

1 Introduction
Apatites are a large family of compounds with the general chemical formula M10(XO4)6Y2, where M represents a divalent cation, XO4 an anion group and Y a monovalent anion. They crystallize mainly in the hexagonal system with the P63/m space group [1] (Fig. 1). The main representative member of the apatite family is the hydroxyapatite HA, Ca10(PO4)6(OH)2, that can be considered as a wonderful compound. Its chemical composition and crystallographic structure similar to that of the mineral phase of bone and teeth make it a material of choice in many medical applications [2–4]. Furthermore, as a result of its ability to accommodate a great number of substitutions: cationic, anionic or both cationic and anionic substitutions, practically a third of the periodic table elements can be incorporated within the apatite lattice [5,6], making the application field of the obtained materials practically unlimited [7–14]. Indeed, this flexibility of the apatite structure to accept a variety of species may confer to the prepared materials very diverse properties, resulting in a variety of applications that are either already planned or are still to be discovered.
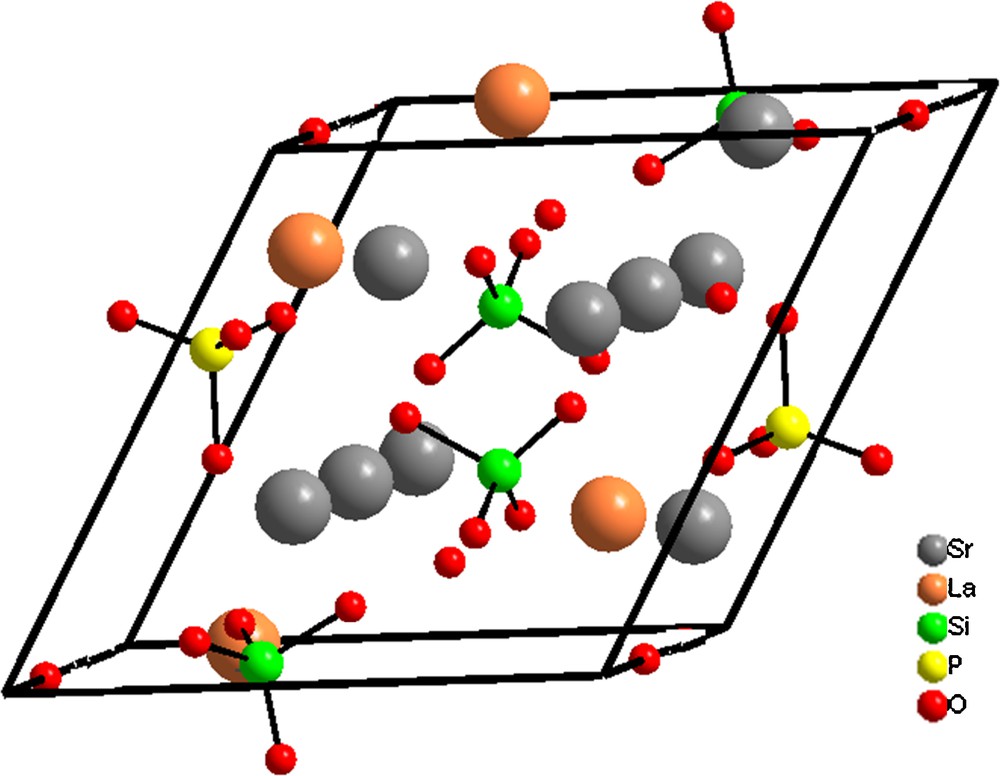
(Color online). Positions of atoms in the strontium oxyapatite unit cell (Sr10(PO4)6O).
For example, simultaneous substitutions can occur in the three sites (M, XO4 and Y). In hydroxyapatite, the substitution of La3+, and O2–for Ca2+, and OH–, respectively leads to new compounds, which have gained considerable interest as oxide ion conductors following the works of Nakayama et al. [15–17]. These compounds are usually prepared by a solid-state reaction at high temperatures. Also, several heat treatments are often required to achieve single-phase materials [18–21]. On the contrary, the mechanochemical synthesis involves only a solid-state reaction at room temperature [22,23]. Therefore, this method appears quite attractive for the preparation of this kind of materials [24,25]. Besides its low energy consumption, simplicity and speed, it is suitable for industrial production. Furthermore, the mechanical treatment leads to an increase in the specific surface area of powders, which should improve the densification of the materials [26].
The aim of this work was to attempt the preparation of the series (Sr10–xLax(PO4)6–x(SiO4)xO, where 0 ≤ x ≤ 6) via the mechanochemical method. The obtained powders were investigated using XRD, FTIR and TGA/DTA. In addition, we have characterized the Sr6La4(PO4)2(SiO4)4O sample by its ionic conductivity.
