

1 Introduction
The design of new multifunctional materials, combining magnetism and non-linear optical activity [1], magnetism and photo-reactivity [2,3] or magnetism and transport properties [4–7], is currently a challenging field of research for molecular chemists, material chemists and physicists. Metal complexes containing bipyridine and analogous ligands such as phenathroline are commonly used in the formation of different complexes with a general variety of transition metals, [8–10] because of their versatile roles at several levels, such as: building blocks for the synthesis of metallo-dendrimers, molecular scaffolding for supramolecular assemblies, in analytical chemistry, in catalysis, in electrochemistry, in the ring-opening metathesis polymerization and biochemistry [11–26]. Such studies are important for better understanding of electron transfer processes, magnetic coupling and magnetic transitions, photochemistry [27–31]. Additionally, owing to an extended π-system, non-covalent π-interactions in biological processes can be mimicked [32]. Azido-bridged complexes have been focused on for molecular magnetism for many years because azide ions have two possible bridging modes: end-to-end (EE) with preferred antiferromagnetic interactions and end-on (EO) with preferred ferromagnetic interactions. However carboxylate-bridged complexes continue to be much investigated due to their interesting coordination chemistry allowing for unusual structural features, leading to various physical and chemical properties (dyes, extractants, drugs, pesticides, catalysts) [33–40]. Indeed, in most cases, it is still difficult to predict the exact structures and compositions of the product of the mixed ligands [41]. As a continuation of our researches on the synthesis of new chiral inorganic coordination complexes using α-hydroxy carboxylates [42–46], we report in this paper the synthesis and crystal structure of three new complexes using α-amino carboxylates (L-cysteic acid) with mixed ligands (dmbpy “4,4’dimethyl-2,2’bipyridine”, N3 “azide”, the Cu(L-cyst)(dmbpy).H2O (1), cis-[Mn(N3)Cl(dmbpy)2] (2) and [Co(dmbpy)3].[CoCl4].7H20.Cl (3).
Only few results have been reported on complexes involving the cysteic acid ((S)-2-amino-3-sulfopropanoic acid) (Scheme 1) [47–51]. It can adopt several coordination modes: monodentate, bidentate, tridentate and tetradentate. Scheme 2 shows the different coordination modes of cysteic acid with the corresponding references.
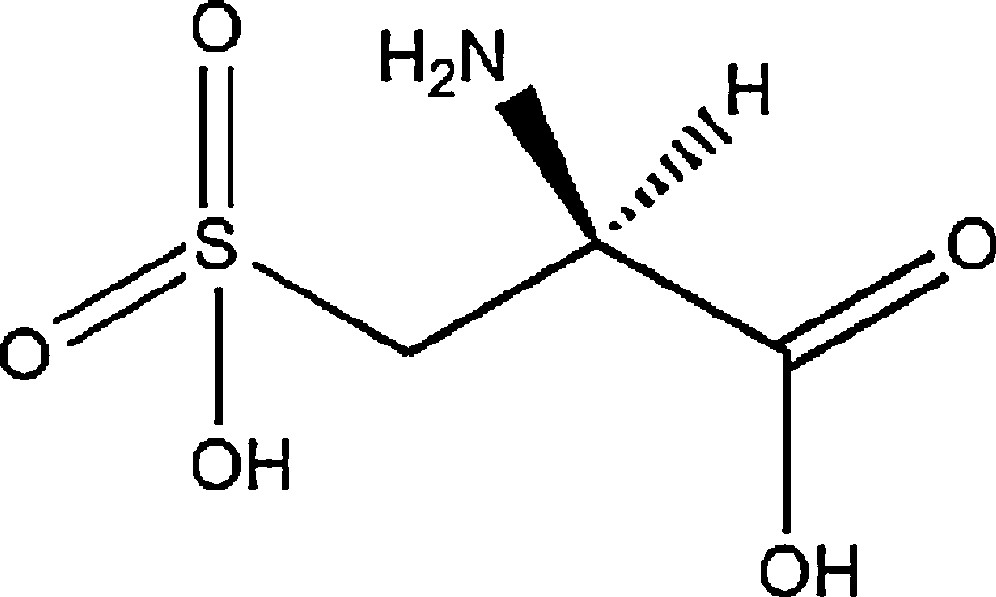
(S)-2-amino-3-sulfopropanoic acid ((S)-cysteic acid).
