
The orange complex 2 [CoII(L1)2(DMF)2] (Fig. 3) crystallizes in the monoclinic centrosymmetric space group C 2/c. Here again, the crystal structure shows that the CoII ion is surrounded by two L1 (with the charge (1) ligands in trans configuration). In this case, the octahedral sphere is completed by two DMF molecules. Selected distances are given in the caption of Fig. 3. Similar complexes have been already obtained with FeII ion11, and also with MnII but in this last case8 a cis- configuration is observed for both ligands and the DMF molecules.

ORTEP view of complex 2 with partial labeling scheme. The ellipsoids enclose 50% of the electronic density. Selected distances (Å): Co-O2: 2.015(1); Co-N2: 2.134(1); Co-O3: 2.174(1); N2-N1: 1.404(2); O2-C7: 1.281(2). Symmetry codes for equivalent positions: a: −x + 3/2, −y + 1/2, −z + 1.
Several syntheses have been made, testing the reactivity of the ligand L2 with CoII salt, with various solvents. Only one new complex was stabilized and characterized with this ligand. The corresponding yellow complex [complex 3·CH2Cl2, CoII(L2)2(Cl)2·CH2Cl2] (Fig. 4) crystallizes in the monoclinic centrosymmetric space group P 21/c. As often observed with such ligands, the crystal structure shows that the CoII ion is surrounded by two neutral L2 ligands in trans situation. The CoII surrounding is (a deformed octahedral with typical distances and angles – selected distances are given in Fig. 4 caption) completed by two Cl− in accordance with the charge + 2 for the metal centre.

ORTEP view of complex 3·CH2Cl2, with partial labeling scheme. The solvent molecule was removed for clarity. The ellipsoids enclose 50% of the electronic density. Selected distances (Å): Co1-O1: 2.057(2); Co1-N1: 1.186(2); Co1-Cl1: 2.446(1); N2-N1: 1.384(3); O1-C7: 1.245(3). Symmetry codes for equivalent positions: a: −x + 1, −y + 1, −z.
2.3 Synthesis and structural characterization of CoIII complexes
Considering the possibility of stabilizing CoIII complexes with these supporting ligands, several tests were done. The target point was to obtain dinuclear CoIII compounds. Firstly, the oxidation process of CoII into CoIII was driven in the presence of air, with a basic solution. With this approach, three new complexes (two mononuclear and one dinuclear) were obtained and structurally characterized. Direct reaction of the ligands with Co(OH)3 freshly prepared (see below) is another way to synthesize these mononuclear complexes as well as the dinuclear compounds (with two equivalents of the ligand), as described in the experimental section. The Co(OH)3 reagent was prepared following this procedure:
| 2CoCl2(aq) + NaOCl(aq) + 4NaOH + H2O → 2Co(OH)3(s) + 5 NaCl |
The general way to obtain the CoIII complexes is described in Scheme 2.
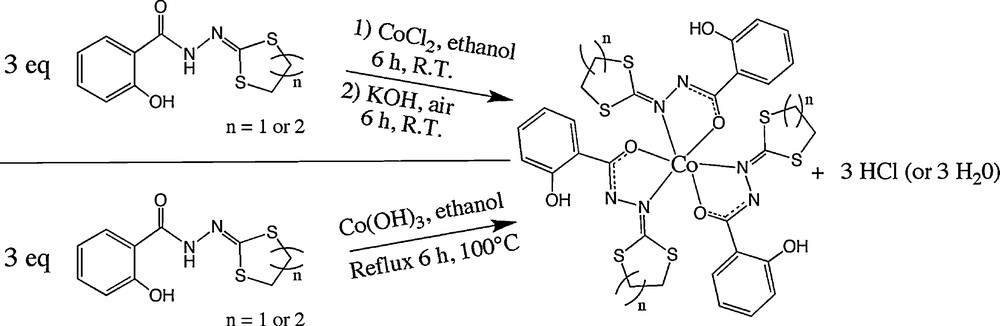
The two different ways for the synthesis of the CoIII complexes.
Both CoIII mononuclear complexes 4·CH2Cl2 and 5·CH2Cl2 supported by L1 and L2 ligands respectively were obtained as single crystals suitable for X-ray diffraction. Their crystal structures are described hereafter.