2 Experimental procedure
2.1 Preparation of powders
The samples were prepared by the mecanochemical process using a Retsch PM200 planetary micro mill. The synthesis reaction occurs as follows:
| (1) |
Appropriate amounts of strontium oxide (SrO), lanthanum oxide (La2O3), silica (SiO2) and strontium diphosphate (Sr2P2O7) in order to obtain (Sr + La)/(P + Si) atomic ratios of 1.67 were ground and homogenized in an agate mortar prior to grinding. Then, the mixture was introduced with several stainless steel balls, 10 mm in diameter in a 50 cm3 stainless steel cell. The weight ratio of the ball-to-powder for all samples was 34:1. The rotating disc speed and cell speed were 500 and 1000 rpm, respectively. Following a previous study [24], the grinding duration was fixed at 25 h. In order to avoid excessive temperature inside the mill cell, milling was carried out in 30 minutes intervals with a 5 minutes pause.
The strontium diphosphate was obtained by heating a mixture of strontium carbonate and di-ammonium hydrogenophosphate at 900 °C for 10 h, while strontium oxide was obtained by calcination of the corresponding carbonate at 1100 °C for 24 h. To avoid deviation from stoichiometry, lanthanum oxide was calcined at 1000 °C for 24 h just before use. Indeed, when La2O3 is exposed to air, there is formation of La2(OH)6–2x(CO3)x, with x ≈ 1 [27,28].
In the following sections, the compositions Sr10(PO4)6O, Sr9La(PO4)5(SiO4)O, Sr8La2(PO4)4(SiO4)2O, Sr6La4(PO4)2(SiO4)4O, Sr5La5(PO4)(SiO4)5O and Sr4La6(SiO4)6O will be labeled Sr10O, Sr9LaO, Sr8La2O, Sr6La4O, Sr5La5O and Sr4La6O, respectively.
2.2 Powder characterization
X-ray diffraction (XRD) patterns of samples were recorded on an X’Pert Pro, PANalytical diffractometer operating with the Cu Kα radiation. The samples were scanned in the 2θ range from 20 to 65° with a step size of 0.01° and a counting time of 1 s per step. The experimental patterns were compared to standards compiled by the Joint Committee on Powder Diffraction and Standards (JCPDS) using the X’Pert HighScore Plus software. The lattice parameters of all samples were determined by the least squared method.
Infrared absorption spectra were recorded in the 400–4000 cm−1 range with a PerkinElmer Spectrum 100 spectrometer, using the KBr pellet technique.
Thermal analysis was conducted in air from room temperature to 1000 °C at a heating rate of 10 °C/min, using a Setaram 92 apparatus.
Impedance measurements were carried out on the Sr6La4O sample using a Hewlett-Packard 4192-A impedance analyzer, operating at frequencies ranging from 5 to 13 MHz. After milling for 25 h, the powder was pelletized under a pressure of 13 tons. Then, the pellets were heated at 900 °C for 24 h. This temperature was imposed by our furnace capacity. The obtained pellet has a thickness (e) of 0.0885 cm and a surface (S) of 1.252 cm2. The geometric factor (e/S) of the pellet is 0.07 cm−1. The relative density was of about 60%. Silver electrodes were painted on the two faces of the pellets with a silver paste, and then the painted pellets were heated at 300 °C for 1 h.
3 Results and discussion
3.1 X-ray diffraction
Fig. 2 shows the XRD patterns of samples ground for 25 h. As seen, all the patterns exhibit only the reflections of an apatite phase, indexed in the hexagonal system (P63/m space group) based on the strontium oxyapatite, Sr10(PO4)6O (JCPDS card No. 00-044-0654), and no evidence of any second phase was found. This indicates that all the prepared samples were single phase. However, the presence of other phosphate compounds as amorphous phase or as impurities in small quantities could not be excluded.