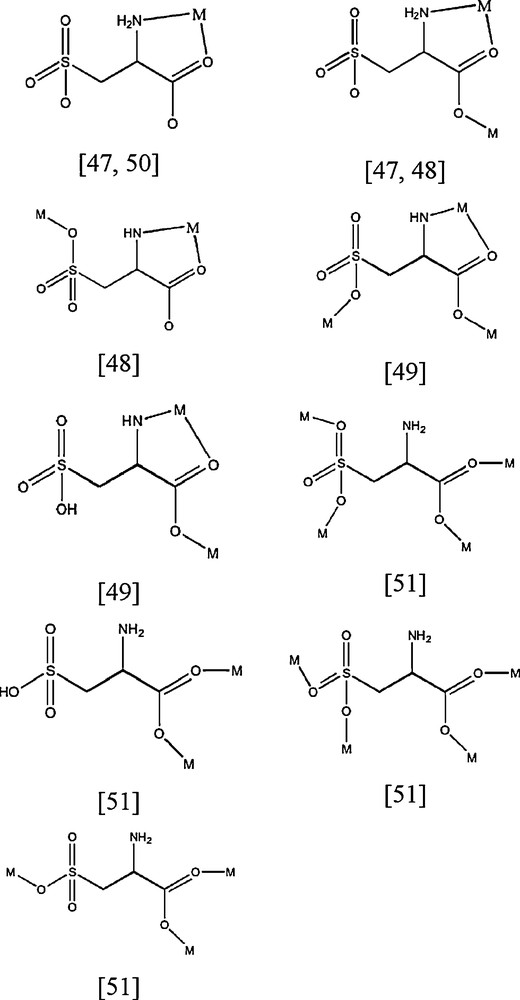
Coordinations modes of (S)-2-amino-3-sulfopropanoic acid ((S)-cysteic acid).
2 Experimental
2.1 Materials
L-Cysteic acid ((S)-2-amino-3-sulfopropanoic acid), 4,4’-dimethyl-2,2’-bipyridine, sodium azide, Cu(NO3)2.3H2O, CoCl2.6H2O, MnCl2.4H2O were purchased from Alfa Aesar. All the reagents and solvents were used as received without further purification. The complexes were obtained at room temperature using a general procedure described below.
2.2 Physical measurement
Elemental C, H, N, and S analyses were performed by the ‘Service de Microanalyses’ of the Institut Le Bel and of the Institut Charles-Sadron (Université de Strasbourg, France). FT-IR studies were performed with an ATI Mattson Genesis computer-driven instrument (0.1 mm thick powder samples in KBr). Thermogravimetric experiments were performed using a Setaram TG92 instrument (heating rate of 5 °C min−1, air stream) [heating in air from 30 °C to 1000 °C at 5 °C min−1, 30 min at 1000 °C to transform into the corresponding metal oxide (the nature of which was checked by X-ray powder diffraction) and cooling to 30 °C at 10 °C min−1]. Crystal structures were determined using a NONIUS MACH3 Kappa CCD diffractometer equipped with graphite-monochromatized MoKα radiation (λ = 0.71073 Å).
2.3 Synthesis of the complexes
2.3.1 Synthesis of [Cu(L-cyst)(dmbpy).H2O] (1)
A methanolic solution containing Cu(NO3)2.3H2O (0.241 g, 1 mmol) and 4,4’-dimethyl-2,2’-bipyridine (0.186 g, 1 mmol) was added with stirring to a methanolic solution containing L-cysteic acid (0.187 g, 1 mmol) and sodium azide (0.195 g, 3 mmol). After few minutes a blue green precipitate appears and was filtrated (after recrystallization of this precipitate by slow evaporation of methanol as solvent, we obtain the product 1). A diluted clear blue solution of the complex in methanol solution was left at room temperature. After several days, X-ray qualified blue single crystals were grown, which were filtrated and characterized (yield: 0.31 g, 71.6% on the basis of [Cu(NO3)2.3H2O]). Anal. Calc. for C15H19CuN3O6S: C: 41.61; H: 4.42; N: 9.70, S: 7.40%. Found: C: 42.01 H: 3.98; N: 9.39; S: 7.72%.
2.3.2 Synthesis of [Mn(dmbpy)2(N3).Cl] (2)
A methanolic solution containing MnCl2.4H2O (0.1 g, 0.5 mmol) and sodium azide (0.098 g, 1.5 mmol) and L-cysteic acid (0.094 g, 0.5 mmol) was stirred for 30 min, and then a methanol solution containing 4,4’-dimethyl-2,2’-bipyridine (0.093 g, 0.5 mmol) was added. After few minutes a beige precipate appears and was filtrated while the solution was left at room temperature. After one month, orange single crystals suitable for XRD-analysis were obtained, which were filtrated and characterized (yield: 0.09 g, 35.92% on the basis of [MnCl2.4H2O]). Anal. Calc. for C24H24ClMnN7: C, 57.54; H,4.82; N, 19.57. Found: C, 56.84; H, 4.37; N, 20.02%.