* The red brown complex 4·CH2Cl2 [CoIII(L1)3·CH2Cl2] (Fig. 5) crystallizes in the monoclinic centrosymmetric space group P 21/a. The crystal structure clearly shows that the CoIII ion is surrounded by three L1 (with the charge −1) ligands inducing conformation chirality for each molecule. As clearly demonstrated in Fig. 5 (insert), two diastereoisomers constitutes the asymmetric unit with a total of four diastereoisomers in the unit cell. Selected distances are given in the Fig. 5 caption.

ORTEP view of one molecule of the asymmetric unit of complex 4·CH2Cl2 with partial labeling scheme. The ellipsoids enclose 50% of the electronic density. Selected distances (Å): Co1-O2: 1.877(4); Co1-O4: 1.872(4); Co1-O6: 1.898(1); Co1-N2: 1.929(5); Co1-N4: 1.918(5); Co1-N6: 1.940(5). Insert: the two diastereoisomers of the asymmetric unit.
Many examples of such a coordination sphere around the CoIII metal ion were reported [12] but only one example with hydrazine derivative ligand was already described; however, it involves a MnIII ion [5].
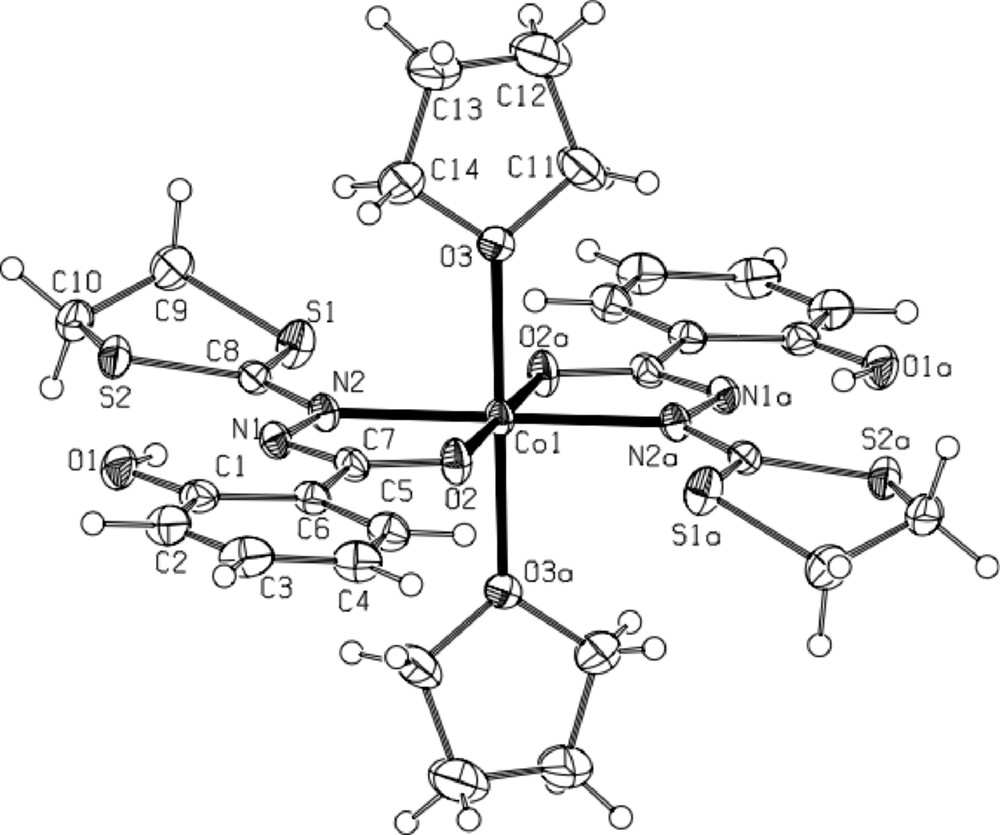
* The red brown complex 5·CH2Cl2 [CoIII(L2)3·CH2Cl2] (Fig. 6) crystallizes in the triclinic centrosymmetric space group P −1. The asymmetric unit contains only one isomer of the octahedral CoIII complex supported by three L2 ligands (with −1 charge), as clearly shown in Fig. 6. All distances are classical and very similar to those occurring in the complex 4·CH2Cl2. Selected distances are gathered in the Fig. 6 caption. To our knowledge, it constitutes the first example of transition metal complex supported by the 2-salicyloylhydrazono-1,3-dithiane ligand (L2).
