



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Plants are the main sources of natural products and have contributed to the discovery of drugs for the treatment of various human diseases [1, 2]. However, the arsenal of specialized metabolites has been recently expanded to include those of microbial origin and, among others, those associated with plants and insects [3, 4, 5]. Indeed, the discovery of penicillin isolated from Penicillium sp. fungus led to the search for many other classes of antimicrobial compounds in the fungal and bacterial kingdoms. Actually, the effectiveness of antimicrobial drugs is decreasing while the resistance of microorganisms is significantly increasing [6]. The rapid spread and emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) bacteria and fungi are considered to be major threats to global public health in the 21st century [7, 8]. Over the last few decades, many of the existing drugs used to treat infectious diseases have become increasingly ineffective because of the global emergence of antimicrobial resistance (AMR). Therefore, there is a constant demand to find new, effective compounds that could help alleviate some of this pressure.
Symbiotic microorganisms have captured the attention of many researchers because they can produce novel bioactive compounds [3, 4, 5], many of which possess broad-spectrum antimicrobial activities [9]. However, while highly promising, research in this area is still in its infancy. Symbiotic microbes produce a plethora of bioactive compounds that promote the health of their host and may lead to the ecological success of holobionts (such as plant and/or insect holobionts) [10, 11]. Therefore, these environmental microorganisms have the ability to produce antimicrobial compounds because of the constant environmental pressure. The selection pressure is even stronger in areas with high biodiversity [12, 13, 14]. Thus, we focused our research efforts in the team Functional Chemistry—Chemical Ecology of Institut de Chimie des Substances Naturelles (ICSN of the Centre National de la Recherche Scientifique (CNRS)) on our library of symbiotic strains from French Guiana, including those associated with insects or plants, from which many antimicrobial compounds have been isolated recently [15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25]. Among several strain extracts tested in a broad range of antimicrobial assays, the SNB-CN88 crude extract showed a minimal inhibitory concentration (MIC) of 8 μg∕mL against methicillin-resistant Staphylococcus aureus (MRSA) without cytotoxicity on healthy human cells (MRC5 lines).
SNB-CN88, isolated from a termite nest, was identified as a strain of Serratia marcescens, a bacterial species widely distributed in the environment and an opportunistic Gram-negative nosocomial pathogen belonging to the Enterobacteriaceae family. This bacterium isolated from a termite nest had already been observed in French Guiana, associated with mosquito colonies [26].
In our study, approaches based on high-resolution mass spectrometry (HRMS) and tandem mass spectrometry (MS/MS) were used to explore the specialized metabolome of the mutualistic bacterial strain SNB-CN88. Molecular diversity was annotated in a first step by exploring the molecular network generated by MS/MS data, finely studying fragmentation patterns, and then proposing putative in silico structures. In a second step, the isolation of the bioactive compounds was performed to confirm their structures and determine their antibacterial activities.
2. Results and discussion
Molecular network (MN) is now considered a standard bioinformatic tool in the metabolomic community. Its paradigm is based on the observation that compounds with a high degree of structural similarity share comparable MS/MS fragmentation patterns. To provide a clear separation between MS/MS spectral clusters, only the most relevant similarity scores are selected using dedicated filtering steps. In this study, we used MetGem [27, 28] to explore the metabolome of Serratia marcescens. This software allows the parallel investigation of two complementary representations of a raw dataset, one based on a classic GNPS-style MN and another based on the t-SNE algorithm [27, 28, 29]. The t-SNE graph preserves the relationships between related groups of spectra, while the MN output allows an unambiguous separation of clusters. This multivariate embedding method focuses on local distances using nonlinear outputs to represent the entire data space. The relevance of the t-SNE output and its ability to accurately map the spectral distribution of the metabolome of Serratia marcescens will be illustrated in the following experiments.
In this study, the crude extract of Serratia marcescens SNB-CN88 strain was fractionated into 18 fractions (F2 to F19) and each fraction was tested against MRSA. Fractions F14 and F15 exhibited the highest quantities (F14 (257 mg), F15 (467 mg)) as well as the highest activities (MIC < 32 μg∕mL, Supplementary Table S1). The activities observed for the fractions were lower than those of the crude extract, suggesting a synergistic or additive effect of the compounds in the mixture. Therefore, we sought to identify most of the metabolites in the SNB-CN88 extract. All fractions were then analyzed by liquid chromatography coupled to tandem high-resolution mass spectrometry (LC-HRMS/MS) in positive ion mode with data-dependent acquisition (DDA) mode. The generated MS/MS data were processed to create a molecular network (MN) using MetGem software and the t-SNE algorithm, which allows visualization of cluster proximity [27, 28]. Molecules with intense MS signals are present in clusters A, B, and C in t-SNE MN. These clusters group fractions identified as the most active against MRSA and mainly belong to fractions F14 and F15 (Figure 1).
