

1 Introduction
Beside the fact that most of the anticancer drugs placed in the market are organic compounds [1], the metal-based chemotherapeutic drugs are now becoming popular because of their different mechanisms of action compared with organic chemotherapeutic agents [1–3]. Cis-platin and recently, its new analogues (carboplatin, lobaplatin, oxaliplatin and nedaplatin) are being marketed to cure cancer patients. This has further led interest in the field of metal-based anticancer drugs. With this recent development, researchers are now interested to explore the biological significance of either the borderline or non-transition metals due to their lower toxicity compared to transition metals. This is also evident by recently compiled reviews [3,4].
Tin is a non-transition and biocompatible metal [5–7] and its complexes have shown significant antibacterial properties [8–22]. However, tin complexes have been rarely studied against cancer [12,23–28]. The current study is an effort to further explore this area of research.
The organotin(IV) complexes show interesting coordination geometries as well as structural diversity, which could expand from simple, monomeric to polymeric structures [8,29–35]. This gives biological significance to the tin complexes where their coordination motives and geometry can be monitored. In addition, based on well-documented X-ray crystallographic study, in some cases, the participation of solvent molecules such as water, acetone and methanol in organotin(IV) complexes influences the overall structure of organotin(IV) complexes including its packing which may enhance their water solubility [31,32,34,36].
In this paper, we report the synthesis and structural characterization of organotin(IV) carboxylate complexes (1–2) derived from 5-amino-2-chlorobenzoic acid (HL) and their in vitro anticancer studies against three cancerous and one non-cancerous (normal) cell lines.
2 Experimental
2.1 General and instrumental
All the reagents, starting materials as well as the solvents were purchased commercially and were used without any further purification. Elemental C, H and N analyses were carried out on a Perkin-Elmer 2400 CHN Elemental Analyzer. Tin was determined gravimetrically by igniting a known quantity of each complex to SnO2. The melting points were determined in an open capillary and were uncorrected. The infrared spectra were recorded using a Perkin-Elmer System 2000 FTIR Spectrophotometer as a KBr disc in the frequency range of 4000–400 cm−1. The spectra for 1H, 13C and 119Sn-NMR were recorded on a Joel JNM-ECX 400 FT-NMR Spectrometer using deuterated d6-DMSO as the solvent and tetramethylsilane, TMS as the internal standard.
Single crystal X-ray diffraction data were collected on Bruker APEX II or APEX II Duo CCD area-detector diffractometer operating at 50 kV and 30 mA using Mo Kα radiation (λ = 0.71073 Å). Diffraction data for complexes 1–2 were collected with the Oxford Cryosystem Cobra low temperature attachment at 100 K [37]. Data collection and reduction were performed using the APEX2 and SAINT software. The SADABS software was used for absorption correction. All structures were solved by direct method and refinement was carried out by the full-matrix least-squares technique on F2 using SHELXTL package [38]. All non-hydrogen atoms were refined anisotropically whereas hydrogen atoms were refined isotropically. All N-bound H atoms were located in difference Fourier maps and fixed at their found positions and refined with Uiso(H) = 1.2 Ueq (N). The hydrogen atoms bound to C atoms were positioned geometrically with Uiso (H) = 1.2 or 1.5 Ueq (C). A rotating-group model was applied for the methyl groups.
2.2 5-Amino-2-chlorobenzoic acid, HL
The parent acid, 5-amino-2-chlorobenzoic acid, HL was purchased from Acros Organics and was used without any further purification. FTIR as KBr disc (cm−1): selected data: ν(OH) 2500–3400, ν(COO)as 1624, ν(COO)s 1371, Δν = 253. 1H-NMR (ppm) (d6-DMSO): δ: benzene protons 6.64 (dd, 2.4 Hz, 8.6 Hz, 1H); 6.91 (d, 3.0 Hz, 1H); 7.06 (d, 9.2 Hz, 1H). 13C-NMR (ppm) (d6-DMSO): δ: benzene carbons 115.8, 117.6, 118.1, 131.3, 131.98, 147.9; COO 166.9.
2.3 Preparation of sodium salt (HL-Na)
The sodium salt of the acid was obtained by heating under reflux a 1:1 molar mixture of sodium hydroxide, NaOH and 5-amino-2-chlorobenzoic acid in ethanol (50 mL) for two hours. After few days, white precipitates were obtained. Sodium salt of 5-amino-2-chlorobenzoic acid: FTIR as KBr disc (cm−1) selected data: ν(COO)as 1565, ν(COO)s 1387, Δν = 178.
2.4 Synthesis of complexes
2.4.1 Preparation of [{5-NH2-2-Cl-C6H3COO(C4H9)2Sn}2O]2 (1)
Complex 1 was obtained by heating under reflux a 1:1 molar mixture of dibutyltin(IV) oxide (1.50 g, 6 mmole) and 5-amino-2-chlorobenzoic acid (1.03 g, 6 mmole). The reaction was carried out in a mixture of ethanol/toluene (1:4, 60 mL) for three hours. A clear transparent solution was isolated by filtration and kept in a bottle. After few days, colorless crystals (2.17 g, 88.0% yield) were collected. Melting point: 200–202 °C. Analysis for C60H92N4O10Cl4Sn4: 42.58; H, 6.66; N, 3.76; Sn, 27.11%. Calculated for C60H92N4O10Cl4Sn4: C, 43.78; H, 5.63; N, 3.41; Sn, 28.84%. FTIR as KBr disc (cm−1): ν(COO)as 1637, 1600, ν(COO)s 1281, 1342, Δν = 356, 258; ν(O–Sn–O)/ν(Sn–O–Sn) 629, ν(Sn-C) 556, ν(Sn-O) 436. 1H-NMR (ppm) (d6-DMSO): δ: benzene protons 6.54 (d, 6.8 Hz, 8H); 6.98 (d, 8.2 Hz, 4H); butyl, CH3 0.75 (t, 7.3 Hz, 12H), 0.79 (t, 7.4 Hz, 12H); CH2 1.15–1.29 (m, 32H); CH2 1.43–1.60 (m, 16H). 13C-NMR (ppm) (d6-DMSO): δ: benzene carbons 116.1, 117.6, 130.2, 13.0, 148.0; butyl 14.0, 14.1, 26.2, 26.9, 27.0, 27.4; COO 172.1. 119Sn-NMR (ppm) (d6-DMSO): δ: –185.7, –230.8.