ORTEP view of complex 5·CH2Cl2 with full labeling scheme. The ellipsoids enclose 50% of the electronic density. Hydrogen atoms are not represented for clarity. Selected distances (Å): Co1-O2: 1.879(3); Co1-O4: 1.884(3); Co1-O6: 1.876(3); Co1-N2: 1.948(4); Co1-N4: 1.955(4); Co1-N6: 1.961(4).
Considering the results obtained with mononuclear complexes, we tried to synthesize the μ-methoxo dinuclear compounds by the way of direct synthesis of Co(OH)3 with ligands L1 and L2, respectively. Unfortunately, the numerous tests made with methanol as reagent as well as solvent did not give any result, probably due to the lack of solubility of Co(OH)3 in methanol. On the other hand, syntheses carried out in ethanol as solvent (Scheme 3) allowed us to isolate a dinuclear complex with hydroxo bridges, but only with the ligand L1. Curiously, with the same synthetic procedure no reaction was observed with the ligand L2.
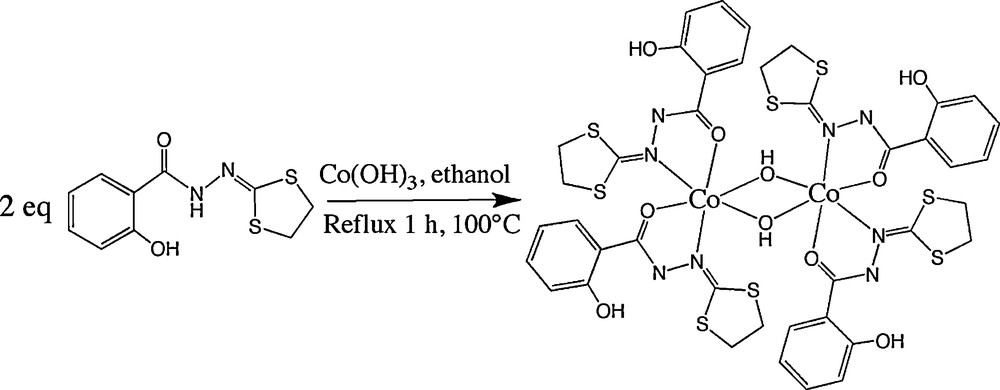
Synthesis of the CoIII μ-hydroxo dinuclear complex (6) supported by the ligand L1.
The dinuclear complex 6 were obtained as single crystals ready for X-ray diffraction by slow diffusion of pentane in CH2Cl2. The corresponding crystal structure is described hereafter.
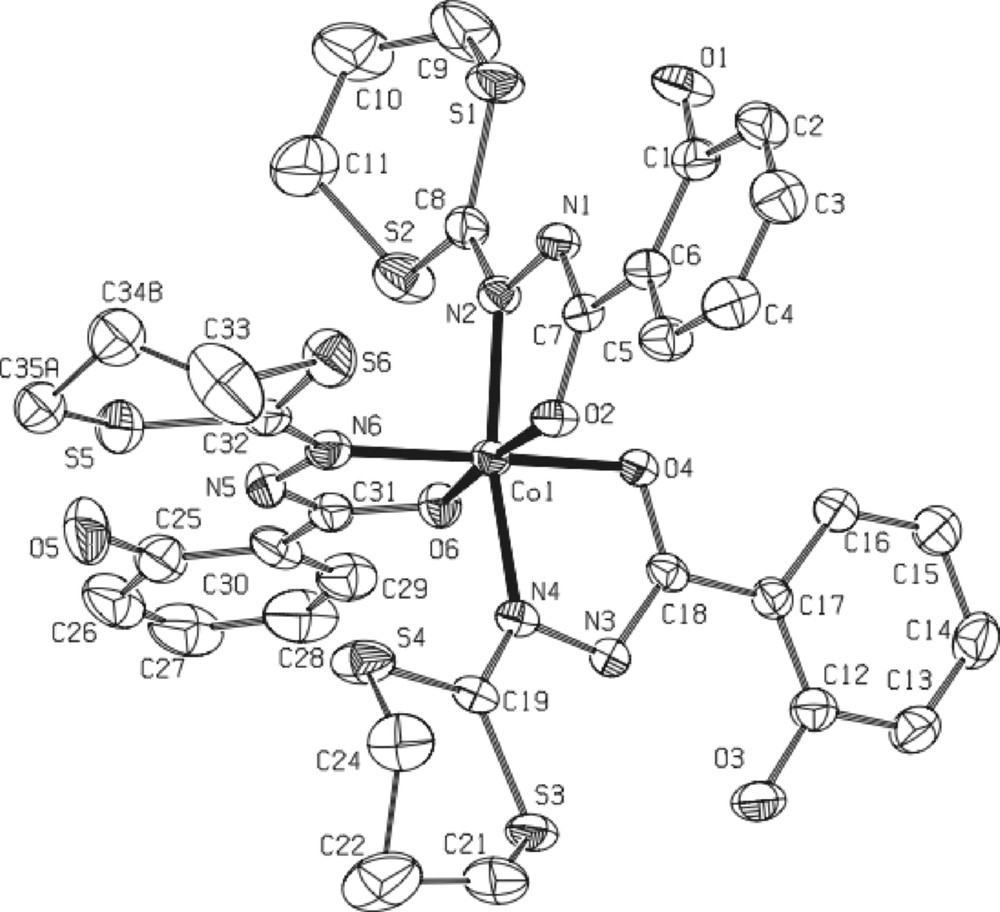