t-SNE molecular network generated from metabolic extract fractions of Serratia marcescens SNB-CN88 colored according to their MIC against MRSA. Dereplicated clusters are circled in black. Dereplicated specialized metabolite clusters with MICs <64 μg∕mL are circled in blue for cluster A, red for cluster B and green for cluster C. Node sizes are relative to the maximum intensity of ions in the whole dataset.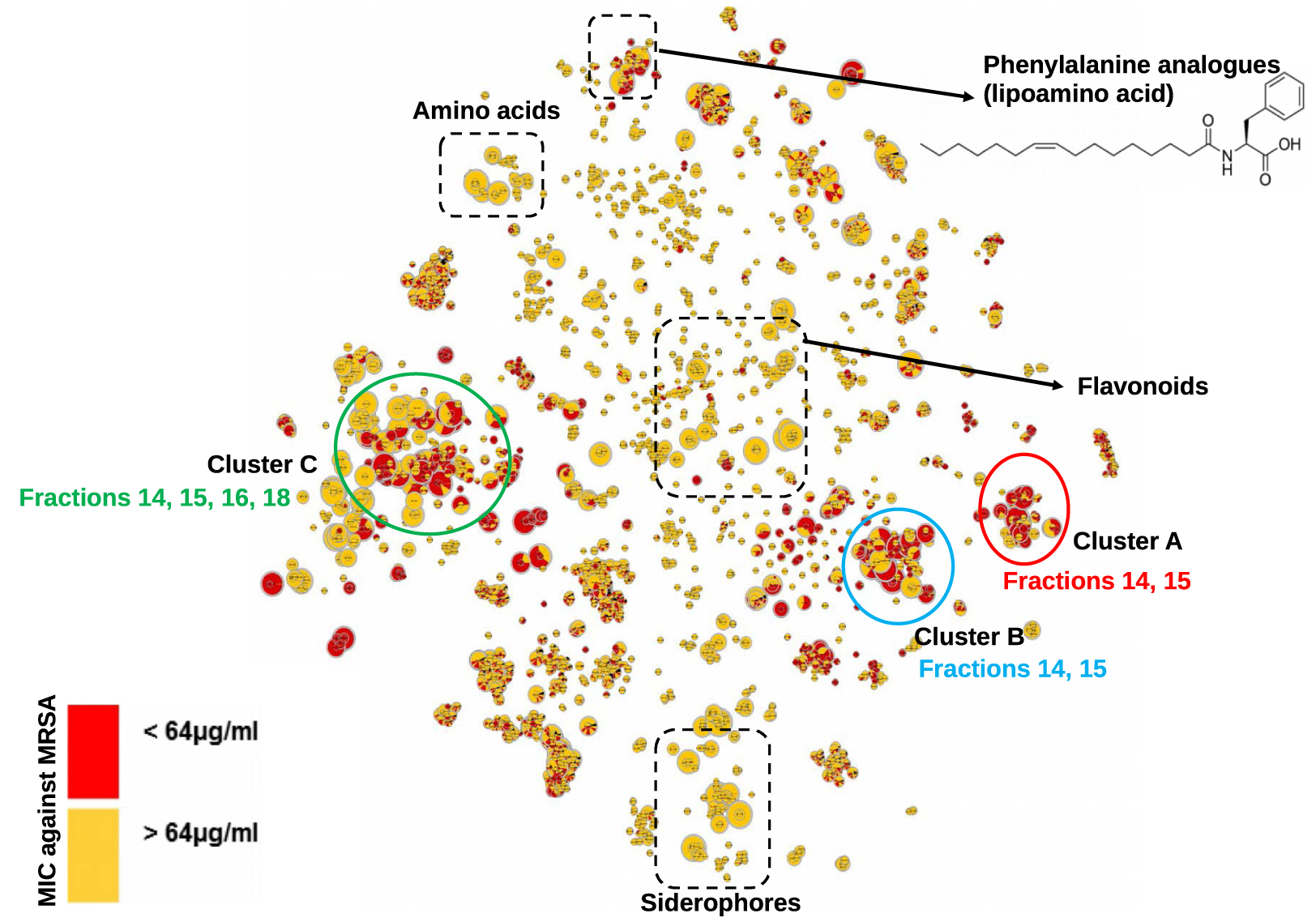
In order to annotate actives molecules, a reduced t-SNE molecular network from only the F14 and F15 fractions was generated from 1221 nodes with m∕z values ranging from 211 to 1361. As expected, clusters A, B, and C were also identified in this reduced MN (Figure 2).
t-SNE molecular network generated from active fractions of F14 and F15 of Serratia marcescens SNB-CN88. Dereplicated specialized metabolite clusters are circled in blue for cluster A, red for cluster B and green for cluster C. Node sizes are relative to the maximum intensity of ions in the whole dataset.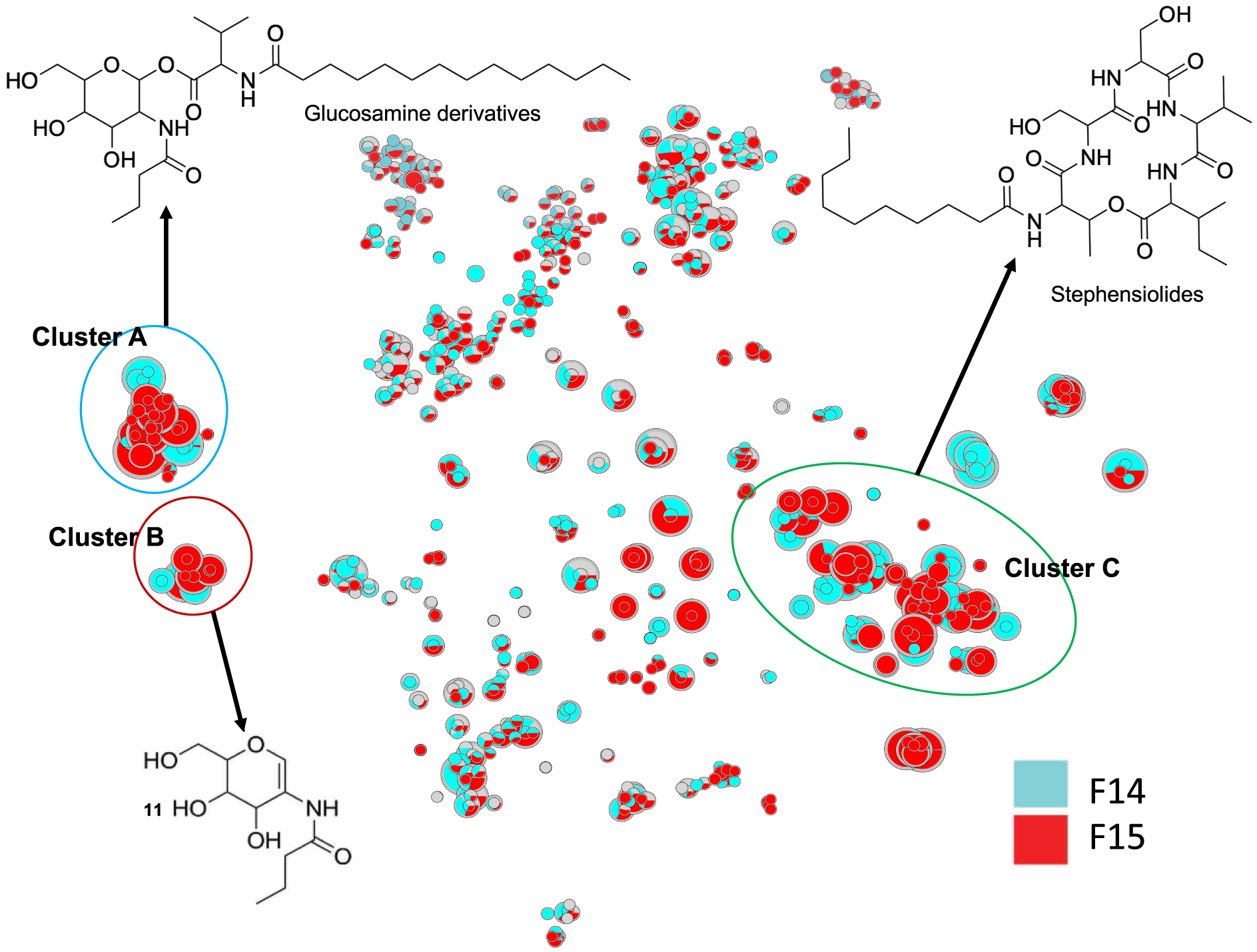
A first annotation was performed by interrogating MS/MS spectral databases. To refine the annotation of the features and to respect the standards of reproducible science, the annotation levels proposed by the Metabolomics Standards Initiative (MSI) were used [29]. Level 0 corresponds to an unambiguous 3D structure obtained from isolated pure compounds, level 1 corresponds to the compound identified in comparison with a standard, level 2 corresponds to a putative annotation (MS/MS library comparison or tentative structure), level 3 includes a chemical class assignment (comparison with literature), and level 4 corresponds to unknown molecules (Supplementary Table S2).