2.3.3 Synthesis of [Co(dmbpy)3].[CoCl4].7H2O. Cl (3)
A methalonic solution containing cobalt (II) chloride hexahydrate (0.119 g, 0.5 mmol) and 4,4’dimethyl-2,2’bipyridine (0.093 g, 0.5 mmol) was added with stirring to a methanol solution containing L-cysteic acid (0.094 g, 0.5 mmol) and sodium azide (0.033 g, 0.5 mmol). After few minutes a red-brown precipate appears, and which filtrated then the clear solution was left at room temperature. After few weeks, X-ray qualified green single crystals were obtained, which were filtrated and characterized (yield: 0.20 g, 41.06% on the basis of [CoCl2.6H2O]). Anal. Calc. for C36H50Co2N6O7Cl5: C: 44.39; H: 5.17; N: 8.62%. Found: C: 44.71; H: 4.92; N: 8.92%.
CAUTION: Although not encountered in our experiments, azido compounds of metal ions are potentially explosive. Only a small amount of the materials should be prepared, and it should be handled with care.
2.4 X-ray diffraction studies
Single crystals of the complexes 1, 2, 3 were mounted on a Nonius Kappa-CCD area detector diffractometer, Mo Kα radiation (λ = 0.71073 Å), using ω−2θ scans at room temperature. The parameters of the data collection (Denzo software) [52] and structure refinements are given in the Table S. The structures were solved using direct methods (SHELXS97) and refined against F2 using the SHELXL97 and CRYSTALBUILDER softwares [53,54]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were generated according to stereo-chemistry and refined using a riding model in SHELXL97. The absorption was not corrected (small and almost isotropic crystal-shapes). Table S gives crystallographic crystal data and final refinement parameters for the complexes 1, 2 and 3. Selected bond lengths and angles are given in corresponding figure captions. Geometrical calculations were carried out with PLATON [55].
3 Results and discussion
3.1 Crystal and molecular structure of [Cu(2,2’-dmbpy)(L-cyst)(H2O)] (1)
The asymmetric units of 1, 2 and 3 with the atom-numbering schemes are illustrated in Figs. 1, 4 and 6, respectively. The complex 1 crystallizes in the monoclinic system with chiral space group P21, contrary to what has been observed on a similar compound namely of [Cu(2, 2’-bpy)(L-cyst)(H2O)] published recently where the structure is centrosymmetric [47]. The Cu(II) ion is coordinated by one 2,2’-dmbpy molecule acting as a bidentate ligand (through the two nitrogen atoms (N2, N3)), and one L-cys moeity (through the oxygen and nitrogen atoms (O1, N1)). The geometry around the metal is a distorted square pyramidal with a water molecule lying in the axial position through O1W. The basal plan is formed by (O1, N1, N2, N3) where the central metal atom is slightly above it (0.255(3) Å). This deviation is longer than observed in Cu(2,2’-bpy)(L-cys)(H2O) (≈0.13 Å). The Cu-N distances of the dmbpy and the amino nitrogen atoms of the L-cys ligand range from 1.970(4) to 2.004(3) Å, while the Cu-O distance of the carboxylate oxygen coordinated to the metal is 1.950(3) Å and the distance Cu-O of the coordinated water molecule is 2.231(4) Å, close to the distances observed in Cu(bpy)(L-cys)(H2O) [47].The packing diagram and hydrogen bonding interactions scheme were analyzed by PLATON software, the packing and hydrogen bonding interactions viewed down the a-axis are shown in Fig. 2. The crystal structure can be described as chains that develops in a zigzag pattern along the b-axis via weak hydrogen bonds type C-H…O between C7-H7…O3 and C10-H10…O5. These chains are interconnected along the c-axis to form a sheet through medium hydrogen bonds via O-H…O between atoms O1W-H1W…O5 (Fig. S4), these sheets are also interconnected along c-axis by medium hydrogen bond type N-H…O and O-H…O through N1-H7A…O4, O1W-H2W…O1 and O1W-H2W…O2.