* The black complex 6·5/2CH2Cl2 [CoIII2(L1)4(OH)2·5/2CH2Cl2] (Fig. 7) crystallizes in the triclinic centrosymmetric space group P -1. As clearly shown on Fig. 7, the molecular structure can be described as a ‘symmetric’ μ-hydroxo bridged dinuclear complex in which both CoIII ions are in a slightly deformed octahedron (each one of them constituted by four oxygen atoms and two nitrogen atoms). The hydrogen atoms connected to the O9 and O10 oxygen atoms were found by Fourier transform, confirming that O9 and O10 are effectively hydroxy type bridges in this dinuclear compound. All distances and angles are classical with such a CoIII complex. The main particularity of this new complex is the ‘symmetric’ conformation observed: it is the first example with ligand L1. In fact, previous works involving dinuclear complexes with this ligand [5,7,13] always reveal a asymmetric configuration for both metal ions, MnIII or FeIII, supported by specific non classical CH-π interactions between the dithiolane group and the phenol ring (see, for instance, Fig. 1 in [5]). Moreover, each one of the octahedral sites is strongly deformed in the asymmetric complexes, as clearly shown in the insert of Fig. 7. In the present CoIII dinuclear complex, no specific intramolecular CH-π interaction has been detected. Only classical H-bonds occur between the OH group of the phenol rings and the hydrazine groups indicating the charge (1 for the four supported ligands.