The molecules in cluster A were searched using PubChem. The proposed structures described in the literature led to the annotation of already reported natural compounds, i.e., glucosamine derivatives (Figure 3) [30, 31]. These compounds are small molecules (m∕z 545 to 627 for [M+H]+ ions) composed of a sugar (glucose, hexose), an amino acid (valine), a fatty acid (C13 to C17), and a butyric or oxo-hexanoic acid. The highly intense fragment ion corresponding to the sugar moiety was observed at m∕z 232.1177, leading to several fragments at m∕z 214.1071 (loss of one water molecule), 196.0981 (loss of two water molecules), 166.0859, 144.0652, and 126.0546 (Supplementary Figures S1–S7). Compound 1 shows a molecular peak at m∕z 559.3950 [M+H]+ (calculated 559.3953, err. 0.5 ppm) corresponding to the molecular formula of C29H55N2O8. This compound has a fatty acid moiety labelled at m∕z 328.2819 (saturated C14 fatty acid chain with a valine residue calculated 328.2840, err. 6 ppm) and a glucose residue bound to butyric acid labelled at m∕z 232.1181 (calculated 232.1176, err. −2 ppm); therefore, its structure is similar to the glucosamine derivative C (1) (Supplementary Figure S1) [30, 31].
(A) Annotated Cluster C obtained for t-SNE molecular network generated. (B) Enlargement of nodes for annotation. The color of the nodes is relative to the representation of the fractions F14 (blue) and F15 (red) and their sizes relative to the maximum intensity of these ions in the whole dataset.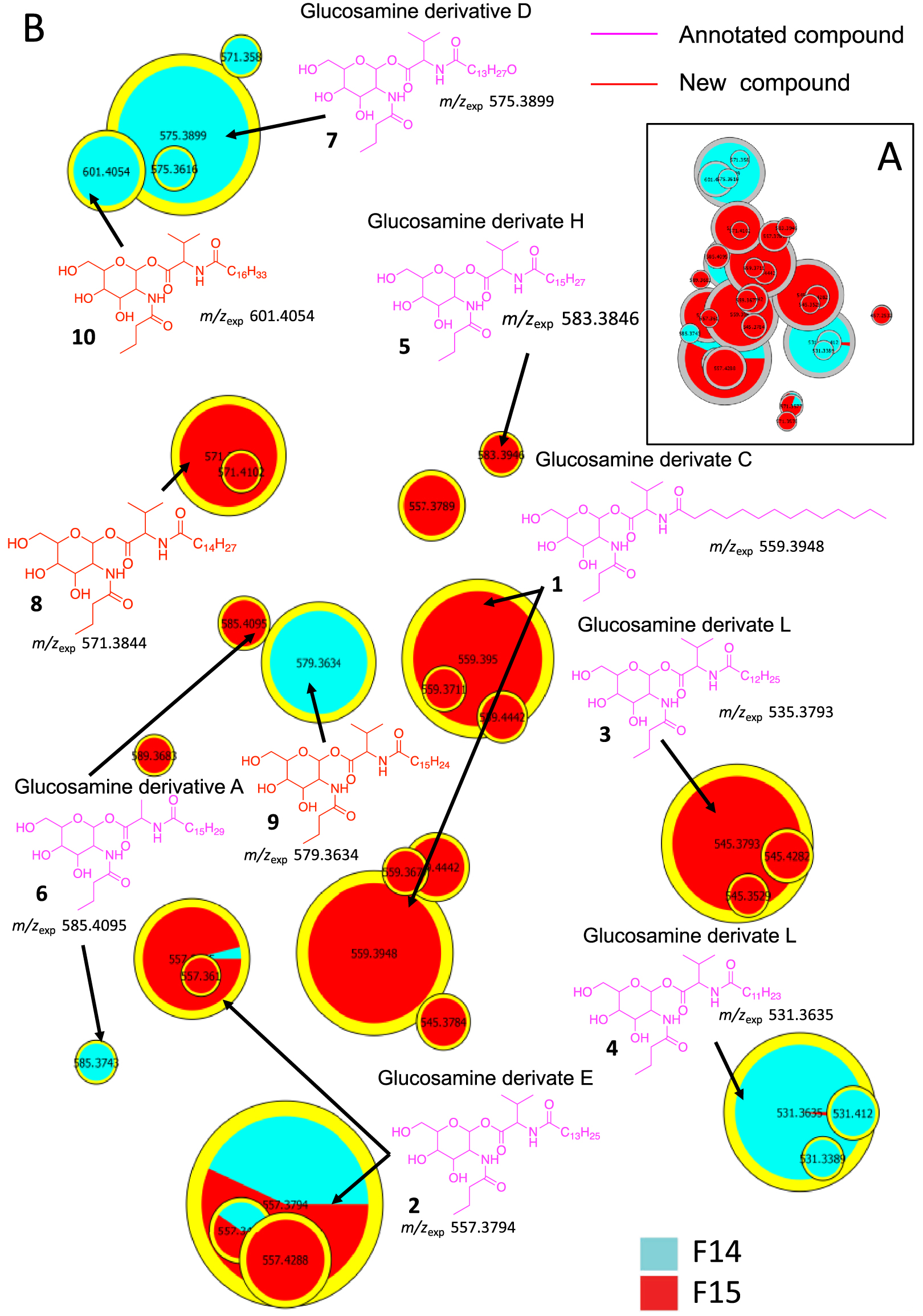
Similar to compound 1, each compound in cluster A exhibits a major fragment at m∕z 232.1 and their fragments (Supplementary Figures S2–S7). Compounds 2–7 have been annotated by comparison with the literature as glucosamide derivatives analogs (Glu) A, C, D, E, F, H and L. MS/MS spectrum of compounds 8, 9 and 10 showed a fragment at m∕z 348.2525, 340.2839 and 370.2944, respectively, corresponding to a valine residue bound to a unsaturated C16:4 for 8 and a C15:1 fatty acid for 9 (Supplementary Figure S7). MS/MS data from compound 10 shows a major fragment corresponding to a valine linked to a saturated C17 fatty acid acyl chain and not to C16:OH as described previously for Glucosamine derivatives L [30]. These three new annotated glucosamine derivatives 8–10 were named glucosamine derivatives O, P and L [30, 31, 32, 33].