2.4.2 Preparation of {5-NH2-2-Cl-Cf6H3COO(C6H5)3Sn}2(CH3)2CO (2)
The title complex was obtained by heating under reflux a 1:1 molar mixture of triphenyltin(IV) hydroxide (1.47 g, 4 mmole) and 5-amino-2-chlorobenzoic acid (0.69 g, 4 mmole) in acetone (50 mL) for two hours. A clear solution was isolated by filtration and kept in a bottle. After few days, transparent crystals (1.56 g, 81.0% yield) were collected. Melting point: 127-129 °C. Analysis for C53H46N2O5Cl2Sn2: C, 57.46; H, 4.17; N, 2.69; Sn, 20.01%. Calculated for C53H46N2O5Cl2Sn2: C, 57.91; H, 4.22; N, 2.55; Sn, 21.59%. FTIR as KBr disc (cm−1): ν(COO)as 1623, ν(COO)s 1355, Δν = 268; ν(Sn–O) 453. 1H-NMR (ppm) (d6-DMSO): δ: phenyl protons 7.36–7.42 (m, 18H); 7.72–7.90 (m, 12H); benzene 6.48 (dd, 3.0 Hz, 8.6 Hz, 2H); 6.68 (d, 3.1 Hz, 2H); 6.93 (d, 8.6 Hz, 2H); (CH3)2CO 2.04 (s, 6H). 13C-NMR (ppm) (d6-DMSO): δ: phenyl carbons Cipso 143.4, Cortho 136.7 (46.0 Hz), Cmeta 128.4 (69.0 Hz), Cpara 128.7; benzene 115.4, 116.4, 117.0, 129.4, 130.6, 147.7; (CH3)2CO 30.0, 207.7; COO 170.2. 119Sn-NMR (ppm) (d6-DMSO): δ: –260.8.
2.5 In vitro anticancer studies
The synthesized compounds were dissolved in DMSO to obtain 10 mM stock solution. For experiment, the compounds were diluted in the indicated culture medium at the indicated concentrations in each test. DMSO (0.1%) was used as a negative control. Source of cell lines, culture conditions, preparation of cell culture and MTT assay were performed according to our previously reported procedure for these cell lines [39–42]. The details of cell culture conditions, preparation of cell culture and MTT assay procedure has been additionally provided in supplementary file.
3 Results and discussion
3.1 Synthesis
In this study, complexes 1–2 have been obtained in solid state. Complexes 1–2 gave a sharp melting points (with a range of less than 3 °C) indicating the isolation of fairly pure complexes. Synthesis of complexes 1–2 has been provided in Scheme 1. The micro-elemental analyses for data obtained were in agreement with the predicted formula for complexes 1–2. Based on the micro-elemental analysis, it was believed that acetone molecule was present in complex 2 which acted as a solvate molecule [31,32,34,36].
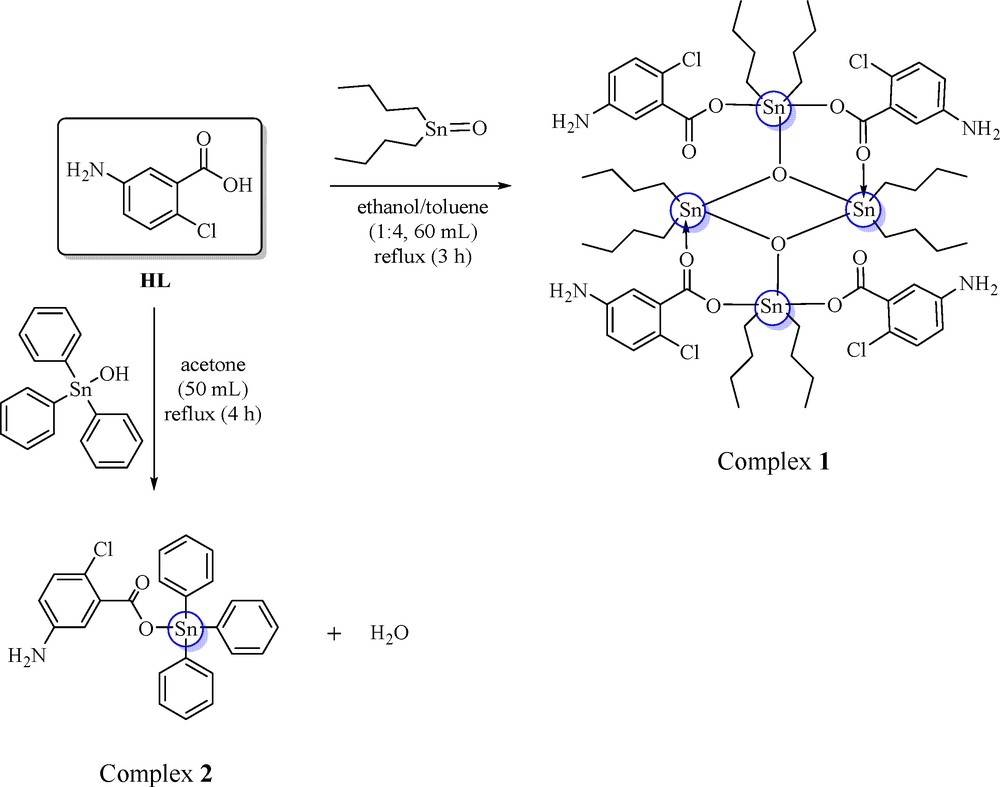
(Color online.) Synthesis of tin complexes 1 and 2.
3.2 Characterization by FTIR spectroscopy
The synthesized complexes (1–2) as well the starting materials (HL and HL-Na) were preliminarily characterized by FTIR to observe the possible spectral changes in complexes than compared to reactants. Fig. 1 represents the overlay spectra of reactants (HL and HL-Na) and complexes (1–2), highlighting the observable changes. The ν(O-H) bands of the acid, HL was absent in the infrared spectra of salt and complexes 1–2 which indicated the deprotonation and possible coordination of the carboxylate anion with the metal cation [43]. In addition, complex 2 revealed that the ν(COO)as shifted to a lower wavelength number compared to the acid, HL which signified that the coordination took place via the oxygen atoms of the carboxylate anion. From the infrared spectrum of 1, the first Δν value (356 cm−1) was larger than the Δν value of the sodium salt while the second Δν value (258 cm−1) was comparable to the sodium salt (194 cm−1) indicating a pair of carboxylate anions coordinated in monodentate manner while another pair coordinated in bidentate manner [44].