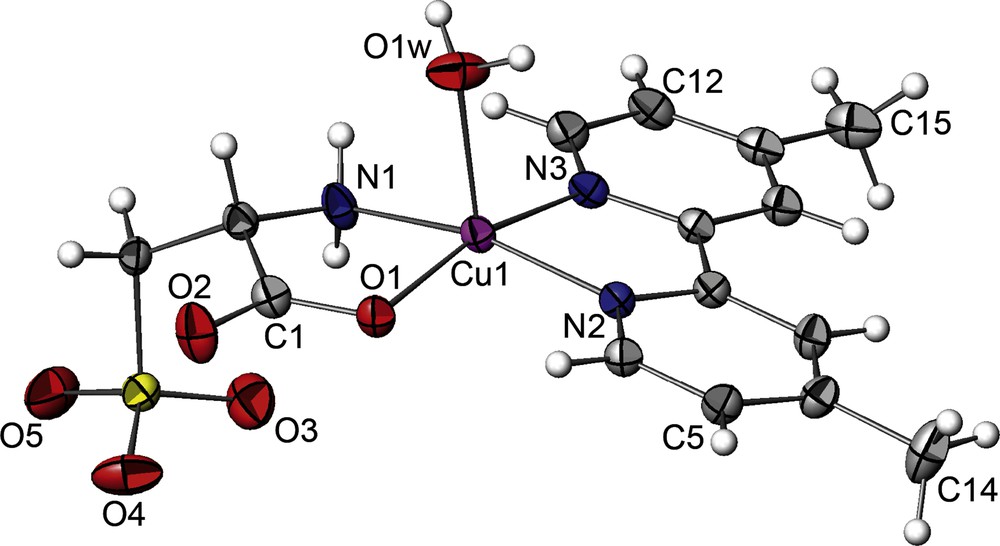
ORTEP view (ATOMS software) of 1 (CuII(L-cyst)(dmbpy)(H2O)) with partial labeling scheme. The ellispsoids enclose 50% of the electronic density. Selected distances (Å) and angles (°): Cu1-O1 = 1.950(3), Cu1-O1W = 2.231(4), Cu1-N1 = 1.970(4), Cu1-N2 = 1.989(3), Cu1-N3 = 2.004(3), S1-O3 = 1.440(3), S1-O4 = 1.424(3), S1-O5 = 1.425(3), S1-C3 = 1.781(4), O1-C1 = 1.263(4), O2-C1 = 1.227(5), N1-C2 = 1.478(5), N2-C8 = 1.349(4), N2-C4 = 1.340(4), N3-C13 = 1.343(4), N3-C9 = 1.356(4), O1-Cu1-O1W = 93.48(11), O1-Cu1-N1 = 83.75(11), O1-Cu1-N2 = 94.05(10), O1-Cu1-N3 = 164.96(11), N1-Cu1-N2 = 165.08(12), O3-S1-O4 = 111.3(2), O3-S1-O5 = 111.9(2).

ORTEP view (ATOMS software) of 2 (MnII(dmbpy)2(N3)(Cl)) with partial labeling scheme. The ellispsoids enclose 50% of the electronic density. Selected distances (Å) and angles (°): Mn1 Cl1 2.4569(15), Mn1-N1 = 2.314(2), Mn1-N2 = 2.262(2), Mn1-N3 = 2.271(2), Mn1-N4 = 2.313(2), Mn1-N5 = 2.173(3), N1-C1 = 1.342(3), N1-C5 = 1.349(3), N2-C6 = 1.343(3), N2-C10 = 1.337(3), N3-C13 = 1.338(3), N3-C17 = 1.345(3), N4-C18 = 1.343(3), N4-C22 = 1.336(4), N5-N6 = 1.109(4), N6-N7 = 1.191(4), Cl1-Mn1-N1 = 168.74(5), Cl1-Mn1-N3 = 93.16(6), Cl1-Mn1-N4 = 89.44(6), N1-Mn1-N2 = 71.39(7), N2-Mn1-N3 = 160.35(8), N3-Mn1-N4 = 71.95(7), N3-Mn1-N5 = 95.48(9), N4-Mn1-N5 = 166.02(9), N5-N6-N7 = 178.8(4).

ORTEP view (ATOMS software) of 3 ([CoII(dmbpy)3].[CoIICl4].Cl.7H2O) with partial labeling scheme. The ellispsoids enclose 30% of the electronic density. Dashed lines indicate inter molecular H-bonds. Selected distances (Å) and angles (°): Co1-N1 = 1.937(3), Co1-N2 = 1.947(3), Co1-N3 = 1.930(3), Co1-N4 = 1.936(3), Co1-N5 = 1.925(3), Co1-N6 = 1.925(3), Co2-Cl1 = 2.2982(16), Co2-Cl2 = 2.267(2), Co2-Cl3 = 2.2790(17), Co2-Cl4 = 2.258(2), N1-Co1-N2 = 83.20(10), N1-Co1-N4 = 93.59(11), N1-Co1-N6 = 176.20(11), N2-Co1-N4 = 175.98(11), N2-Co1-N5 = 89.23(10), Cl3-Co2-Cl4 = 109.85(6), Cl1-Co2-Cl4 = 110.04(7), Cl1-Co2-Cl2 = 113.69(6), Cl2-Co2-Cl3 = 110.20(6), Cl2-Co2-Cl4 = 106.54(6).