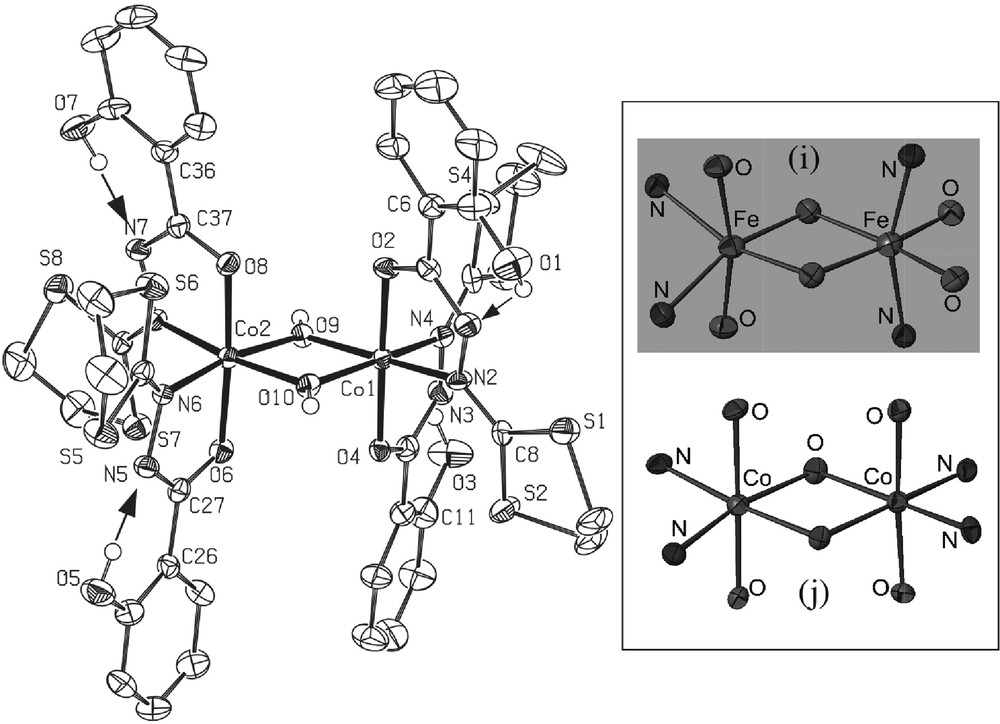
Left: ORTEP view of complex 6·5/2CH2Cl2 with partial labeling scheme. The ellipsoids enclose 50% of the electronic density. Arrows indicate the intramolecular hydrogen bonds. Selected distances (Å) and angles (°): Co1-O4: 1.878(4); Co1-O9 1.887(4); Co1-O2 1.893(4); Co1-O10 1.920(5); Co1-N4 1.926(6); Co1-N2 1.935(5); Co1-Co2 2.8695(12); Co2-O9 1.879(4); Co2 O8 1.885(5); Co2 O6 1.889(5); Co2 N8 1.916(5); Co2 O10 1.934(4); Co2 N6 1.939(6); Co1-O10-Co2: 96.2(2); Co2-O9-Co1: 99.3(2). Insert: cores of the dinuclear iron complex (i)13(asymmetric) and of the dinuclear cobalt complex (symmetric, present work).
2.4 Concluding remarks
In this article, we have presented the results of CoII and CoIII chemistry with the ligands 2-salicyloylhydrazono-1,3-dithiolane ligand (L1) and 2-salicyloylhydrazono-1,3-dithiane (L2). Thus, six new complexes were synthesized and structurally characterized. Both ligands considered in this work have very similar behavior with regard to the stabilization and crystal structures of the mononuclear compounds. On the other hand, only one dinuclear CoIII complex could be identified and structurally characterized with the supported ligand L1. In addition, this new dinuclear μ-oxo dinuclear CoIII complex has, in contrast to the closly related compounds containing manganese and iron ions, a ‘symmetrical’ structure considering the configuration of the two ligands around each CoIII ion. Despite the fact that the complex is diamagnetic, this point is very interesting for our work within the framework of molecular magnetism. Indeed, for the strongly ferromagnetic compound containing manganese +III ion, we have shown that the origin of the strong ferromagnetic interaction is related to the perpendicularity of both dz2 orbitals of the metal ion, i.e., related to the asymmetric situation in that complex. The challenge becomes, in that case, the possible control of the magnetic coupling by the synthesis, either of a symmetrical complex, awaited antiferromagnetic, or of asymmetrical complex awaited to be ferromagnetic. We currently work in that direction within our group.
3 Experimental
3.1 General procedures
All reactions and manipulations, excepted for ligands L1 and L2, were carried out under inert atmosphere of argon (AirLiquide, U-grade) using standard Schlenk tube techniques. Solvents were dried under nitrogen prior to use: tetrahydrofuran over sodium-benzophenone and dimethylformamide over sodium hydride. Elemental C, H, N, and S analyses were performed by the ‘Service de Microanalyses’ of Le Bel Institute and Charles Sadron Institute (ULP, Strasbourg, France). Infrared spectra were recorded as films on KBr disks, on a Perkin-Elmer 1600 series FTIR spectrometer. The 1H NMR spectra were recorded on a 300 MHz on a Bruker AVANCE 300 instrument.