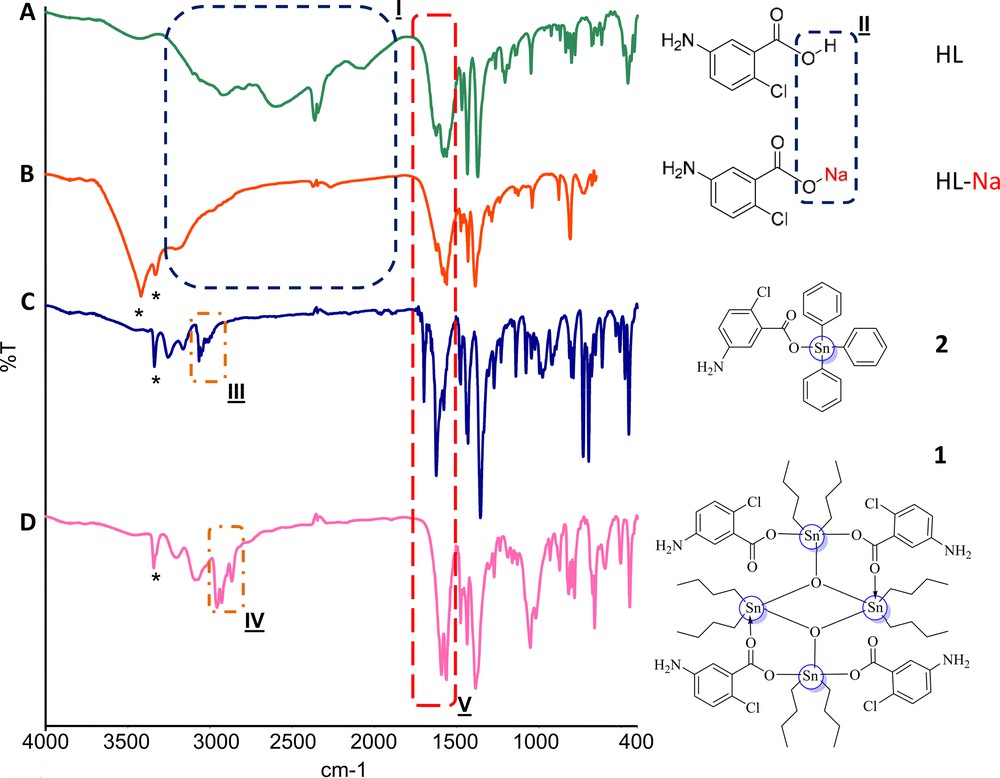
(Color online.) FTIR spectral features of HL, HL-Na, 1 and 2. Spectrum A (for HL) shows that a broad vibrational band (2500–3400 cm−1 for COOH) vanished in spectrum B (for HL-Na) due to the replacement of proton with sodium ion indicating the possible salt formation. In spectrum B two “*” symbols as enhanced vibrational bands (3332 and 3422 cm−1) indicated the presence of primary amine. These vibrations remained suppressed in HL, perhaps due the presence of either intermolecular H-bonding or overlapped by COOH vibrations. Also, changes in the patterns of these NH2 vibrations in the complexes 1 and 2 might be due to stronger intermolecular H-bonding as can be seen in the crystal packing of these complexes (Supplemental). Furthermore, in spectrum C, appearance of highlighted vibrations III (3000–3100 cm−1) and in D, IV (2800–3000 cm−1) verified the enhanced aromatic rings (C–Harom) in 1 and incorporation of alkyl groups (C–Halkyl) in 2, respectively. In all the spectra (A–D), the highlighted region V indicates the observed changes for ν(COO)as before and after coordination as described in the text of the article.
For complexes derived from triphenyltin(IV) carboxylate, Δν greater than 200 cm−1 would be expected for the monodentate bonding carboxylate anions [45]. Hence, the carboxylate anion in complex 2 would be expected to bond to the tin(IV) atom in monodentate manner since the Δν is above 200 cm−1. For further evidence of the coordination to tin(IV) atom via oxygen atoms revealed by the presence of the ν(O–Sn–O)/ν(Sn–O–Sn) and ν(Sn–O) stretching bands in the spectra of 1–2.
3.3 Characterization by 1H, 13C, 119Sn-NMR spectroscopies
The 1H-NMR spectra of 1–2 exhibited similarities to the acid, HL. The only exceptional differences were in the upfield regions of the 1H-NMR spectrum of 1 which showed the signals of the butyl protons of the organotin(IV) at 0.83 ppm and in the range of 0.75–1.60 ppm. Whereas for 2, the resonances appeared as two well separated sets of multiplets in the regions centering around δ ≈ 7.39 and 7.81 ppm (downfield) with the integration values of 9:6 respectively, ascribed to the aromatic protons of the phenyl group [46]. Furthermore, based on the 1H-NMR spectral studies of 2, the proton resonances originating from the acetone molecule occurred at δ = 2.04 ppm and based on the integration, only one acetone molecule was expected to present in 2.
Evidences of the formation of the complexes were further observed in the 13C-NMR spectra. The 13C-NMR spectra of complexes 1–2 showed that the δ(COO) signals shifted to the downfield region compared to that of the acid, HL indicating the carboxylate anion were bonded to tin(IV) atoms. Complex 1 is derivative of organodistannoxane dimer types which exhibited two sets of signals corresponding to the butyl groups in the 13C-NMR spectra. These two sets of signals were assigned to the butyl groups linked to the exo- and endocyclic tin(IV) atoms, respectively [47]. Complex 2 revealed the chemical shifts of the δ(13C)ipso at 143.4 ppm indicative of a penta-coordinated tin(IV) atom thus indicating that the tin(IV) atom in complex 2 was five-coordinated and has a trans-trigonal bipyramid geometry [48–50]. In addition, in 13C-NMR spectrum of 2 the signals due to the presence of acetone molecule were located at δ = 30.0 and 207.7 ppm.