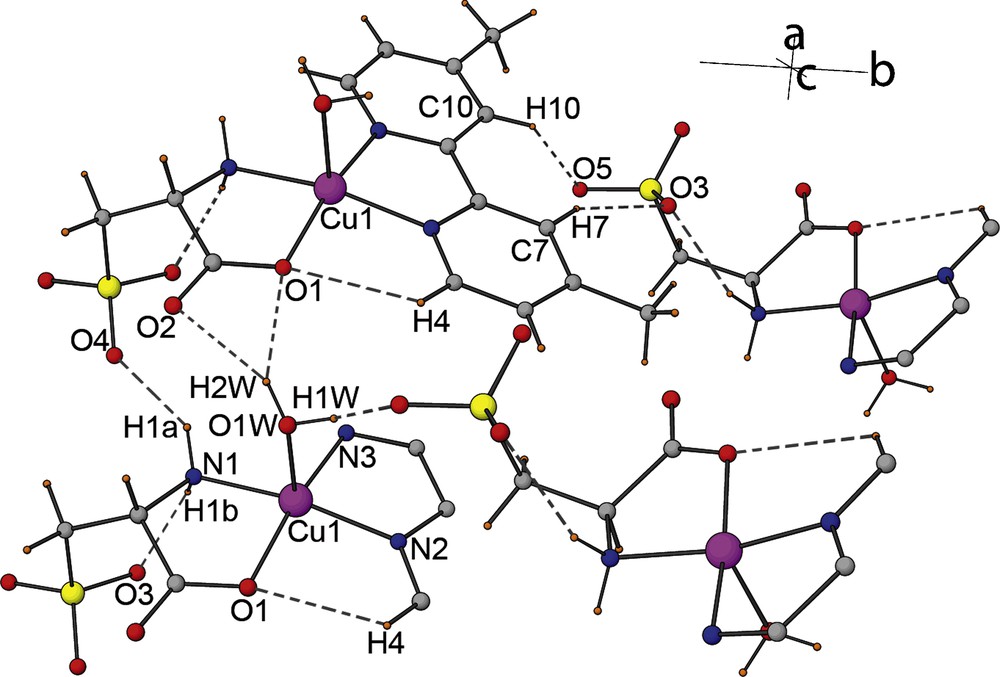
Partial view of a selected part of the crystal structure of 1 showing hydrogen bonds. Dashed lines indicate inter- and intramolecular H-bonds.
In addition to these hydrogen interactions, we also note that the cohesion in the crystal is ensured by weak interactions type π…π stacking, observed between centroid (Cg1) of the N2-C8 pyridil ring and the centroid (Cg2) of the N3-C13 pyridil ring of the adjacent molecule, with a Cg1…Cg2 distance of 3.765(3) Å (Fig. 3) [47]. The racemic mixture in the crystal structure is then not observed, on the contrary to what has been observed for complexes described with the cysteic acid and 2,2’ bpy [47,48,50].
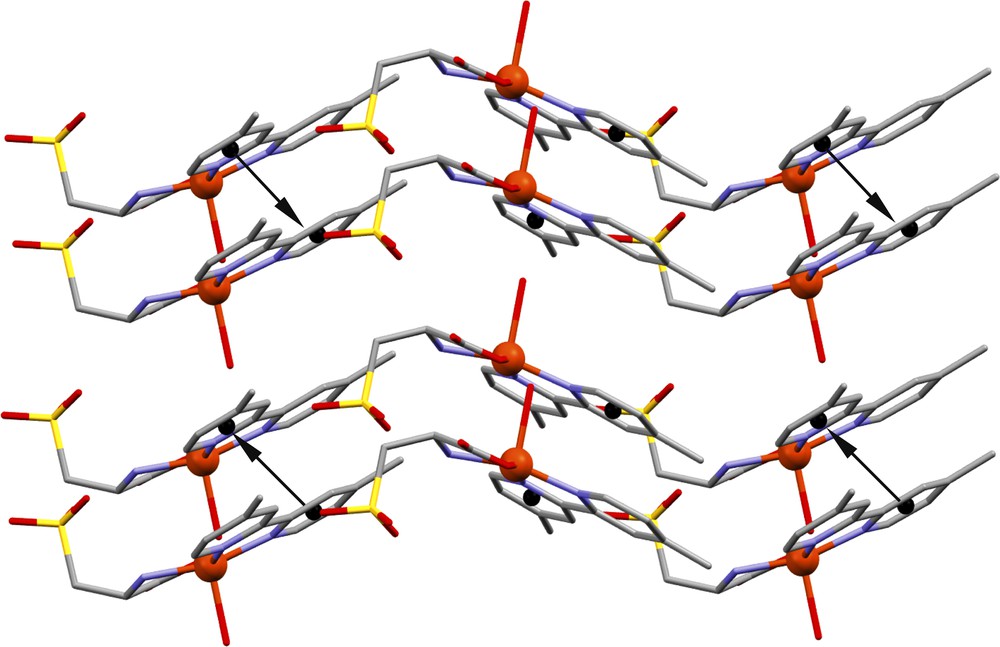
Partial view of the crystal structure of 1 showing π…π stacking interactions.