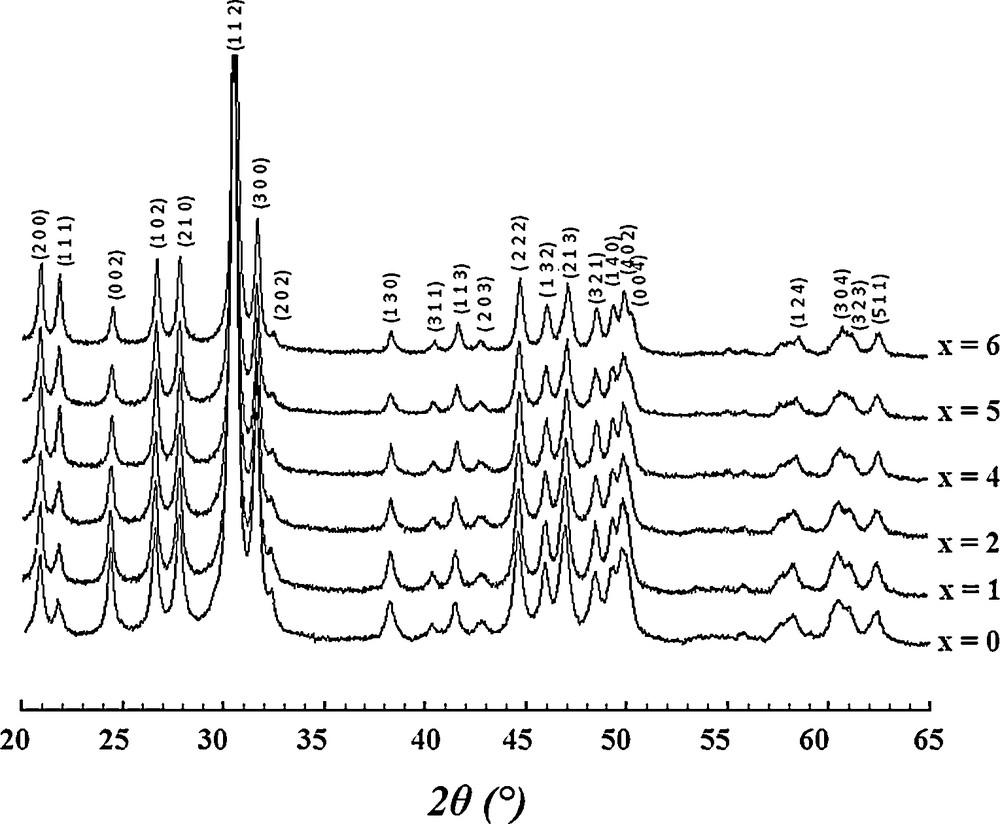
XRD patterns of all the samples milled for 25 h.
3.2 Infrared spectroscopy
The IR absorption spectra of the samples milled for 25 h are displayed in Fig. 3. The assignments of the different absorption bands were performed according to the literature [29–32]. The similarity of the vibration modes of the two tetrahedral groups ( and ) has made the identification of the bands somewhat difficult. The assignments of the observed vibrational frequencies of the prepared samples are summarized in Table 1.
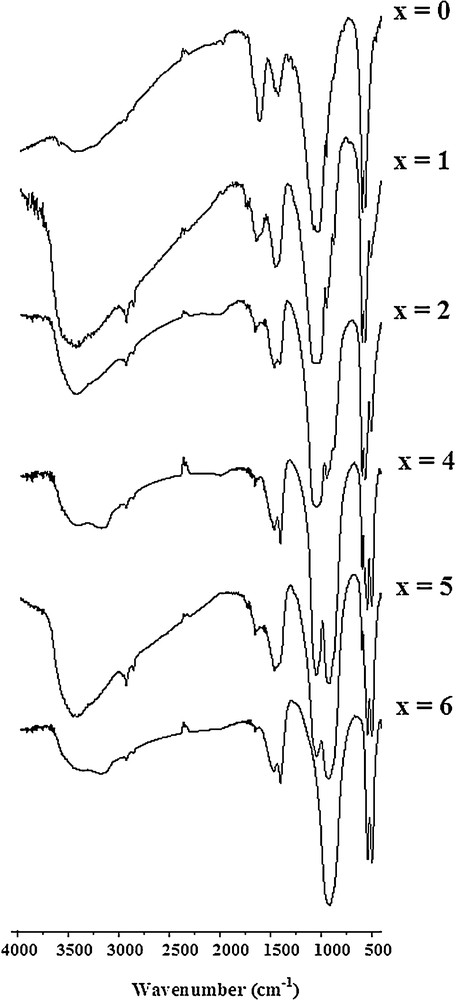
FTIR spectra of all the samples milled for 25 h.
Assignment of the IR absorption bands for the samples milled for 25 h.