The δ(119Sn) values of the four-coordinated complexes fall in the range between +200 to –60 ppm; the five-coordinated complexes between –90 to –190 ppm and the six-coordinated complexes between –210 to –400 ppm [51]. The organodistannoxane dimer type complexes usually exhibit two well resolved δ(119Sn) signals (complex 1 = –185.3, –230.8 ppm). Based on the 119Sn-NMR spectra, a pair of tin(IV) atoms were five-coordinated and another pair of tin(IV) atoms were six-coordinated in complex 1. Normally, the δ(119Sn) value of triphenyltin(IV) complexes lie in the range between –180 to –260 ppm was believed to be five-coordinated and in the distorted trigonal bipyramid geometry [Ph3SnX•L (L is a monodentate ligand)]. Complex 2 showed that the δ(119Sn) value at –260.88 ppm which lies slightly upfield in the range of –180 to –260 ppm indicated the tin(IV) atom was penta-coordinated and possessed a trans-trigonal bipyramid geometry [49,50]. From the 119Sn-NMR study, the acetone molecule (coordinating solvent) was expected to be coordinated to the tin(IV) atom in complex 2 resulting the tin(IV) atom being penta-coordinated.
3.4 Characterization by X-ray crystallography
3.4.1 Structural elucidation of complex 1
Single crystal X-ray determination revealed that the crystal system of complex 1 is triclinic with space group Pī and unit cell parameters a = 12.2010(6) Å, b = 12.2891(6) Å, c = 13.7860(7) Å, α = 108.029(1)°, β = 95.744(1)° and γ = 107.168(1)°. The summary of the crystallography data of 1 is shown in Table 1 while the selected bond lengths (Å) and angles (°) are listed in Table 2. The hydrogen bond geometry (Å,°) is given in Table 3. Fig. 2 represents the molecular structure of 1 with its numbering scheme while the molecular packing diagram is depicted in Fig. 1S.
Crystallography data of complexes 1 and 2.
| Parameter | 1 | 2 |
| Empirical formula | C60H92Cl4N4O10Sn4 | 2(C25H20ClNO2Sn)·C2H6O |
| Formula weight | 1645.94 | 1099.20 |
| Temperature (K) | 297.0 | 100.0 |
| Radiation/wavelength | Mo Kα/0.71073 Å | Mo Kα/0.71073 Å |
| Crystal system, space group | Triclinic, | Orthorhombic, Pbcn |
| Unit cell dimensions | a = 12.2010(6) Å, α = 108.029(1)° b = 12.2891(6) Å, β = 95.744(1)° c = 13.7860(7) Å, γ = 107.168(1)° | a = 14.6013(3)Å, α = 90° b = 20.2960(5)Å, β = 90° c = 15.8675(4)Å, γ = 90° |
| Volume (Å3) | 1835.83(16) | 4702.30(19) |
| Z, Calculated density (Mg/m3) | 1, 1.489 | 4, 1.553 |
| Absorption coefficient (mm−1) | 1.54 | 1.230 |
| F(000) | 828 | 2208 |
| Crystal size (mm) | 0.43 × 0.30 × 0.21 | 0.52 × 0.25 × 0.18 |
| Crystal morphology and color | Block/colourless | Block/colourless |
| θ range for data (°) | 2.8–30.9 | 2.4–32.7 |
| Limiting indices | –17 ≤ h ≤ 17, –17 ≤ k < = 17, –19 ≤ l ≤ 19 | –21 ≤ h ≤ 22, –24 ≤ k ≤ 30, –24 ≤ l ≤ 23 |
| Reflections collected/unique | 41859/11516 | 64943/8650 |
| R(int.) | 0.027 | 0.032 |
| Completeness | 98.5% | 99.5% |
| Max. and min. transmission | 0.741, 0.558 | 0.810, 0.570 |
| Goodness of fit | 1.03 | 1.05 |
| R[F2 > 2σ(F2)], wR(F2) | 0.037, 0.117 | 0.024, 0.055 |
| Largest diff. peak and hole (e.Å−3) | 1.66 and –0.92 | 0.52 and –0.53 |
Geometry parameter, bond lengths (Å) and angles (°) of complex 1.