3.2 Crystal and molecular structure of [Mn(2,2’-dmbpy)2(N3)(Cl)] (2)
An ORTEP view of the molecular structure of complex 2 in the crystal is depicted in Fig. 4. The manganese(II) ion is surrounded by two cis-coordinated dmbpy ligand, coordined by nitrogen atoms (N1, N2) of the first ligand (Mn1-N1 = 2.315(2) Å, Mn1-N = 2 2.261(2) Å) and the atoms (N3, N4) for the second ligand (Mn1-N3 = 2.270(2) Å, Mn1-N4 2.312(2) Å), and the N5 atom provided by the azide ligand (Mn1-N5 = 2.174(3) Å), the coordinated sphere is completed by the chlorine anion Cl1 (Mn1-Cl1 = 2.4567(15) Å). The coordination is a distorted octahedral, with an irregular equatorial (N2-N3-N4-N5) plane.
The molecular structure of complex 2 is stabilized by non standard weak hydrogen-bonds. In fact, C-H…Cl intermolecular hydrogen bonds are observed via C(4)- H(4)…Cl(1), C(21)- H(21)…Cl(1) and C(23)- H(23)…Cl(1). The dihedral angle between the pyridil rings of the first dmbpy ligand is 22.24(5)°, and 3.95(4)° for the second dmbpy ligand (Fig. 5).
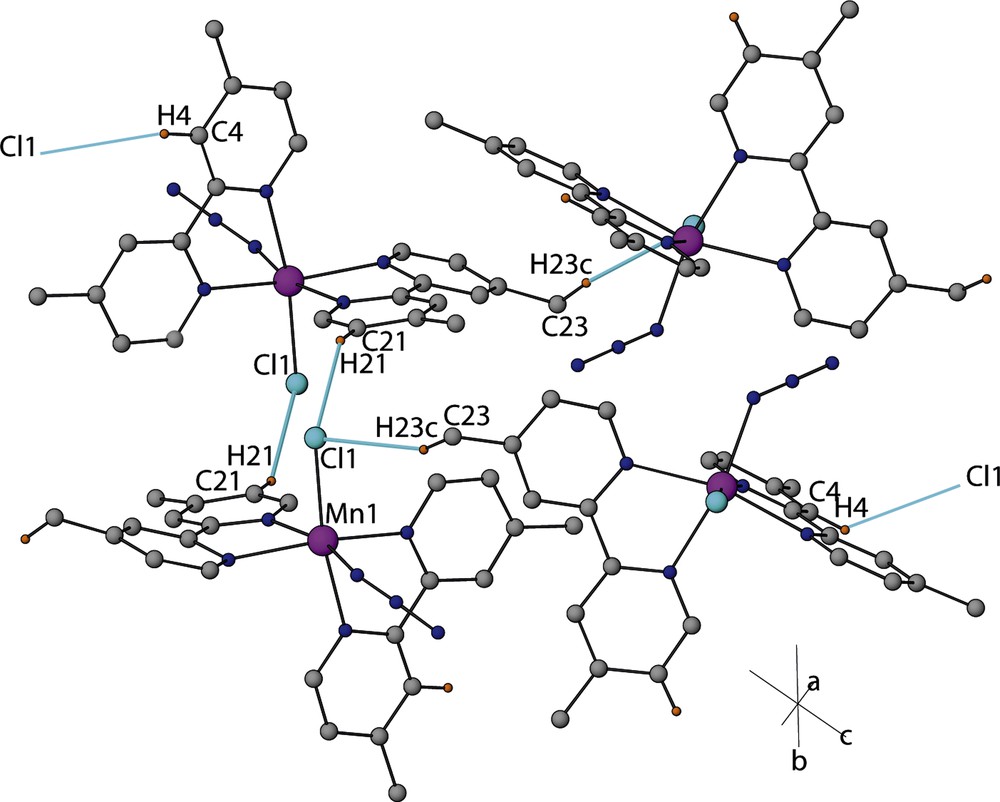
Partial view of the crystal structure of 2 showing the three-dimensional network of the hydrogen bonds (in green) via C-H…Cl atoms.