| Samples | PO43– | SiO44– | ||||
| ν1 (cm−1) | ν3 (cm−1) | ν4 (cm−1) | ν1 (cm−1) | ν3 (cm−1) | ν4 (cm−1) | |
| Sr10O | 946 | 1042 1073 | 591 561 | – | – | – |
| Sr9LaO | 942 | 1041 | 591 563 | 871 | 916 | 504 543 |
| Sr8La2O | 942 | 1043 | 593 564 | 872 | 916 | 505 544 |
| Sr6La4O | 943 | 1042 | 562 595 | 874 | 915 | 499 542 |
| Sr5La5O | 942 | 1046 | 596 – | 877 | 920 | 498 544 |
| Sr4La6O | – | – | – | – | 922 | 540 496 |
For the non-substituted sample, the bands at 1073–1042 and 946 cm−1 correspond to P–O stretching vibration modes, while the doublet at 591–561 cm−1 corresponds to the OPO bending mode. The doublet at 1453 and 1406 cm−1, and the band at about 863 cm−1 were assigned to the ν3 and υ2 vibration modes of groups. These bands indicate that some groups were partly replaced by ones in the apatite structure (B-type substitution). Therefore, an equivalent amount of SiO2 was expected to remain in its original form in the powder. It was not detected probably due to its low quantity. The weak band at around 3570 cm−1 can be attributed to the stretching mode of the hydroxyl group, indicating that some OH–groups were inserted into the apatite lattice. Notice the lack of the band at 630 cm−1 corresponding to the bending mode of this latter group. Thus, although the grinding was carried out by a dry process, air moisture may result in the incorporation of hydroxyl groups into the apatite structure. It is well known that phospho-oxyapatite is rarely obtained in a pure state [33]. The bands around 3422 cm−1, as well as the band at 1606 cm−1, are related to the adsorbed water on the solid surface. For the substituted samples with 1 ≤ x ≤ 5, additional bands appeared at 920, 872 and 500–545 cm−1, they were assigned to the group. The band located at 920 cm−1 is assigned to the asymmetric stretching mode (ν3) of the SiO bond, while that at 872 cm−1 corresponds to the symmetric stretching mode (ν1). Finally, the bands between 500 and 545 cm−1 are attributed to the asymmetric deformation mode (ν4). Furthermore, with the increase of the silica amount in the initial mixture, the intensities of the bands increased, whereas those of the bands decreased. This suggests that the ions substituted for the ones into the apatite lattice. However, as some carbonate ions were incorporated into the apatite structure, an equivalent amount of SiO2 would not participate in the reaction. The bands associated with the group disappeared completely for the totally silicated sample (x = 6).
3.3 Thermal behavior
TG/DTA curves of the Sr6La4O sample milled for 25 h are given in Fig. 4. According to equation (1), the heat treatment must not be accompanied by any weight loss. However, when carbonate groups are incorporated into the apatite structure, in accordance with the charge balance, a certain amount of lanthanum oxide or strontium oxide would not participate in the reaction [24]. It is well known that the lanthanum oxide is very sensitive to moisture and carbon dioxide of air. So, after a prolonged exposure to air, it turns into a mixture of oxide, hydroxide and amorphous hydrated hydroxycarbonate [27,34]. Furthermore, as the FTIR spectroscopic analysis showed that the prepared apatites were carbonated, weight losses should occur during heat treatment. Panteix studying the sintering of La9.33(SiO4)6O2 observed that although the pellets are sufficiently densified, they degraded when they are left long enough in the air. He attributed this phenomenon to the very high hydrophilicity of the unreacted La2O3 and to its sensitivity to air carbon dioxide [27].
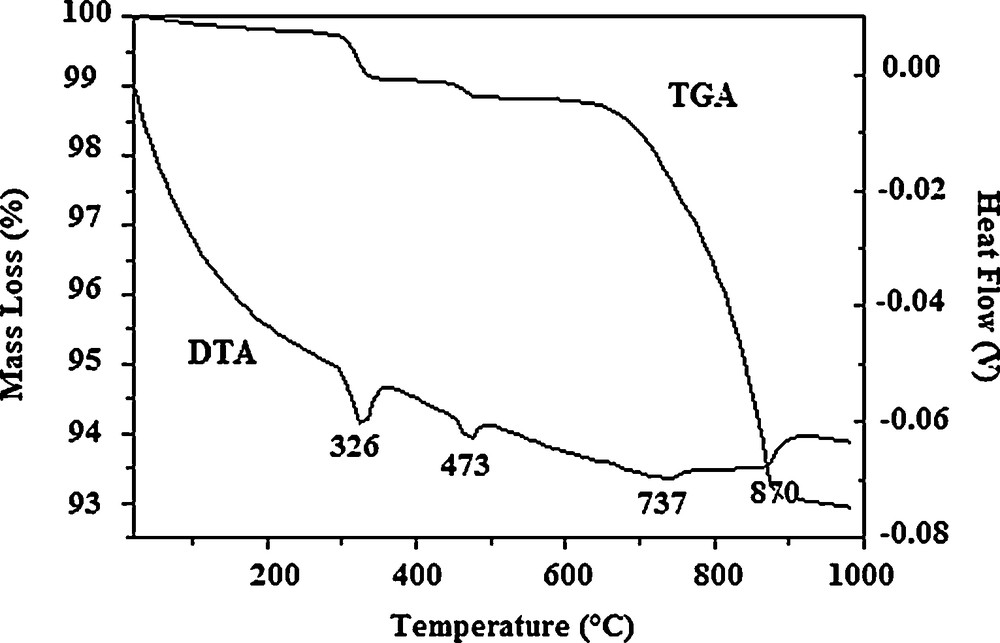
Thermal analysis of the sample Sr6La4O milled for 25 h.