| Bond lengths | |||
| Sn(1)–O(3) | 2.039 (2) | Sn(2)–O(3) | 2.0103 (19) |
| Sn(1)–C(15) | 2.115 (10) | Sn(2)–C(23A) | 2.042 (15) |
| Sn(1)–C(19) | 2.120 (8) | Sn(2)–C(27) | 2.080 (16) |
| Sn(1)–C(15A) | 2.13 (3) | Sn(2)–O(4) | 2.115 (3) |
| Sn(1)–O(3)a | 2.1505 (19) | Sn(2)–C(27A) | 2.153 (12) |
| Sn(1)–C(19A) | 2.16 (4) | Sn(2)–C(23) | 2.166 (12) |
| Sn(1)–O(1) | 2.240 (2) | O(3)–Sn(1)a | 2.1505 (19) |
| Bond angles | |||
| O(3)–Sn(1)–C(15) | 114.0 (2) | O(3)–Sn(1)–C(19A) | 115.1 (13) |
| O(3)–Sn(1)–C(19) | 113.3 (3) | C(15)–Sn(1)–C(19A) | 130.8 (13) |
| C(15)–Sn(1)–C(19) | 132.6 (4) | C(19)–Sn(1)–C(19A) | 1.8 (16) |
| O(3)–Sn(1)–C(15A) | 110.8 (8) | C(15A)–Sn(1)–C(19A) | 134.0 (16) |
| C(15)–Sn(1)–5(15A) | 3.3 (10) | O(3)a Sn(1)–C(19A) | 98.5 (12) |
| C(19)–Sn(1)–C(15A) | 135.9 (9) | O(3)–Sn(1)–O(1) | 74.34 (8) |
| O(3)a–Sn(1)–O(3)a | 73.35 (9) | C(15)–Sn(1)–O(1) | 93.3 (4) |
| C(15)–Sn(1)–O(3)a | 98.2 (4) | C(19)–Sn(1)–O(1) | 96.5 (2) |
| C(19)–Sn(1)–O(3)a | 97.7 (3) | C(15A)–Sn(1)–O(1) | 93.3 (13) |
| C(15A)–Sn(1)–O(3)a | 96.5 (12) | O(3)a–Sn(1)–O(1) | 147.64 (8) |
| C(19A)–Sn(1)–O(1) | 96.5(10) | Sn(1)–C(19A)–H(19B) | 107.1 |
| O(3)–Sn(2)–C(23A) | 119.5 (6) | Sn(1)–C(19A)–H(19C) | 107.1 |
| O(3)–Sn(2)–C(27) | 110.0 (6) | C(16)–C(15)–Sn(1) | 115.0 (6) |
| C(23A)–Sn(2)–C(27) | 123.0 (8) | Sn(1)–C(15)–H(15A) | 108.5 |
| O(3)–Sn(2)–O(4) | 81.32 (9) | Sn(1)–C(15)–H(15B) | 108.5 |
| C(23A)–Sn(2)–O(4) | 107.0 (4) | C(24)–C(23)–Sn(2) | 116.8 (8) |
| C(27)–Sn(2)–O(4) | 106.6 (5) | Sn(2)–C(23)–H(23A) | 108.1 |
| O(3)–Sn(2)–C(27A) | 109.4 (4) | Sn(2)–C(23)–H(23D) | 108.1 |
| C(23A)–Sn(2)–C(27A) | 124.0 (7) | C(24A)–C(23A)–Sn(2) | 115.0 (10) |
| C(27)–Sn(2)–C(27A) | 1.1 (8) | Sn(2)–C(23A)–H(23B) | 108.5 |
| O(4)–Sn(2)–C(27A) | 105.6 (3) | Sn(2)–C(23A)–H(23C) | 108.5 |
| O(3)–Sn(2)–C(23) | 107.1 (3) | C(28)–C(27)–Sn(2) | 124.3 (13) |
| C(23A)–Sn(2)–C(23) | 13.3 (6) | Sn(1)–C(15A)–H(15 C) | 107.8 |
| C(27)–Sn(2)–C(23) | 136.1 (6) | Sn(1)–C(15A)–H(15D) | 107.8 |
| O(4)–Sn(2)–C(23) | 101.5 (4) | C(20)–C(19)–Sn(1) | 116.9 (6) |
| C(27A)–Sn(2)–C(23) | 137.1 (5) | Sn(1)–C(19)–H(19A) | 107.6 |
| C(7)–O(1)–Sn(1) | 118.7 (2) | Sn(1)–C(19)–H(19D) | 108.7 |
| Sn(2)–O(3)–Sn(1) | 123.30 (9) | Sn(2)–C(27)–H(27A) | 106.3 |
| Sn(2)–O(3)–Sn(1)a | 130.03 (10) | Sn(2)–C(27)–H(27D) | 106.3 |
| Sn(1)–O(3)–Sn(1)a | 106.65 (8) | C(28A)–C(27A)–Sn(2) | 115.6 (8) |
| C(14)–O(4)–Sn(2) | 106.9 (2) | Sn(2)–C(27A)–H(27B) | 108.4 |
| Sn(1)–C(19)–H(19A) | 104.1 | Sn(2)–C(27A)–H(27C) | 108.4 |
| Sn(1)–C(19A)–H(19D) | 109.4 |
a Symmetry code: –x + 1, –y + 1,–z + 1.
Hydrogen bond geometry (Å,°) of complex 1.
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N1···O5a | 0.79 | 2.22 | 2.986 (7) | 166 |
| N2—H1N2···O2b | 0.83 | 2.42 | 3.195 (8) | 157 |
| C24—H24A···O5 | 0.99 | 2.48 | 3.312 (13) | 142 |
| C28—H28A···O5 | 0.99 | 2.57 | 3.31 (2) | 132 |
a Symmetry codes: − x + 1, − y + 2, − z + 2.
b Symmetry codes: x − 1, y − 1, z.
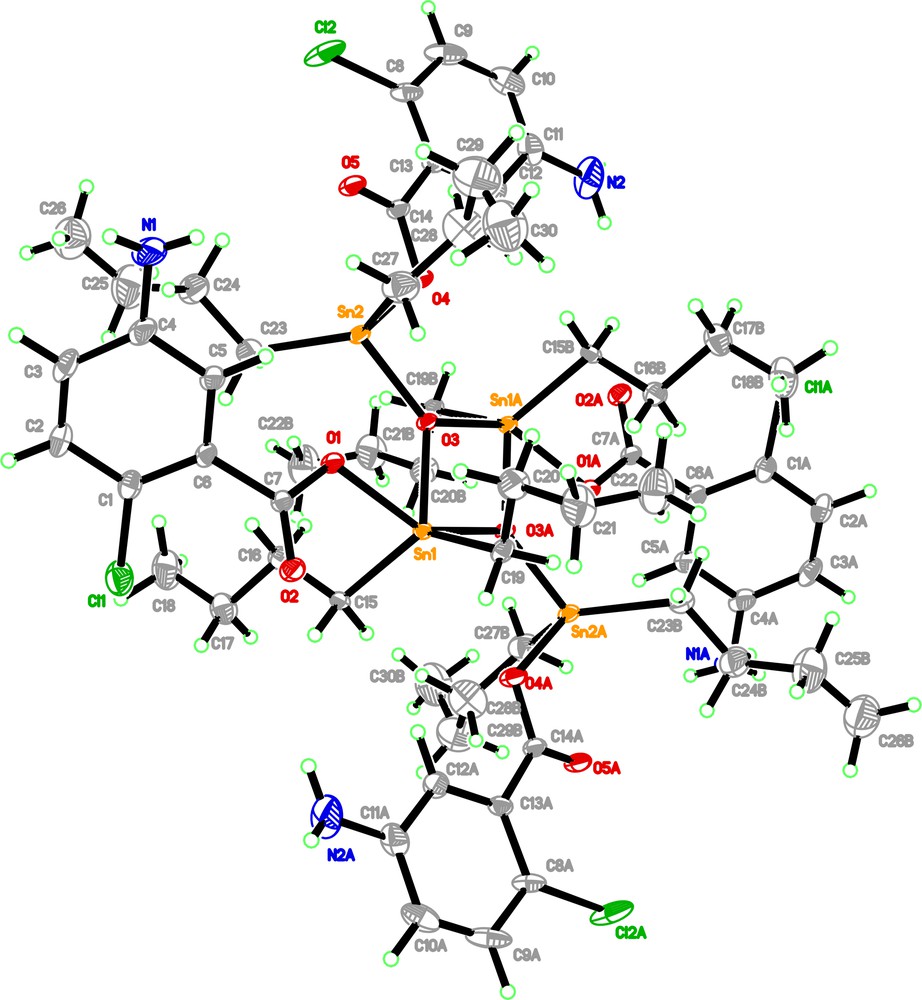
(Color online.) Molecular structure of complex 1 showing 20% probability displacement ellipsoids and its atomic numbering. Minor disorder component has been omitted for clarity.