Cluster B contains compounds related to N-acetyl-D-glucosamine and was annotated via the GNPS databases (Figure 4). Indeed, the only dereplicated compound of cluster B is the potential precursor of the compounds of cluster A. This molecule exhibits a m∕z value at 232.1177 and corresponds to the calculated chemical formula of C10H18NO5 (err. 1.1 ppm, Supplementary Figure S8). Compound 11 was dereplicated by MS data as N-(2-ethyl-3,4-dihydroxy-3,4-dihydro-2H-pyran-5-yl)butyramide.
(left) Dereplicated N-acetyl-D-glucosamine (m∕z 232.1177) in Cluster B, (right) comparison between database and experimental mass spectra.
The different nodes observed in cluster B suggest that different sugars may be present in our extract. Thanks to the use of the t-SNE algorithm, we were able to annotate the glucosamine precursor 11, observed as a solitary node when using classical GNPS-like representation [30, 31].
The size of the nodes is relative to the maximum intensity of these ions in the data set, thus allowing to estimate the amount of each molecule in the extract or fractions of S. marcescens. The main compounds found in fraction F14 are molecules 1, 2, 3 and 11, and in fraction 15 compounds 2, 5, 6 and 11. Only compound 2 (GluE) is found in high proportion in these two fractions. All these metabolites may be partly responsible for the observed antibacterial activity. Analogs of glucosamine derivatives have been reported in the literature to have antibacterial activity [30, 31, 32, 33, 34, 35]. Bioassays on compound 1 (GluC derivative), previously isolated from a strain of Serratia sp., showed weak activity against Mycobacterium diernhoferi which is probably due to the complexity of its cell wall [32]. Several other glucosamine analogs have been annotated in the metabolic profiles of Serratia marcescens strains and some of them (GluC and GluA) showed MIC values of 156 μg∕mL against S. aureus [30, 31]. Therefore, the observed biological activity of our strain extract is due to GluC, D, E, F and L or either to the synergistic or additive effect of the glucosamide mixture or to other compounds. This is why we were also concerned with Cluster C.
In cluster C, 6 stephensiolides were annotated by comparing the MS data with the chemical formulas in the literature [30, 31, 34, 35]. The stephensiolides (Stp) were described with five amino acid residues (Thr-Ser-Ser-Val/Ile-Val/Ile) linked to a 𝛽-hydroxy fatty acyl residue (C8–C14) [30, 31, 36]. Therefore, MS/MS spectra were used for preliminary screening of stephensiolide analogs (Supplementary Figures S9–S15). Compounds 12–18 were dereplicated as stephensiolides C, D, E, F, G, I and R, respectively (Figure 5). Two analogs 19 and 20 were proposed by comparison with MS/MS spectra database. The difference in m∕z between compound 15 and compound 19 is 2.0152 (calculated 2.0157, err. 2.48 ppm), which corresponds to a double bond located on the C9 alkyl chain from the MS/MS fragmentation spectra (Supplementary Figure S16). Then, the difference in mass between compounds 20 and 15 is 42.047 Da, which may correspond to 3 additional methylene groups (calculated 42.047, err. 0 ppm), i.e. an unsaturated C12 alkyl chain (Supplementary Figure S17). Then, these two annotated compounds were named by comparison with the literature stephensiolides U2 (19) and dehydrostephensiolide F (20). Finally, the use of t-SNE algorithm for molecular network visualization allows us to observe that on the right of cluster C (separated by a dotted line in Figure 5) amino acid residues 5 are always valines, while on the left of the same cluster Ile/Leu residues are always found (Supplementary Figures S9–S17). These data suggest that the sequence of the different amino acids of stephensiolides may vary as reported by Clements-Decker et al. (Figure 5) [30, 31]. As a result, we were able to identify molecules 21 and 22 as stephensiolides K and Q [30, 31].