3.2 Synthesis
3.2.1 General method for ligands
To a well-stirred solution of salicylhydrazide (9.13 g, 60 mmol) and NaOH (4.8 g, 120 mmol) in absolute ethanol (150 mL) at room temperature, CS2 (4.3 mL, 72 mmol) was added and the mixture was left under stirring overnight. Followed by the addition of 1,2 dibromoethane (5.2 mL, 60 mmol) (L1) or 1,3 dibromopropane (6.1 mL, 60 mmol) (L2). Stirring was continued for 8 h and the mixture was poured into ether. The resulting white precipitate was filtered and washed with ether. Recristallization was performed from absolute ethanol.
3.3 Ligand L1: 2-salicyloylhydrazono-1,3-dithiolane
Yield: 76% (11.58 g).
1H NMR (300 MHz, DMSO-d6, 300K) δ/ppm: 11.81(br s, 1H, OH), 10.99 (br s, 1H, NH), 7.95–7.93 (d, 1H, Ar), 7.44–7.38 (t, 1H, Ar), 7.01–6.94 (m, 2H, Ar), 3.77–3.73(m, 2H, SCH2CH2S), 3.62–3.58 (m, 2H, SCH2CH2S).
Elemental analysis: anal. Calc. for C10H10N2O2S2: C, 47.23; H, 3.96; N, 11.01; Found: C, 47.40; H, 4.10; N, 11.20.
IR ν (cm−1): (OH) 3018 cm−1, (NH) 2920 cm−1, (CO) 1716 cm−1, (CN) 1629 cm−1.
3.4 Ligand L2: 2-salicyloylhydrazono-1,3-dithiane
Yield: 65% (10.45 g).
1H NMR (300 MHz, DMSO-d6, 300K) δ/ppm: 11.75 (br s, 1H, OH), 11.40 (br s, 1H, NH), 7.96–7.93 (d, 1H, Ar), 7.42–7.37 (t, 1H, Ar), 7–6.94 (m, 2H, Ar), 3.26–3.21(m, 2H, SCH2CH2CH2S), 3.18–3.13(m, 2H, SCH2CH2CH2S), 2.19–2.10 (m, 2H, SCH2CH2CH2S).
Elemental analysis: anal. calc. for C11H12N2O2S2 + H2O: C, 46.14; H, 4.93; N, 9.78; Found: C, 46.20; H, 4.98; N, 9.75.
IR ν (cm−1): (OH) 3017 cm−1, (NH) 2922 cm−1, (CO) 1714 cm−1, (CN) 1629 cm−1.
3.5 General method for CoII complexes
To a solution of ligand (2 mmol) in THF, DMF or CH2Cl2 (≈ 35 mL), anhydrous CoCl2 (0.13 g, 1 mmol) was added. The mixture was stirred at room temperature overnight. The precipitate was filtered and washed with ether.
3.6 Complex CoII(L1)2(THF)2
X-ray quality crystals have been obtained by slow diffusion of pentane in THF.
Yield: 75% (0.42 g).
Elemental analysis: anal. calc. for C28H34CoN4O6S4: C, 47.38; H, 4.83; N, 7.89; Found: C, 47.29; H, 4.57; N, 8.02.
Mass spectroscopy (ESMS) m/z: calc: 709.07 Found: (M + H).708.13.
IR ν (cm−1): (OH) 3026 cm−1, (CN) 1631 cm−1.
3.7 Complex CoII(L1)2(DMF)2
X-ray quality crystals have been obtained by slow diffusion of pentane in DMF.
Yield: 71% (0.42 g).
Elemental analysis: anal. calc. for C26H32CoN6O6S4: C, 43.87; H, 4.53; N, 11.81; Found: C, 43.39; H, 4.67; N, 11.85.
Mass spectroscopy (ESMS) m/z: calc: 711.06 Found: (M + H) 710.79.
IR ν (cm−1): (OH) 3025 cm−1, (CN) 1625 cm−1.
3.8 Complex CoII(L2)2Cl2
X-ray quality crystals have been obtained by slow diffusion of pentane in CH2Cl2.
Yield: 67% (0.40 g).