The core geometry of 1 consists of a centrosymmetric, planar, four-membered Sn2O2 unit with the Sn(1)–O(3)–Sn(1A) and O(3)–Sn(1)–O(3A) angles of 106.65(8)° and 73.35(9)° respectively, as observed from the related structure. The Sn2O2 unit was found to be connected to a pair of exocyclic Sn atoms via the bridging oxygen atoms such that the oxygen atoms [O(3) and O(3A)] were tri-coordinated. The exocyclic tin atom, Sn(2), formed four primary bonds: one to the O(3) atom, one to the carboxylic oxygen atoms, O(4) and two to the tin-bound butyl groups. The endocyclic Sn(1) tin atom was also penta-coordinated and existed in a distorted trigonal bipyramid geometry, with the atoms C(15), C(19), O(3), O(3A) and O(1). Based on the crystal structure of 1, the Sn(2A) tin atom was exocyclic and had a similar coordination mode to Sn(2) whereas the Sn(1A) tin atom was similar to Sn(1). The chloro- and amine groups did not participate in the coordination to the Sn centres and were directed away from the central unit.
In the crystal structure, the molecules are linked to form a two-dimensional network (Fig. S1) parallel to the (1 –1 0) by CH···O and NH···O hydrogen bonds (Table 3).
Based on the spectroscopy studies, it was expected that a pair of carboxylate anions is coordinated in monodentate manner whereas the other pair of carboxylate anions is coordinated in bidentate manner. However, based on the crystallography structure studies of the 1, all the four carboxylate ligands were found to be connected to each of the exo- and endocyclic Sn(IV) atoms through one oxygen atom of the carboxylate anions. From this study, it has been confirmed that a new type of organodistannoxane dimer has been found and characterized where all the carboxylate anions are bonded in monodentate manner resulting a pair of tin(IV) atoms are tetra-coordinated and another pair of tin(IV) atoms are penta-coordinated.
3.4.2 Structural elucidation of complex 2
Complex 2 was isolated as a colorless single crystals and the structure of the complex was postulated and determined by single crystal X-ray crystallographic technique. Our X-ray study revealed that the crystal system of 2 is orthorhombic, with space group Pbcn and unit cell parameters a = 14.6013(3) Å, b = 20.2960(5) Å, c = 15.8675(4) Å; α = β= γ = 90°. The molecular structure of 2 with atomic labeling is depicted in Fig. 3. The summary of the crystallography data is shown in Table 1 while the data of the bond lengths (Å) and angles (°) are listed in Table 4. The hydrogen bond geometry (Å,°) is given in Table 5 and the molecular packing diagram is depicted in Fig. S2.

(Color online.) The asymmetric unit of complex 2, showing 50% probability displacement ellipsoids and its atomic numbering.
Selected bond lengths (Å) and angles (°) of complex 2.
| Bond lengths | |||
| Sn(1)–C(13) | 2.1246 (14) | C(26)–C(27)a | 1.5009 (19) |
| Sn(1)–C(7) | 2.1253 (13) | C(27)–H(27C) | 0.9800 |
| Sn(1)–C(1) | 2.1380 (13) | C(27)–H(27D) | 0.9800 |
| Sn(1)–O(2) | 2.1250 (10) | C(27)–H(27A) | 0.9800 |
| O(3)–C(26) | 1.2220 (3) | ||
| C(26)–C(27)a | 1.5009 (19) | ||
| Bond angles | |||
| C(13)–Sn(1)–C(7) | 124.45 (5) | C(14)–C(13)–Sn(1) | 117.64 (10) |
| C(13)–Sn(1)–O(2) | 98.30 (5) | C(18)–C(13)–Sn(1) | 123.25 (10) |
| C(7)–Sn(1)–O(2) | 103.74 (4) | O(3)–C(26)–C(27)a | 121.68 (9) |
| C(13)–Sn(1)–C(1) | 112.14 (5) | O(3)–C(26)–C(27) | 121.67 (9) |
| C(7)–Sn(1)–C(1) | 118.04 (5) | C(27)a–C(26)–C(27) | 116.65 (19) |
| O(2)–Sn(1)–C(1) | 90.11 (4) | C(26)–C(27)–H(27C) | 109.5 |
| C(19)–O(2)–Sn(1) | 114.80 (9) | C(26)–C(27)–H(27D) | 109.5 |
| C(6)–C(1)–Sn(1) | 123.19 (10) | H(27C)–C(27)–H(27D) | 109.5 |
| C(2)–C(1)–Sn(1) | 118.52 (10) | C(26)–C(27)–H(27A) | 109.5 |
| C(12)–C(7)–Sn(1) | 120.62 (10) | H(27C)–C(27)–H(27A) | 109.5 |
| C(8)–C(7)–Sn(1) | 120.37 (10) | H(27D)–C(27)–H(27A) | 109.5 |
a Symmetry code: –x + 2, y, –z + ½.
Hydrogen bond geometry (Å,°) of complex 2.
| D-H···A | D-H | H···A | D···A | D-H···A |
| N1-H1N1···O3a | 0.916 (18) | 2.32(19) | 3.2177(17) | 165.3(17) |
| N1-H1N2···O2b | 0.87(2) | 2.24(2) | 3.0854(16) | 163.0(18) |
a Symmetry code: x – ½, y–½, –z + ½.
b Symmetry code: –x + 1, y, –z + ½.
Based on the crystal structure, depicted in Fig. 3, the tin atom was found to be tetra-coordinated as generally found for triphenyltin(IV) carboxylate complexes [30,52]. It is believed that the geometry occupied by the tin atom of the title complex is a distorted tetrahedral [30,52]. However, from the molecular structure diagram, the tin atom of complex 2 was bonded to an oxygen atom (Sn–O) of the carboxylate group while the other three bonds were attributed to the bonding between the carbons of the phenyl group to the tin atom (Sn–C), resulting in the tin atom being tetra-coordinated. The bond lengths and angles of complex 2 have normal values and agree with those found for related structures [30,52].
In the crystal packing (Fig. S2), molecules are linked via intermolecular N-H···O2 hydrogen bonds (Table 5) into chains propagating in [001].