The TG profile (Fig. 4) indicated that the weight loss occurred in three temperature ranges. The first weight loss of 0.9% observed in the range from 280 to 350 °C is due to the dehydration of La(OH)3 and La2(OH)4CO3·nH2O to LaOOH and La2O2CO3, respectively. The second weight loss of 0.4% achieved at 480 °C was attributed to the departure of water resulting from the transformation of LaOOH into La2O3 [27,28,34]. These dehydrations are related to the two first endothermic peaks positioned on the DTA curve at 326 and 473 °C, respectively. The third weight loss of 5.6% occurring between 640 and 900 °C is related to the decarbonation of La2O2CO3 [27,34] and the apatite, The departure of CO2 is illustrated on the DTA curve by the third and the fourth effects at 737 and 870 °C. It is worth noting that no traces of unreacted oxides or carbonates were detected on the XRD patterns, due probably to their amounts that were below the detection limit of XRD.
The XRD pattern of Sr6La4O sample milled for 25 h and calcined at 900 °C for 24 h shows that the powder was formed by a pure apatite phase, and no secondary phases were detected (Fig. 5). As observed in Fig. 6, the heat treatment did not cause any significant change in the IR spectrum. It indicates only a decrease in the intensity of bands associated with the carbonate ions.
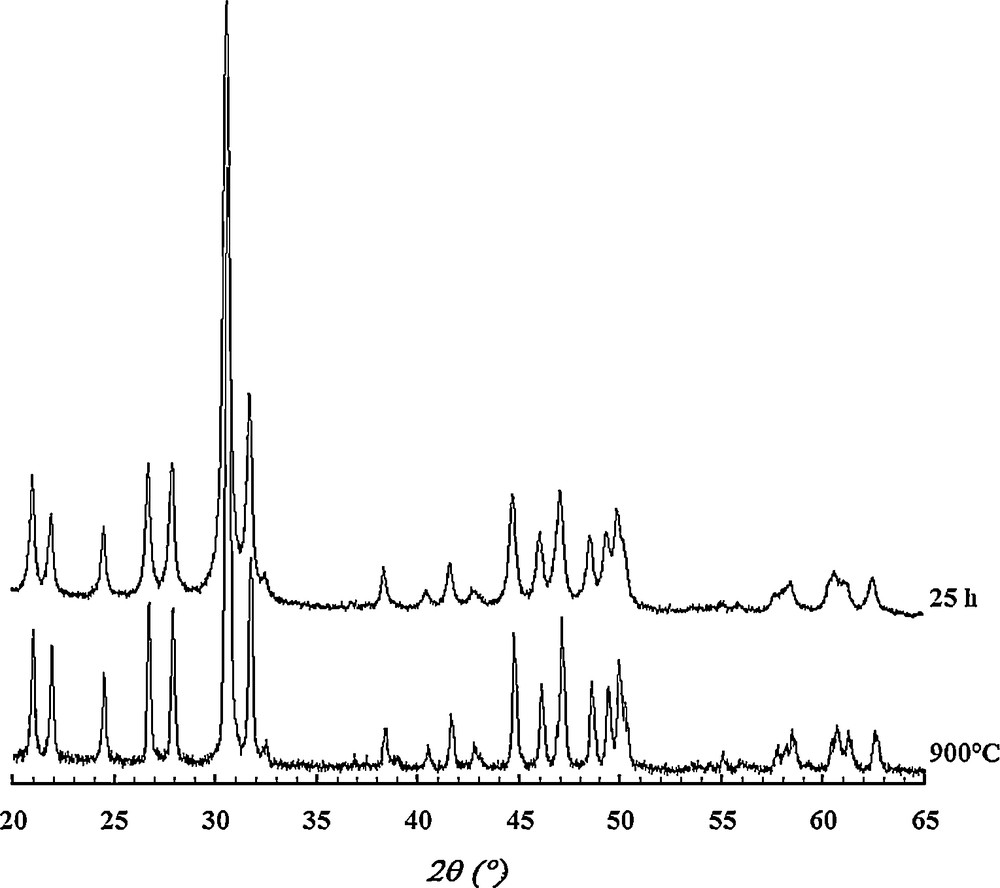
XRD patterns of the sample Sr6La4O milled and calcined at 900 °C.