(A) Annotated Cluster C obtained for the generated t-SNE molecular network. (B) Enlargement of nodes for annotation, part surrounded by dotted lines. The color of the nodes is relative to the representation of the fractions F14 (blue) and F15 (red) and their sizes relative to the maximum intensity of these ions in the whole dataset.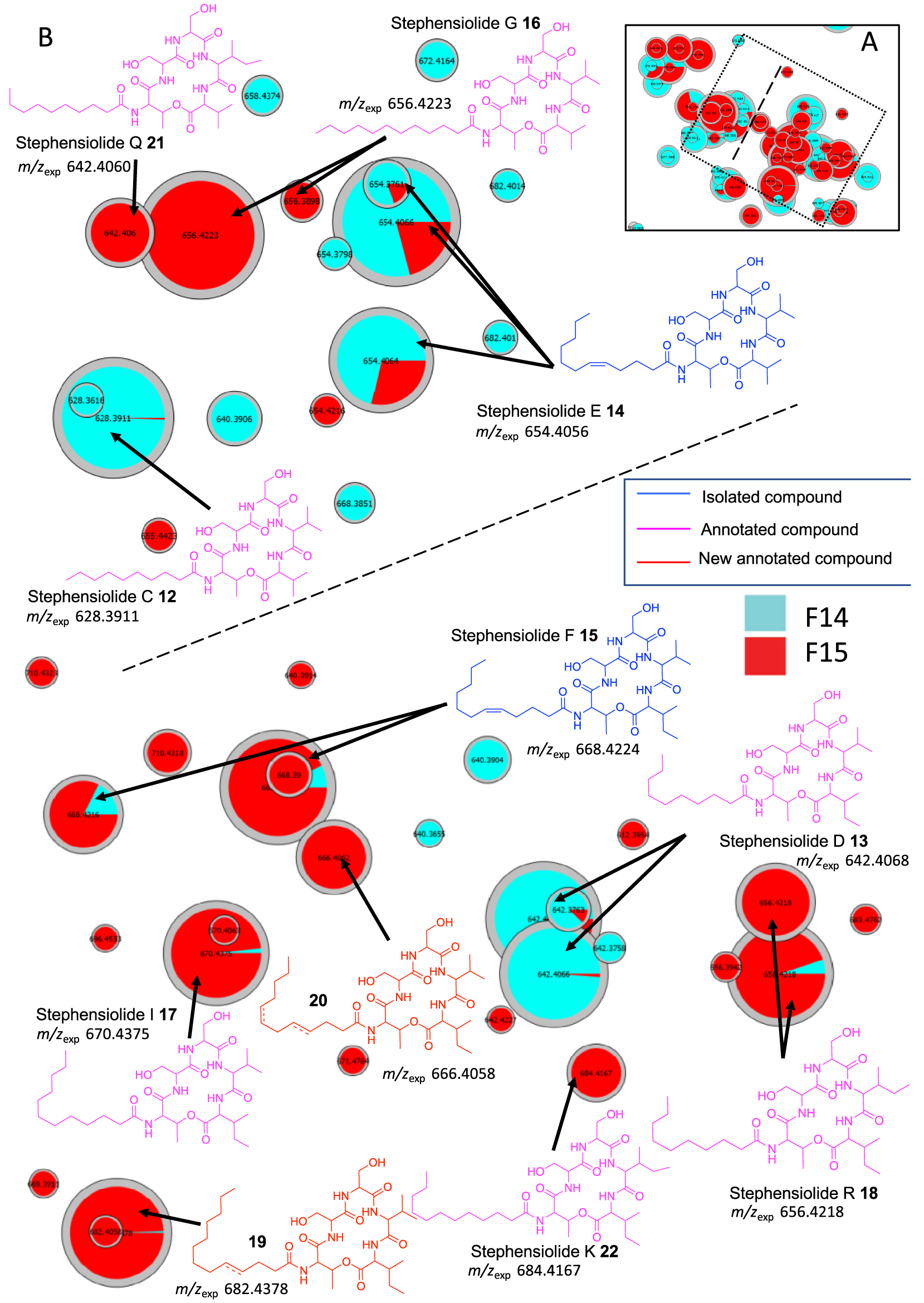
In order to confirm annotation of the major compounds, fraction F14 and F15 were re-fractionated to isolate pure molecules. Seven subfractions from fraction F14 and 5 subfractions form F15 were obtained by flash chromatography (Supplementary Table S1). All these fractions were biologically tested showing that fractions F14.F and F15.E are the most active with MIC at 32 and 8 μg∕mL, respectively (Supplementary Table S1).
The purification of fraction F14.F allowed us to isolate the major compound 14. The HR-ESI-MS analyses show a molecular peak at m∕z 654.4108 [M+H]+ corresponding to the calculated molecular formula of C32H56N5O9 (calculated 654.4078, err. 4.58 ppm). Analysis of the 1D and 2D NMR spectra and LC-MS/MS indicated the presence of five amino acids: threonine and two valine and serine residues (Supplementary Figures S18–S24). A large aliphatic peak in the 1H NMR spectrum of compound 14 suggested the presence of a long alkyl chain typical of lipopeptides. HMBC correlations indicated that the nitrogen atom of the threonine was acylated with this alkyl chain, whereas the shift of Thr H-2 suggested that the hydroxyl group of threonine was esterified (Figure 6). Further HMBC correlations show that threonine is linked to valine via an ester bond and to serine through an amide bond. The positions of the remaining serine and valine residues were determined by COSY correlations between NH and H-1 and HMBC correlations between NH/H-1/H-2 and their respective carbonyls (Figure 6). Purification of the F15.E fraction allowed us to isolate the major compound 15. HR-ESI-MS analyses show a molecular peak at m∕z 668.4224 [M+H]+ (calculated 668.4229, err. 0.8 ppm) corresponding to the calculated molecular formula of C33H57N5O9. MS and NMR analysis of this compound determined the presence of the following 5 amino acids: threonine, valine, isoleucine, and two serine residues, as well as the presence of an alkyl chain (Supplementary Figures S25–S30). The amino acid residue chain was determined in the same way as that for compound 14 (Figure 6). The masses of these two compounds allow us to determine that we have C12 fatty acid chains. The olefinic signals in the 1H NMR spectra for 14 and 15 and two carbons in 13C NMR observed at 130.1/128.9 and 131.9/129.6 ppm, respectively, suggest the presence of a double bond within the lipid side chain. The location of these double bonds for 14 and 15 were determined by HMBC correlations to be between the C-5 and C-6 carbons in the acyl chain. The 13C shifts of the vinyl carbons along with constants between the sp2 proton (10.9 Hz, Supplementary Figure S26) suggest the Z isomer [37, 38, 39]. All the experimental 1D and 2D NMR data and comparison with the literature allowed us to determine that stephensiolides E (14) and F (15) were isolated [30, 31, 32, 33, 34, 35, 36].