3.3 Crystal and molecular structure of [Co(dmbpy)3].[CoCl4].7(H2O).(Cl) (3)
The crystal structure of 3 is composed of [Co(dmbpy)3]2+ cation, [CoCl4]2− anions, uncoordinated Cl− anion and seven solvatation water molecules; one is an hydronium cation (H3O+) (Fig. 6). The cobalt atom of the cation (Co1) is coordinated to six nitrogen atoms (N1; N2; N3; N4; N5; N6) which belong to three different bipyridyl ligands forming a distorted octahedron. The Co1—N distance range from 1.925(3)–1.947(3)Å, and dmbpy rings are planar, while the [CoCl4] anion, exhibit tetrahedral geometry, with the Co(2) ion surrounded by four Cl atoms, with Cl—Co—Cl angles ranging from 106.53 (4) to 113.69 (3)°. The mean Co—Cl bond length, 2.27 (7) Å, is close to those observed in similar complexes [57–61].
The structure is stabilized by an extensive network of O—H…O, O—H…Cl and C—H…Cl hydrogen bond interactions [Supplementary material]. The cation [Co (dmbpy)3]2+ interacts with the [CoCl4]2− anion and two water molecules (O6W; O7W) through intermolecular weak hydrogen bonds type C—H…Cl and C—H…O respectively through C2-H2…O6W, C9—H9…O7W, C7—H7…Cl4, C19—H19…Cl1, C33—H33…Cl2 and C35—H35A…Cl4 as shown in Fig. 6. However, the water molecules’ hydrogens are involved in intermolecular O-H…Cl interactions with the two anion [CoCl4]2−; Cl− via Cl1—H71…O7W, Cl2—H12…O1W, Cl3—H21…O2W and Cl5—H62…O6W as shown in Fig. 7.We also note the presence of weak C-H…π interactions between the centroid (Cg1) of the N2-C10 pyridil ring and H12A (3.657(6)Å; 99.86(2)°), and also between the centroid (Cg2) of the N6-C34 pyridil ring and H12A (3.852(7)Å; 107.08(2)°).

Partial view of anion [CoCl4]−2 in complex 3 showing the hydrogen bonds in dashed lines.
3.4 Spectroscopic properties
The IR spectrum for compound 1 exhibits characteristic bands of the cysteate. The frequency difference between the symmetrical (νCOsym = 1384 cm−1) and antisymmetrical (νCOasym = 1616 cm−1) carboxylate vibrations, Δν = 164 cm−1, is smaller than that observed with the corresponding acid (Δνi ≈ 270 cm−1) in agreement with the monodentate coordination mode of the carboxylate groups [56]. The presence of broad νN-H bands in the range 3223–3254 cm−1 is related to the cysteate NH moieties bonded to the metal ions, and the large band observed at 3430 cm−1 is assigned to the coordinated water molecule (Fig. S1). For compound 2, strong absorption bands at 2040 cm−1 and 1299.9 cm−1 in the IR spectra correspond to νas (N3) and νs (N3), and some characteristic bands for 2,2’-dmbpy in the region of 1041, 1612, 1550.7, 1685.7, 1446, 914 and 833 cm−1 (Fig. S2). The compound 3, show some characteristic bands for 2,2’-dmbpy in the region: 1041.6, 1558, 1620, 1444, 931 and 831 cm−1. The large band observed in the region: 3200 - 3500 cm−1 corresponds to the solvation water molecules (Fig. S3).
3.5 Thermogravimetric analysis
The thermal stability of the compounds was evaluated by thermogravimetric analysis between ambient temperature and 600 °C in air (5 °C min−1) for compound 1 and ambient temperature and 1000 °C for compound 3.
The TGA of compound 1 (see trace in Fig. 8) revealed that the decomposition takes place in three steps. The first is observed at 150 °C (−5%) and indicates the loss of the coordinated water molecule. The second loss is observed at 250 °C (−13.78%) and indicates the decomposition of the (COO−) and (NH2) groups. Then, the very exothermic final weight loss of 58% starting at 430 °C, is associated to the decomposition of the dmbpy (C12N2H12) and (C2H3SO3−) into CuO.

TGA and TDA curves of 1.