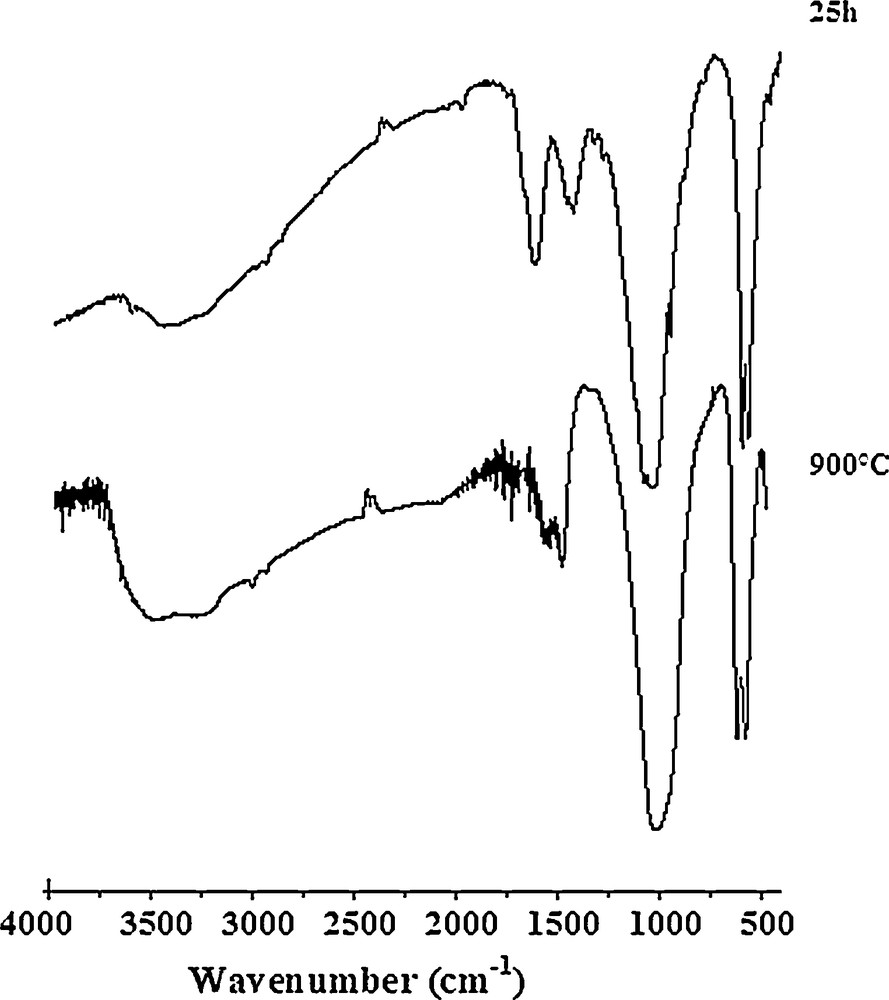
FTIR spectra of the sample Sr6La4O milled and calcined at 900 °C.
3.4 Lattice parameters
The lattice parameters are given in Table 2. For the non-substituted sample, the values of a = 9.749(2) Ǻ and c = 7.270(3) Ǻ are lower than those reported in the JCPDS card No. 00-044-0654 (a = 9.7670 Å and c = 7.2890 Å). With the substitution, the a parameter increased, while c decreased. The changes in the lattice parameters confirm once again that the pair (La3+, ) has entered the apatite structure [24]. However, the interpretation of the variation of the parameters with substitution remains difficult because of the difference in size of the substituent ions (Table 3) [35]. Furthermore, after the insertion of some carbonate ions into the apatite lattice, as it was indicated by IR absorption spectroscopy, the interpretation becomes more complicated. However, it is possible to draw some conclusions. In the apatite lattice, the a-axis is rather influenced by the size of the M(2) site, the anionic tetrahedron and the anion of the tunnel, while the c-axis is mainly influenced by the size of the M(1) site. In the present study, the group (the average length of P–O is 1.51 Å) is replaced by both a more and a less voluminous groups, (the average length of Si-O is 1.62 Å) and (the average length of C–O is 1.25 Ǻ) [35]. In addition, Sr2+ ( = 1.21 Å; = 1.31 Å) was substituted by a less voluminous ion La3+ ( = 1.10 Å; = 1.216 Ǻ) [35].
Lattice parameters for the samples milled for 25 h.
| Samples | a (Å) | c (Å) | V(Å3) |
| Sr10O | 9.749 (2) | 7.270 (3) | 599.06 (3) |
| Sr9LaO | 9.752 (1) | 7.269 (3) | 599.35 (1) |
| Sr8La2O | 9.756 (3) | 7.265 (2) | 599.51 (3) |
| Sr6La4O | 9.758 (2) | 7.263 (2) | 599.59 (2) |
| Sr5La5O | 9.762 (3) | 7.260 (4) | 599.83 (3) |
| Sr4La6O | 9.767 (4) | 7.258 (2) | 600.28 (2) |
The ionic radii of the different ions.