Major NMR correlations observed for compounds 14 and 15, COSY correlations are shown in bold and HMBC correlations by arrows.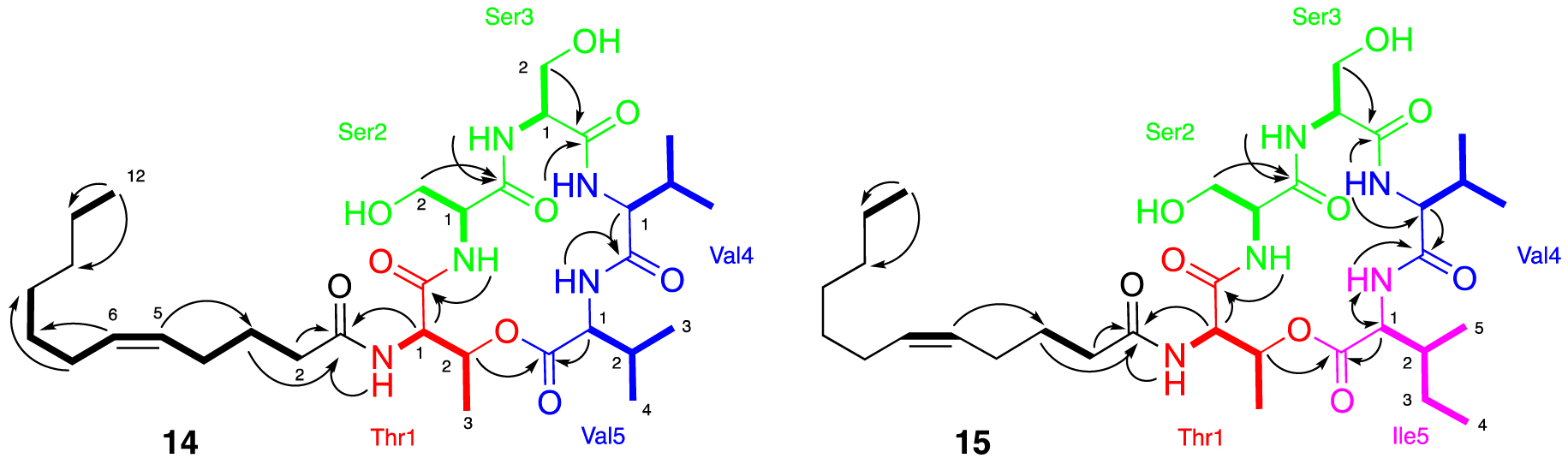
The compounds predominantly found in fractions F14 (12–14) and F15 (14–20) seem to be responsible for the observed activities. The major compounds 14 and 15 isolated from fractions F14.F and F15.E showed antimicrobial activity against MRSA (MIC = 4 and 8 μg∕mL, respectively). Only StpE (14) is found in a large proportion in these two fractions. This result can explain the observed antimicrobial activity of the Serratia marcescens strain. Compounds 14 and 15 were previously isolated from Serratia sp. and Lecanicillium sp. strains and showed activity against Bacillus subtilis and MRSA (MICs < 32 μg∕mL) [34, 35]. The metabolites 12–17 named stpC, D, F, G, and I were also isolated from the Lecanicillium sp. strain and showed activity against MRSA with MIC values of 128, 32, 32, 16 and 4 μg∕mL, respectively, whereas weak or no activity was observed in a previous study for other stephensiolides (StpQ, StpR, StpK) against S. aureus, with MIC higher than 300 μg∕mL [31].
3. Conclusion
The molecular network approach using the t-SNE algorithm allowed us to easily annotate new target compounds responsible for the observed biological activity on MRSA. Our study highlighted that Serratia marcescens SNB-CN88, isolated from a termite nest, could produce diverse stephensiolides and glucosamine analogs with antimicrobial activity against methicillin-resistant Staphylococcus aureus (MRSA). Moreover, 22 compounds were annotated; 2 stephensiolides and 3 glucosamine derivatives were identified for the first time in this study, and two major compounds, including new metabolites from active fractions of SNB-CN88 crude extract, were isolated and structurally characterized. Compounds 14 and 15 were identified as known stephensiolides E and F (for which 13C and 1H NMR are described for the first time, see Supplementary Table S3). All identified compounds contributed to the observed biological activity of the crude extract and its fractions. First, stephensiolide E and F showed antimicrobial activity against MRSA with MIC⩽8 μg∕mL. Other compounds that were not isolated in this study could also have antimicrobial activities. In particular, the glucosamine derivative E (2), which is present in large amounts in the fractions, has never been isolated, characterized, or tested biologically. This compound may possess antibacterial activity similar to that of its analog (glucosamine derivative C).
Preliminary data suggest that this strain SNB-CN88 of Serratia marcescens biosynthesizes different compounds from the strain of the same species published by Clements-Decker et al. [30, 31] and suggested another operational chemical unit (OCU) of S. marcescens, as proposed by Barthélemy et al. [40]. Clardy’s team determined the biosynthetic origin of stephensiolides by sequencing the genome of Anopheles-associated Serratia sp. and demonstrated that biosynthesis differed according to the media used in vitro, which appear to differ from the natural environment [36]. Consequently, the strain origin may explain the molecular composition differences observed with those previously published in the literature.
Stephensiolides may play a protective role against entomopathogenic bacteria and viruses [30, 31, 36]. This study suggests that termite mounds (workers, soldiers, tunnel and nest, here called meta-holobiont) can be putatively protected from biotic aggressions of the environment by association with a Serratia sp. strain. In return, this association would allow Serratia sp. strains in association with the termite meta-holobiont to be easily propagated in the environment because of the termites’ way of life (e.g. swarming, feeding, prospecting …).
The Gram-negative bacterium Serratia marcescens is also known to be an opportunistic pathogen responsible for a wide range of human diseases, including ocular, uropathogenic, and respiratory infections [41, 42]. Studying the termite microbiome could help to understand the transmission of this human infectious disease and improve our understanding of the complex interspecies relationships that contribute to the transmission of vector-borne infectious agents.
4. Materials and methods
4.1. General experimental procedure
NMR spectra were recorded on Bruker 300, 500, 600, and 700 MHz spectrometers (Bruker, Rheinstetten, Germany). The chemical shifts (δ) are reported as ppm based on the solvent signal, and the coupling constants (J) are in hertz. Preparative HPLC was conducted using a Gilson system equipped with a 322 pumping device, a GX-271 fraction collector, a 171 diode array detector, and a prep ELSII. All solvents were HPLC grade, purchased from Sigma-Aldrich (Saint-Quentin-Fallavier, France).
4.2. Isolation and identification of termite mutualistic microorganisms
The mutualistic bacterial strain was isolated in 2009 from an environment (termite aerial nest of Nasutitermes similis) sampled in Piste de Saint-Elie in French Guiana (ABSCH-CNA-FR-240495-2). The strain SNB-CN88 from the strain library collection at ICSN was identified as Serratia marcescens. Taxonomic marker analyses were externally performed by BACTUP, France. Bacterial isolates were identified on the basis of 16S rDNA sequence analysis. The sequences were aligned with DNA sequences from GenBank, NCBI (http://www.ncbi.nlm.nih.gov, accessed on 7 June 2021), using BLASTN 2.2.28. The sequences were deposited in GenBank under the registry number KJ023777.