The carboxylate anions were found to be chelated to the Sn(1) tin atom asymmetrically via one oxygen atom of the carboxylate group in a monodentate manner. As a result, the geometry of the Sn(1) tin atom was a highly distorted tetrahedral (Fig. 3). Moreover, based on the bond lengths (Å) and angles (°) listed in Table 5, it was found that the Sn(1)–O(2), Sn(1)–C(1), Sn(1)–C(7) and Sn(1)–C(13) bond length values were 2.1250(10), 2.1380(13), 2.1253(13) and 2.1246(15) Å, respectively, resulting in the structure being a distorted tetrahedral. Based on Table 5, the angles occupied by C(13)-Sn(1)-C(7), C(13)–Sn(1)-O(2) and O(2)–Sn(1)-C(1) were 124.45, 98.30 and 90.11° respectively, which was the main factor attributing to the distorted tetrahedral geometry of the tin atom moiety. In addition, occurrence of one acetone molecule sandwiched in between two triphenyltin(IV) carboxylate complexes was clearly observed on the crystal structure of 2. Referred to the similar structure documented by Yeap and Teoh in 2003 [45]; the structure obtained in 2003 was a simple monomeric type without any participation of solvent molecules. However, in the current study by altering the solvent system; an acetone molecule is participate in the crystal lattice and showed no coordination to the any tin(IV) atoms.
Based on the infrared spectral studies and the single crystal structure determination, the tin(IV) atom of 2 was tetra-coordinated and existed in a distorted tetrahedral geometry. The four bonds were attributed to the bonding of Sn–O and Sn–C, respectively. However, the NMR studies which were carried out in solution form, indicated that the tin(IV) atom of 2 was penta-coordinated and may due to the d6-DMSO/acetone being coordinated to the tin(IV) atom. However, in crystallographic study acetone molecule was found to be present in the crystal packing but uncoordinated as shown in Fig. 2. This might be due to different behavior of complex 2 in solvent system (NMR) and crystalline nature (single crystals).
3.5 In vitro anticancer studies
Cancer patients and mortality rate is consistently increasing worldwide because of rapid growth of world population and adoption of cancer causing behaviors specifically smoking in developing countries [53]. According to GLOBOCAN 2008 estimates, about 7.6 million cancer deaths occurred worldwide [53]. Researchers are consistently striving to explore the therapeutic ways to treat cancer. Among the different therapeutic options like chemotherapy, radiotherapy and surgery [54], Metallodrugs in chemotherapy has recently got attention. Discovery of cis-platin by Rosenberg [55] and its new marketing analogues (carboplatin, lobaplatin, oxaliplatin, nedaplatin) have further created interest in metal-based anticancer drugs [1,56]. Tin complexes have been studied in detail as antibacterial agents [9,15,17–19,28] and recently against cancerous cell lines [12,26,57] other than colon cancer.
In the current study, complexes 1–2 were tested against three cancerous cell lines (human colon cancer HCT 116, breast cancer MCF-7, leukemia K562) and a non-cancerous cell line (3T3-L1). The IC50 values have been compiled in Table 6. Both the complexes (1–2) showed potential efficacies in a dose dependent manner (Fig. 4A) against HCT 116 cells with the same IC50s 0.2 μM that is about 40 and 18 times better than the standard drugs (5-FU, IC50 8.1 μM and cis-platin, IC50 = 3.7 μM, respectively).
IC50 values of complexes 1–2 tested against cancerous and non-cancerous cell lines.
| Carcinoma cell lines | Normal | |||
| Cell lines | HCT 116 | MCF 7 | K562 | 3T3-L1 |
| Complexes | ||||
| 1 | 0.2a | 86.5b | 22.9b | 2.0a |
| 2 | 0.2a | 53.4b | 49.6b | 16.0a |
| Standards | 5-Flourouracil | Tamoxifen | Betulinic acid | Betulinic acid |
| 8.1a | 8.7a | 15.0a | 44.0a | |
| ← cis-platin → | cis-platin | |||
| 3.7a | 11.9a | 3.3a | 38.3a |
a μM.
b nM.
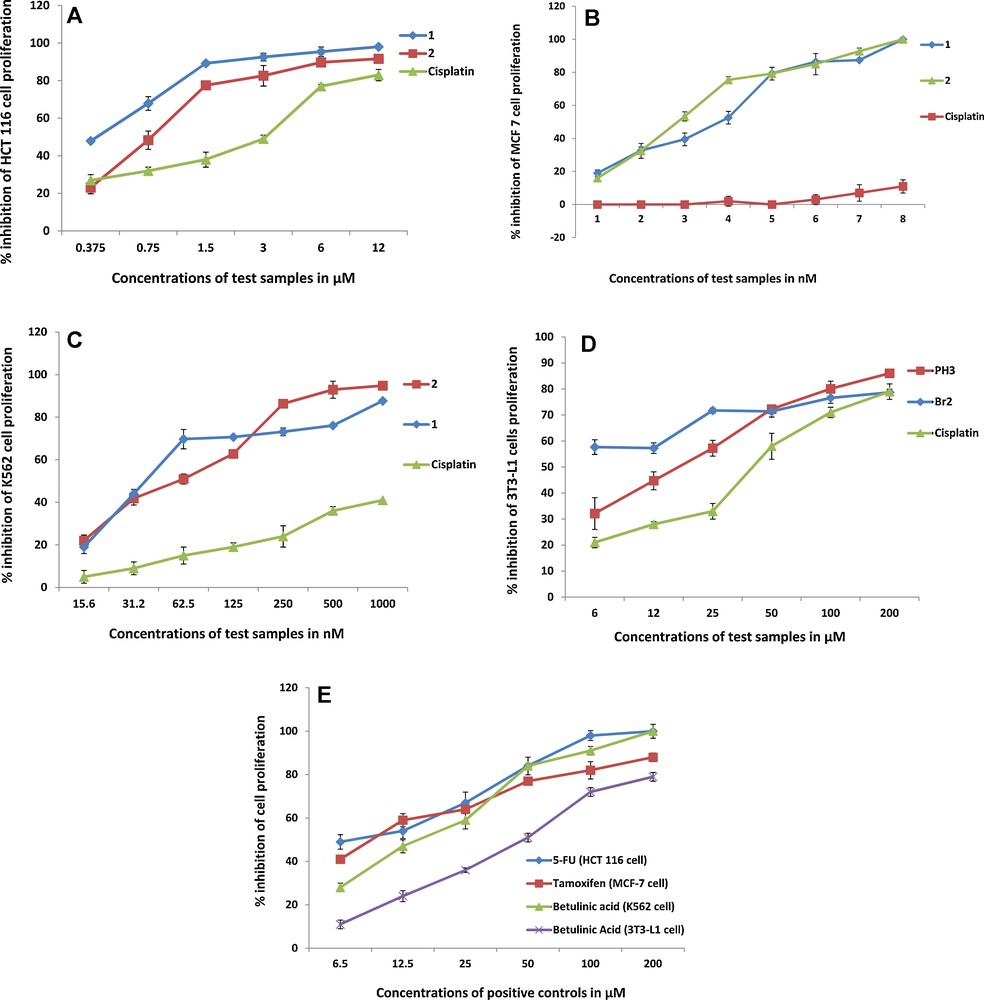
(Color online.) A–E: The figures A–E show that the antiproliferation efficacies of cancer cell lines, HCT 116 (4A); MCF-7 (4B); K562 (4 C)and a non-cancerous cell line 3T3 (4D), increased with the increase in sample concentrations in the similar manner as that for the standard drugs (4E).