| Sr2+ | La3+ | P5+ | Si4+ | C4+ | |
| CN7/CN9 | CN7/CN9 | CN4 | CN4 | CN4 | |
| Ionic radii (Å) | 1.21/1.31 | 1.10/1.22 | 0.17 | 0.26 | 0.15 |
As was shown in ref. [24], following these substitutions, there was creation of vacancies in the cationic sites. In the apatite structure, the cations are distributed between two nonequivalent crystallographic sites. Four cations per unit cell occupying the (4f) sites are arranged along the three-fold axis. The six other cations located in the (6 h) sites are positioned in the summit of two alternated equilateral triangles, centered on a helical six-fold axis of the structure. The alignment in columns, and the shorter distances between the (4f) sites cause strong repulsions between the La3+ ions located in these sites, while the higher distances between the (6 h) sites make them more suitable to minimize the repulsions. Therefore, these latter sites should be fully occupied by the cations and the vacancies should be present only at the (4f) sites. This assumption is supported by the Rietveld refinement of La9.33(SiO4)6O2, which showed that all of the vacancies are present at the (4f) sites [36]. Vacancies were also created in the anionic tunnel following the substitution of O2–for OH–. Thus, it appears that the contraction of the c parameter is due to the replacement of Sr2+ by a smaller ion, La3+, on the one hand, and to the presence of vacancies along this axis, on the other hand, while the increase of the a parameter is due to the contraction following the c-axis. The explanation of the effect of substitution on the a-axis is not as obvious because of the different sizes of the substituted ions. The comparison of the obtained parameter values with those determined by K. Boughzala for the same compounds, prepared at high temperature [37], shows the same trend as a function of x for the two parameters. It is worth noting that these compounds are free of carbonates. Our values are weaker probably because of the incorporation of carbonates into the apatite structure (see § 3.2.).
3.5 Ionic conductivity
Typical complex impedance plots of the Sr6La4O sample taken in the 525–800 °C range are illustrated in Fig. 7. As seen, the impedance response of the sample consists of semicircular arcs. The value of resistance at different temperatures has been obtained from the intercept of the semicircular arcs on the real axis, and the electrical conductivity at different temperatures was calculated using the following equation: σ = e/R·s, where R is the resistance determined from impedance plots, and e and s are the thickness and the area of the sample, respectively. The obtained values increased with increasing temperature, varying from 6.76 × 10−8 to 1.52 × 10−6 S·cm−1 for 525 and 800 °C, respectively, indicating a thermally activated conduction mechanism. These values correspond to the total conductivity because of the difficulty to separate the grain and bulk boundary contribution from the diagrams.
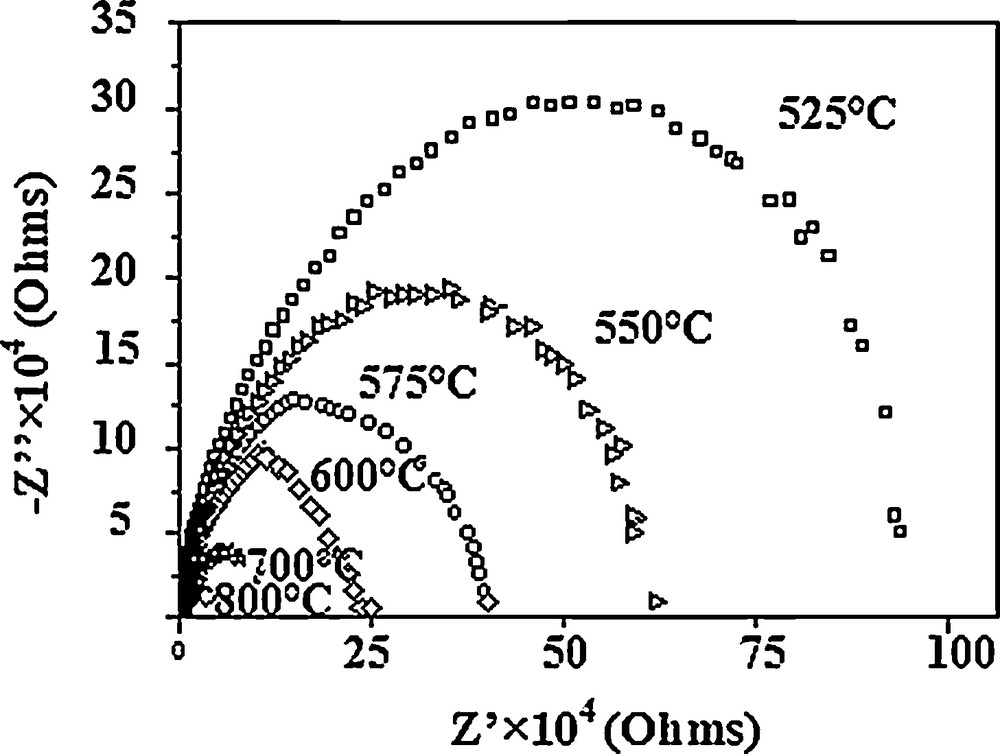
The impedance plots of Sr6La4O at different temperatures.
The temperature dependence of the ionic conductivity has been analyzed using an Arrhenius equation: σ T = A exp(–Ea/kT), where σ, A, Ea, k and T are the conductivity, pre-exponential factor, activation energy, Boltzmann constant and absolute temperature, respectively. Fig. 8 shows the Arrhenius plot of the sample. The activation energy Ea, determined from the slope of the previous plot, is 0.85 eV.