4.3. Culture and extraction of microorganisms
4.3.1. General cultivation and extraction procedure
SNB-CN88 was cultivated on 330 Petri dishes (14 cm diameter) at 28 °C for 15 days on potato dextrose agar (PDA) medium (Dominique Dutscher SAS, Brumath, France). The culture medium containing the mycelium was cut into small pieces and macerated three times at room temperature with ethyl acetate (EtOAc) on a rotary shaker (70 rpm) for 24 h. The contents were extracted with 10 L of EtOAc using a separatory funnel. Insoluble residues were removed via filtration, and the organic phase was washed three times with an equivalent volume of water (H2O), dried with anhydrous solid Na2SO4, and then evaporated using a rotary evaporator under reduced pressure and a temperature of 30 °C to yield a crude extract (2.7 g).
4.3.2. Fractionation of SNB-CN88
The crude extract (2.7 g) was fractionated using reverse-phase flash chromatography (Grace Reveleris, Grace, Maryland, USA) with a C18 column and UV and DEDL detectors. A 10-min step gradient of H2O/formic acid (99.9/0.1)-acetonitrile/formic acid (99.9/0.1) (v/v 95/5, 75/25, 50/50, 20/80, 0/100) with a flow rate of 80 mL/min was performed, followed by a second gradient 50% acetonitrile/formic acid (99.9/0.1)-methylene chloride for 15 min and then 100% methylene chloride in 15 min with a flow rate of 80 mL/min was performed to generate 18 fractions, named F2 (38.8 mg), F3 (19.4 mg), F4 (13.9 mg), F5 (26.6 mg), F6 (83.0 mg), F7 (5.6 mg), F8 (96.9 mg), F9 (62.2 mg), F10 (46.7 mg), F11 (34.6 mg), F12 (76.5 mg), F13 (90.7 mg), F14 (257 mg), F15 (467 mg), F16 (165 mg), F17 (76.5 mg), F18 (26.2 mg) and F19 (586 mg).
The F14 fraction was fractionated by flash reverse phase chromatography using a C18 column and UV and ELSD detectors. A 6.5-min step gradient of solvent A (H2O/formic acid (99.9/0.1)) and solvent B (acetonitrile) (v/v 75/15, 0/100) followed by a second gradient 50% acetonitrile (B) and 50% dichloromethane (A) for 6.7 min and finished with a third gradient 100% isopropyl alcohol for 10 min with a flow rate of 30 mL/min were performed to generate 7 fractions, named F14.A (2.1 mg), F14.B (0.9 mg), F14.C (1.8 mg), F14.D (3.4 mg), F14.E (17.0 mg), F14.F (42.8 mg), F14.G (0.7 mg).
The F15 fraction was fractionated by flash reverse phase chromatography (CombiFlash NextGen300, Teledyne Technology, USA) using a C18 column and UV and ELSD detectors. A 5.9-min step gradient of solvent A (H2O/formic acid (99.9/0.1)) and solvent B (acetonitrile) (95:5) followed by a second gradient 57.6% acetonitrile (B) and 42.4% dichloromethane (A) for 8.3 min and a third gradient 54.4% acetonitrile (B) and 45.6% dichloromethane (A) for 2. 7 min with a flow rate of 30 mL/min were performed to generate 5 fractions, named F15.A (2.0 mg), F15.B (5.9 mg), F15.C (2.0 mg), F15.D (4.5 mg), F15.E (212.0 mg).
Fractions F14.F and F15.E (20 mg) were re-purified by preparative HPLC (Agilent InfinityLab Proshel 120 × 4 SB-C18, 21.2 × 1500 mm) with the following parameters: flow rate: 21 mL/min, eluents: H2O+0.1% formic acid (A) and ACN (B), gradient (using isocratic elution): 40% B to 55% B (in 4 min), 55% B (in 9 min), 55% B to 100% B (in 9 min), 100% B (in 10 min) to give compound 14 (tR 12.8 min, 2.3 mg) and compound 15 (tR 14.8 min, 1.2 mg), respectively.
4.3.3. Molecules isolation and characterization
Stephensiolide E (14) white powder; 1H NMR (700 MHz, DMSO-d6) δH Threonine1: 7.94 (1H, d (J = 9.3 Hz), NH), 4.46 (1H, m, H-1), 5.29 (1H, m, H-2), 1.15 (3H, m, H-3), δH Serine2: 7.52 (1H, d (J = 7.6 Hz), NH), 4.25 (1H, m, H-1), 3.47 (1H, m, H-2a), 3.51 (1H, m, H-2b), δH Serine3: 7.84 (1H, d (J = 7.6 Hz), NH), 4.13 (1H, m, H-1), 3.52 (1H, m, H-2a), 3.69 (1H, m, H-2b), Valine4: 7.72 (1H, d (J = 7.3 Hz), NH), 4.13 (H, m, H-1), 2.02 (2H, m, H-2), 0.85 (3H, m, H-4), 0.84 (3H, m, H-3), δH Valine5: 8.39 (1H, d (J = 6.4 Hz), NH), 4.19 (1H, m, H-2), 2.02 (1H, m, H-3), 0.86 (3H, m, H-5), 0.83 (3H, m, H-4), δH Acyl: 5.36 (1H, m, H-5), 5.35 (1H, m, H-6), 2.27 (2H, m, H-2), 2.02 (2H, m, H-4), 1.99 (2H, m, H-7), 1.29 (2H, m, H-8), 1.59 (2H, m, H-3), 1.25 (4H, m, H-9 and H-11), 1.24 (2H, m, H-2, H-10), 0.85 (3H, m, H-12); 13C NMR (700 MHz, DMSO-d6) δC Threonine1: 168.7 (C, C=O), 55.8 (CH, C-1), 69.9 (CH, C-2), 17.5 (CH3, C-3); δC Serine2: 170.5 (C, C=O), 61.3 (CH2, H-2), 54.6 (CH, C-1), δC Serine3: 170.6 (C, C=O), 59.7 (CH2, H-2), 55.2 (CH, C-1), δC Valine4: 170.7 (C, C=O), 57.8 (CH, C-1), 28.9 (CH, C-2), 19.1 (CH3, C-3), 18.4 (CH3, C-4), δH Valine5: 170.1 (C, C1), 57.5 (CH, C-1), 30.0 (CH, C-2), 18.7 (CH2, C-3), 18.3 (CH3, C-4), δH Acyl: 172.9 (C=O), 130.1 (CH, C-6), 128.9 (CH, C-5), 34.9 (CH2, C-2), 31.1 (CH2, C-10), 29.1 (CH2, C-8), 28.3 (CH2, C-9), 26.6 (CH2, C-7), 26.3 (CH2, C-4), 25.4 (CH2, C-3), 22.0 (CH2, C-11), 13.9 (CH3, C-12). ESI–HRMS m∕z [M+H]+ 654.4108 (calcd for C31H56N5O9H+, 642.4078 err. 4.5 ppm).