Elemental analysis: anal. calc. for C22H24CoCl2N4O4S4: C, 39.64; H, 3.63; N, 8.41; Found: C, 40.70; H, 3.59; N, 9.27.
Mass spectroscopy (ESMS) m/z: calc: 664.94 Found: (M + H) 663.79.
IR ν (cm−1): (OH) 3023 cm−1, (CN) 1624 cm−1.
4 General method for CoIII complexes
Method 1: To a solution of ligand (3 mmol) in ethanol (30 ml), was added anhydrous CoCl2 (0.13 g, 1 mmol). The mixture was stirred at room temperature overnight followed by the addition of KOH (0.17 g, 3 mmol). The solvent was removed under vacuum and the complex was extracted with dichloromethane.
Method 2: A solution of ligand (3 mmol) and Co(OH)3 (0.11 g, 1 mmol) in ethanol (30 ml) was heated to 373K (100 °C) over 6 h. The solvents were removed under vacuum and the complex was extracted with dichloromethane.
4.1 Complex CoIII(L1)3
X-ray quality crystals (red brown) were obtained by slow diffusion of pentane in CH2Cl2.
Yield: 46% (0.38 g) method 1, 70% (0.57 g) method 2.
1H NMR (300 MHz, DMSO-d6, 300K) δ/ppm: 11.14 (br s, 3H, OH), 8.14–8.02 (d, 3H, Ar), 7.35–7.26 (m, 3H, Ar), 6.97–6.81 (m, 6H, Ar), 3.73–3.36 (m, 12H, SCH2CH2S).
Elemental analysis: anal. calc. for C30H27CoN6O6S6: C, 44.00; H, 3.32; N, 10.26; Found: C, 44.15; H, 3.35; N, 10.39.
Mass spectroscopy (ESMS) m/z: calc: 817.965 Found: (M + H) 818.972.
IR ν (cm−1): (OH) 3028 cm−1, (CN) 1638 cm−1.
4.2 Complex CoIII(L2)3
X-ray quality crystals (red brown) were obtained by slow diffusion of pentane in CH2Cl2.
Yield: 39% (0.34 g) method 1, 66% (0.57 g) method 2.
1H NMR (300 MHz, DMSO-d6, 300K) δ/ppm: 11.07 (br s, 3H, OH), 8.13–8.02(d, 3H, Ar), 7.35–7.25(m, 3H, Ar), 6.96–6.79(m, 6H, Ar), 3.21–2.02 (m, 18H, SCH2CH2CH2S).
Elemental analysis: anal. Calc. for C33H33CoN6O6S6: C, 46.04; H, 3.86; N, 9.76; Found: C, 45.98; H, 3.91; N, 9.69.
Mass spectroscopy (ESMS) m/z: calc: 860.012 Found: (M + H) 860.023.
IR ν (cm−1): (OH) 3036 cm−1, (CN) 1640 cm−1.
4.3 Complex CoIII2(L1)4(μ-OH)2
A solution of 2-salicyloylhydrazono-1,3-dithiolane (0.51 g, 0.2 mmol) and Co(OH)3 (0.11 g, 0.1 mmol) in absolute ethanol (60 ml) was heated to 373K (100 °C) over 1 h and then the solution was dry under vacuum. The complex was extracted with dichloromethane and the resulting solution was dry under vacuum to obtain the complex.
X-ray quality crystals (black) have been obtained by slow diffusion of pentane in CH2Cl2.
Yield: 21% (0.25 g).
1H NMR (300 MHz, DMSO-d6, 300K) δ/ppm: 11.31 (br s, 4H, OH), 8.20–8.06 (m, 4H, Ar), 7.48–7.33 (m, 4H, Ar), 7.05–6.89 (m, 8H, Ar), 3.81–3.42 (m, 16H, SCH2CH2S).
Elemental analysis: anal. calc. for C40H38Co2N8O10S8: C, 41.23; H, 3.29; N, 9.62; Found: C, 41.12; H, 3.10; N, 9.73.
Mass spectroscopy (ESMS) m/z: Calc: 1163.914; Found: (M + Na): 1186.903.