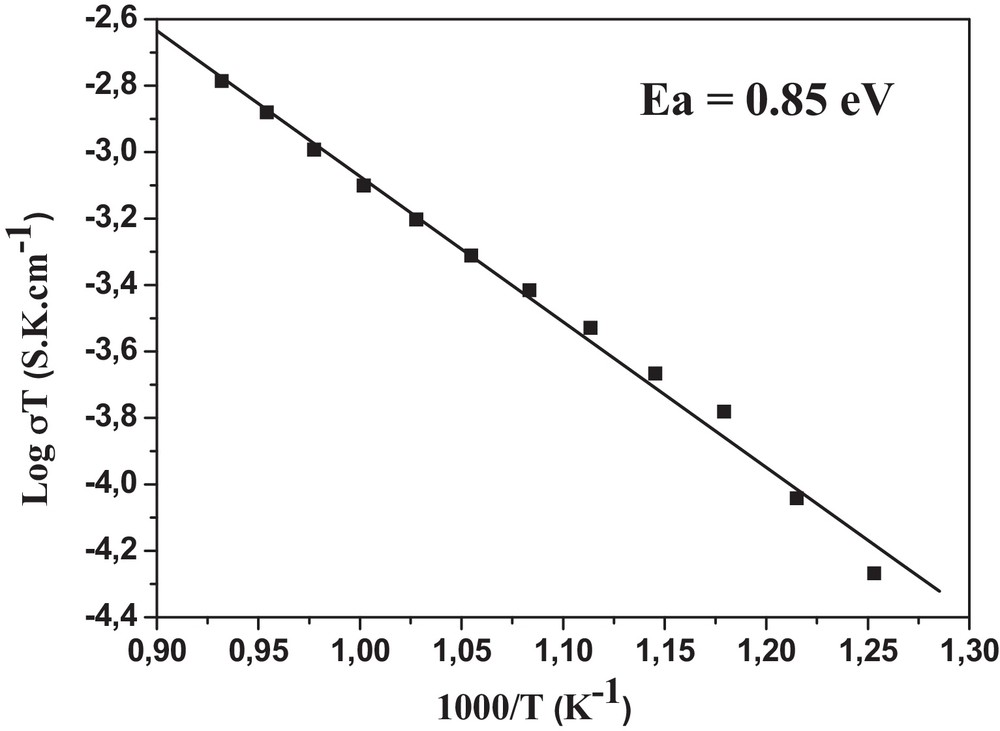
Plot of log σT (S·K·cm–1) versus 1000/T (K–1) for Sr6La4O.
It is well known that for the rare earth silicate oxypatites, the highest conductivity is observed for the samples containing cationic vacancies and/or an oxygen excess [38,39]. Compared to the lanthanum silicate oxyapatite (La9.33(SiO4)6O2) [40], the studied material has a lower conductivity. This can be explained, on the one hand, by the high porosity of the material (relative density = 60%) and on the other hand, by its structure features. As was revealed by IR spectroscopy analysis, all carbonates have not been completely eliminated after heat treatment at 900 °C. Therefore, with respect to the oxyapatite-type La9.33SiO26, the carbonate ions, and those of phosphate contracted the a-axis, in accordance with their respective sizes. It results a contraction of the tunnel containing oxide ions, limiting therefore the movement of these latter ions. Indeed, it was shown that a high conductivity is characterized by larger channels, in addition to the cationic vacancies [41]. Panteix et al. studying the influence of anionic vacancies on the ionic conductivity of the lanthanum silicate oxyapatite observed that the conductivity is enhanced and the activation energy is decreased for the low levels of oxygen vacancies, while for the high levels, the conductivity is lower and the activation energy is higher [42]. Therefore, the anionic vacancies due to the substitution of O2–by OH–contributed to lowering the conductivity for our sample. In contrast to the fully stoichiometric sample, Sr6La4(PO4)4(SiO4)2O2 [40], the compound investigated in the present work had a higher conductivity, confirming once again that this latter contained cationic vacancies, obviously in addition to the anionic vacancies.
4 Conclusion
Oxyapatites with the chemical formula Sr10–xLax(PO4)6–x(SiO4)xO (where 0 ≤ x ≤ 6) have been prepared from SrO, Sr2P2O7, La2O3 and SiO2 using the mechanochemical process. The obtained powders have been characterized by X-ray diffraction, infrared spectroscopy, and thermogravimetric and differential thermal analysis. The results indicated that the powders were single phase. However, they were carbonated, due to the air carbon dioxide. The electrical properties of the Sr6La4(PO4)2(SiO4)4O sample was investigated by using complex impedance analysis. The highest ionic conductivity of 1.52 × 10−6 S·cm−1 was obtained at 800 °C and the ionic jump activation energy is of 0.85 eV.