The TGA trace of compound 3 is shown in Fig. 9; a first weight loss is observed between 60–120 °C (−15%) and corresponds to the removal of the seven solvatation water molecules (−7H2O) and the chlorine anion (Cl−). The second loss observed around 190 °C (−20%) corresponds to the tetrachlorocobaltate anion [CoCl4]2−, while the third loss is observed at 350 °C (−20%) which corresponds to the decomposition of one molecule of dmbpy (C12N2H12). It is worth noting that this decomposition occurs at quite high temperature and that the combustion of the two other molecules of (dmbpy) is very exothermic, leading to an overflow of the temperature regulation. The final total weight loss of 35% at 550 °C corresponds well to the decomposition total of Complex into Co3O4.
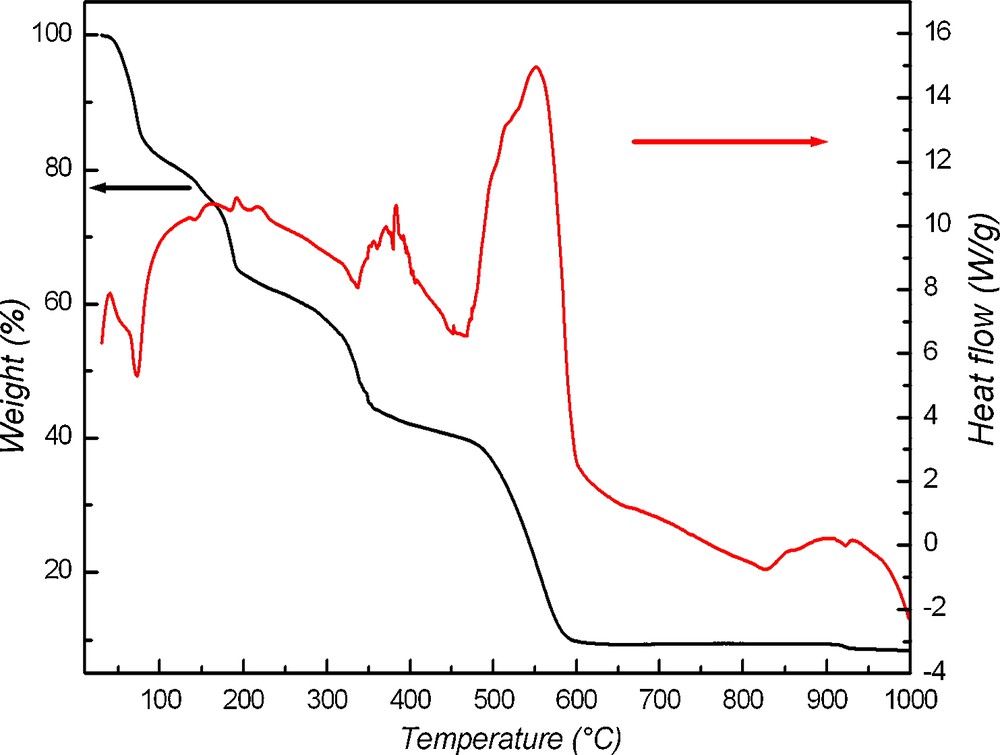
TGA and TDA curves of 3.
For compound 2 the measure has not been realized because it contains the “explosive” azide group (N3).
4 Conclusion
Three new coordination complexes [Cu(Cyst)(dmbpy)(H2O)] (1), [Mn(dmbpy)2(N3)(Cl)] (2) and H2O.Cl[Co(dmbpy)3].[CoCl4] (3) based on mixed ligand with different structure motifs are synthesized and structurally characterized. The complexes 1 and 2 are mononuclear, while the complex 3 is dinuclear. The molecular structure of complex 1 consists in copper metal center in pyramidal square base chelated to one L-cyst acid and one dmbpy and one water molecule in apical position. The complex 2 is hexa coordinated by two dmbpy, one azide and Cl- anions. The complex 3 is composed of [CoII(dmbpy)3] cation, [CoIICl4] anion, Cl− and seven water molecules. Thermal decomposition of complex 1 and 3 reveals that the Cu complex decomposes in three steps whereas the Co complex decomposes in four steps and transforms into the corresponding metal oxide.
Future studies will focus on the construction of new metal–organic coordination polymers from the cysteic acid and other co-ligands for the design of new chiral magnetic systems. This approach is very promising to investigate the complicated issue of the coupling between magnetism and chirality.
Acknowledgements
The authors thank Prof. J.C. Daran (LCC, Toulouse, France), Dr. Pierre Rabu (IPCMS, Strasbourg, France) for their help with structural discussions, Didier Burger from IPCMS, Strasbourg (France), for their contribution to thermogravimetric and A. Touil from département de chimie, université Mentouri-Constantine (Algeria) for IR data measurements. A.L. thanks the Algerian MESRS for the financial support.