4.4 Single crystal X-ray determination
Single crystals of ligand L2, complexes 1, 2, 3·CH2Cl2, 4·CH2Cl2, 5·CH2Cl2 and 6·5/2CH2Cl2 were mounted on a Nonius Kappa-CCD area detector diffractometer (Mo Kα λ = 0.71073 Å). The complete conditions of data collection (Denzo software) [14] and structure refinements are given in Tables 1 and 2. The cell parameters were determined from reflections taken from one set of 10 frames (1.0° steps in phi angle), each at 20 s exposure. The structures were solved using direct methods and refined against F2 using the SHELXL97 software [15]. The absorption was non-corrected.
Crystal data and refinement details for L2 and complexes 4·CH2Cl2, 5 and 6·5/2CH2Cl2.
| Compound | Complex 4·CH2Cl2 | Complex 5·CH2Cl2 | Complex 6·5/2CH2Cl2 |
| Formula | C31H29Cl2CoN6 O6S6 | C34H35Cl2CoN6O6S6 | C42.5H43Cl5Co2N8O10S8 |
| Formula weight | 903.79 | 945.87 | 1377.44 |
| Crystal system | Monoclinic | Triclinic | Triclinic |
| Crystal color | Red brown | Red brown | Black |
| Crystal size | 0.20 × 0.10 × 0.10 | 0.15 × 0.12 × 0.10 | 0.10 × 0.10 × 0.10 |
| Space group | P 21/a | P-1 | P-1 |
| a (Å) | 17.862(5) | 10.1690(3) | 14.4720(5) |
| b (Å) | 20.055(9) | 11.9820(4) | 14.8090(8) |
| c (Å) | 22.136(7) | 17.1820(7) | 15.3940(7) |
| α (°) | 90.00 | 72.475(2) | 61.840(2) |
| β (°) | 103.06 | 88.260(2) | 77.604(3) |
| γ (°) | 90.00 | 89.590(2) | 70.858(3) |
| V (A3) | 7725(5) | 1995.45(12) | 2740.7(2) |
| Z | 8 | 2 | 2 |
| Density (g.cm−3) | 1.554 | 1.574 | 1.669 |
| mu (Mo Kα) (mm−1) | 0.957 | 0.930 | 1.216 |
| F(000) | 3696 | 972 | 1402 |
| Data collection | |||
| Temperature (K) | 173(2) | 173(2) | 173(2) |
| Theta (min – max) | 1.39–30.03 | 1.24–27.50 | 1.49–27.49 |
| Dataset [h, k, l] | −25/24, 0/28, 0/31 | −13/13, −15/15, −22/19 | −18/18, −16/19, 0/19 |
| Tot., Uniq. Data, R(int) | 69715, 22568, 0.0000 | 19381, 9140, 0.0626 | 15257, 12489, 0.0000 |
| Observed data (> 2σ(I)) | 6808 | 6541 | 6491 |
| Nreflections, Nparameters | 22568, 937 | 9140, 512 | 12489, 675 |
| R2, R1, wR2, wR1, Goof | 0.2554, 0.0873, 0.2148, 0.1725, 0.841 | 0.1067, 0.0746, 0.2127, 0.1869, 1.140 | 0.1622, 0.0799, 0.1984, 0.1507, 0.987 |
| Max. and Av. Shift/Error | 0.001, 0.000 | 0.000, 0.000 | 0.000, 0.000 |
| Min, Max. Res. (e.Å−3) | −1.046, 1.528 | −0.972, 2.424 | −1.306, 1.714 |
All non-hydrogen atoms (excepted for CH2Cl2 in 6·5/2CH2Cl2) were refined anisotropically and hydrogen atoms were introduced as fixed contribution (SHELXL97) except for all hydrogen atoms of the ligand L2 and for the hydrogen atom of the hydroxyl group for all complexes (found by Fourier differences).
Crystallographic data (excluding structure factors) have been deposited in the Cambridge Crystallographic Data Centre as Supplementary publication no. CCDC 742826 to CCDC 748332. Copies of these data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44)1223-336-033; e-mail: deposit@ccdc.cam.ac.uk).
Acknowledgements
We thank the CNRS and the ministère de la Recherche (Paris) for the PhD grant of Nicolas Clément and the Agence nationale de la recherche (ANR contract n° ANR-06-JCJC-0008) for funding.