Furthermore, both the complexes showed an exceptional antiproliferation in the breast cancer (MCF-7) cells. The IC50 values of complexes were calculated in nanomoles (86.5 nM for 1 and 53.4 nM for 2) which are many folds better than the respective standard drug (Tamoxifen, IC50 8.7 μM and cis-platin, IC50 11.9 μM). Similarly the complexes also showed IC50 values in nano moles (22.9 nM for 1 and 49.6 nM for 2) for leukemia cell line (K562) whereas respective standard drugs (butelinic acid and cis-platin) showed IC50 values in micro moles (15.0 μM and 3.3 μM, respectively). This indicates that the synthesized tin complexes have many folds better anticancer activity than compared to the standard drugs tested in parallel. Also, the complexes have been tested against a normal cell line (3T3-L1) and showed mild toxicity (IC50 2.0 μM for 1 and 16.0 μM for 2). Complex 2 remained safer against normal cell line, however showed cytotoxicity (against cancer cells) comparable to 1. Hence, overall complex 2 remained better than 1. It might be due to its slower rate of releasing tin metal compared to 1, where triphenyl could hold tin metal ion longer due to its electron withdrawing effect. It has been reported that the complexes which release metal ions at slower rate may induce better cytotoxicity in cancerous cells [1]. The standard drugs (Betulinic acid and cis-platin) have also been tested against the normal cells which induced a comparatively mild cytotoxicity (betulinic acid, IC50 44.0 μM and cis-platin, IC50 38.3 μM) compared to that of the cancerous cell lines. The dose dependent graphs have been shown as Fig. 4A-E and photomicrographic images of the cell lines have been presented as Fig. 5.

Photomicrographic images of the cell lines, taken under an inverted phase-contrast microscope at × 200 magnification using a digital camera at 48 hours after treatment with the 1 and 2. All the cells treated with the vehicle (0.1% DMSO) showed confluent layer of aggressively proliferating cells. Whereas, treatment with 1 showed significant (P < 0.01) inhibitory effect on proliferation of HCT 116, MCF-7 and K562 cancer cell lines with IC50 0.2 μM, 86.5 nM and 22.9 nM, respectively. The pictures revealed that the population of the treated cells is reduced drastically when compared to that of negative control. Treatment with 2 also displayed potent antiproliferative activity on HCT 116, MCF-7 and K562 cancer cell lines with IC50 0.2 μM, 53.4 nM and 49.8 nM, respectively. However, both the compounds demonstrated less cytotoxicity towards normal cell line (3T3-L1), as the selective index (SI: ratio of the IC50 obtained from the test on normal cell (3T3-L1) versus the IC50 for cancer cell) was more than 10. The SI values of the compound 1 for HCT 116, MCF-7 and K562 cell lines were 10, 23 and 87, respectively. The complex 2 displayed strong antiproliferative activity as the SI values for HCT 116, MCF-7 and K562 cell lines were 84, 300 and 321, respectively. Both complexes 1 and 2 showed more pronounced antiproliferative effects than the respective standard reference drugs. The results showed that 5-fluorouracil showed significant cytotoxicity with IC50 8.1 μM against HCT 116 cells. The standard reference tested against MCF-7 cells was tamoxifen, which demonstrated considerable cytotoxicity with IC50 8.7 μM. Whereas, betulinic acid showed significant cytotoxicity against K562 cells with IC50 15.0 μM. The use of cis-platin as a standard drug against all the cancerous and non-cancerous cell lines has also been included which showed inhibition values in μM (Table 6). Cis-platin induced drastic antiproliferative effect against HCT 116 and K562 cell lines compared to MCF-7 whereas remained relative safer for the normal cells (3T3-L1).
4 Conclusion
In the current study, the complexes 1–2 have been successfully synthesized and characterized by spectroscopic techniques. The molecular structures as well as the coordination number of tin(IV) ion in 1–2 have been successfully characterized by X-ray crystallography. Both the complexes were tested against three cancerous and one non-cancerous cell lines. On the basis of results obtained, it can be concluded that tin complexes may be potential ‘low dose’ chemotherapeutic drugs. The fine tuning of coordinating ligands which could facilitate the slower release of metal ion in the biological system may be more feasible to further improve their efficacy. Further work on caspases to understand the pro-apoptotic potential of the reported complexes is in progress and will be reported in due course.
Acknowledgement
The authors would like to thank the Ministry of Higher Education of Malaysia for the Fundamental Research Grant Scheme (Project No. FRGS/1/2012/SG01/UTAR/02/1), Universiti Tunku Abdul Rahman and the Institute of Postgraduate Studies, Universiti Sains Malaysia for financial support as well as for technical assistance and facilities. Dr. Muhammad Adnan Iqbal is the recipient of a USM Postdoctoral Fellowship in research from RUI Grant No. 1001/PKIMIA/811260. The biological activity was conducted using RUT Grant No. 1001/PFARMASI/851001. The article was compiled by MAI.
Appendix A Supplementary data
Crystallographic data for the reported structures in this paper has been deposited with Cambridge Crystallographic Data Center as CCDC 983679 and CCDC 983680. These data can be obtained free of charge at http://www.ccdc.cam.ac.uk/const/retrieving.html [or from the Cambridge Crystallographic Data Center (CCDC), 12, Union Road, Cambridge CB2 1EZ, UK; e-mail: deposit@ccdc.cam.ac.uk.; fax: +44 0 1223 336408].