Stephensiolide F (15) white powder; 1H NMR (700 MHz, MeOD) δH Threonine1: 7.07 (1H, d (J = 8.9 Hz), NH), 4.72 (1H, m, H-1), 5.56 (1H, q (J = 6.5 Hz), H-2), 1.29 (3H, m, H-3), δH Serine2: 7.28 (1H, d, (J = 7.2 Hz), NH), 4.36 (1H, m, H-1), 3.78 (2H, m, H-2), δH Serine3: 6.96 (1H, d (J = 7.2 Hz), NH), 4.36 (1H, m, H-1), 3.96 (1H, m, H-2a), 3.82 (1H, m, H-2b), δH Valine4: 7.12 (1H, d (J = 8.9 Hz), NH), 4.24 (1H, t (J = 8.9 Hz), H-1), 2.13 (1H, m, H-2), 0.96 (6H, m, H-3 and H-4), δH Isoleucine5: 6.96 (1H, d (J = 6.8 Hz), NH), 4.24 (1H, t (J = 6.8 Hz), H-1), 1.82 (1H, m, H-2), 1.51 (1H, m, H-3a), 1.21 (1H, m, H-3b), 0.96 (3H, m, H-4), 0.91 (3H, m, H-5), δH Acyl: 5.42 (1H, m, H-5), 5.41 (1H, m, H-6), 2.36 (2H, m, H-2), 1.71 (1H, m, H-3), 1.29-1.38 (12H, m, H-4, H7, H8, H9, H10, H11), 0.96 (3H, m, H-12); 13C NMR (700 MHz, MeOD) δC Threonine1: 171.9 (C, C=O), 71.5 (CH, C-2), 57.5 (CH, C-1), 19.7 (CH3, C-3); δC Serine2: 173.3 (C, C=O), 62.9 (CH2, H-2), 56.2 (CH, C-1), δC Serine3: 173.3 (C, C=O), 56.1 (CH, C-1), 61.0 (CH2, H-2), δC Valine4: 170.7 (C, C=O), 57.8 (CH, C-1), 28.9 (CH, C-2), 19.1 (CH3, C-3), 18.4 (CH3, C-4), δH Isoleucine5: 172.5 (C, C=O), 59.8 (CH, C-1), 37.6 (CH, C-2), 27.0 (CH2, C-3), 15.6 (CH3, C-4), 14.0 (CH3, C-5); δH Acyl: 176.9 (C=O), 131.9 (CH, C-6), 129.6 (CH, C-5), 23.6-36.6 (8CH2, C-2, C-3, C-4, 7-5, C-8, C-9, C-10, C-11), 11.0 (CH3, C-12). ESI–HRMS m∕z [M+H]+ 668.4241 (calcd for C33H56N5O9H+, 668.4229 err. 0.8 ppm).
4.4. Metabolomic analysis
4.4.1. LC-MS/MS analyses
Crude extract and fractions of SNB-CN88 were prepared at 1 mg/mL in methanol and filtered on a 0.45 μm PTFE membrane. LC-MS/MS analyses were performed with an HPLC 1260 Prime chromatography chain (Agilent Technologies, Waldbrom, Germany) coupled with an Agilent 6546 Q-ToF mass spectrometer (Agilent Technologies, Walbronn, Germany). LC separation was performed using an Accucore RP-MS column (100 × 2.1 mm, 2.6 μm, Thermo Scientific, Les Ulis, France) with a mobile phase of H2O/formic acid (99.9:0.1) and acetonitrile/formic acid (99.9:0.1). The column oven was set at 45 °C. The compounds were eluted with a flow rate of 0.4 mL/min with a gradient from 5% B to 100% B in 20 min and then 100% B for 3 min. The injection volume was 5 μL for all analyses. For the electrospray ionization source, the mass spectra were acquired in positive ionization mode with the following parameters: temperature of the drying gas 325 °C, flow rate of the focalizer gas 10 L/min, capillary voltage 3500 V, nozzle voltage 500 V, fragmentor voltage 130 V, skimmer voltage 45 V and RF voltage of the octopole 1750 V. An internal calibration was performed using two standards: purine and hexakis(1H,1H,3H-tetrafluoropropyxy)phosphazene (m∕z 121.0509 and m∕z 922.0098), allowing high-resolution masses with less than 5 ppm error. MS/MS data-dependent acquisition was performed on the 5 most intense ions detected by a full MS1 scan in the m∕z 200–1000 range, above a threshold of 1000 counts. The selected precursor ions were fragmented with a collision energy set at 30 eV with an isolation window of 1.3 amu. The mass range of the precursor and fragment ions was set between 50 and 1000 m∕z.
4.4.2. Data processing analysis
The raw data files were converted from .d format (Agilent Technologies) to .mzML format using MSConvert software, which is part of the Proteo Wizard 3.0 package [43]. All .mzML files were reprocessed using MZmine v. 2.53 software. Mass detection was performed using an MS/MS background noise level of 1000 for MS1 and 50 for MS/MS. The ADAP chromatogram builder was used with a minimum group size of 3, minimum group intensity of 1000, and m∕z tolerance of 0.005 (or 15 ppm). Chromatogram devolution was performed with the ADAP wavelet algorithm using the following parameters: a threshold signal-to-noise ratio of 30, a minimum component peak height of 50 000, a threshold coefficient/area ratio of 10, a peak duration of 0.001–1 min, and a wavelet retention time range of 0.01–0.08 min. Isotope clustering was performed using the isotope peak clustering algorithm with an m∕z tolerance of 0.005 (or 15 ppm) and a retention time tolerance of 0.1 min. Peaks were filtered using the filter function of the component list to remove all components associated with no MS/MS spectrum. Peak alignment was performed using the attached alignment function, with an m∕z tolerance of 0.005 (or 15 ppm), a weight for m∕z of 80, a retention time tolerance of 0.1 min, and a weight for retention time of 20. The “export/import” function was used to generate .mgf and .csv files. MetGem version 1.3.4 was used to generate the molecular networks. To add a “network” visualization, the spectra were filtered by keeping the 10 most intense peaks in a sliding window in the spectrum of ± 50 Da. Peaks present in the range of ± 17 Da around the m∕z of the precursor ion were removed. The m∕z tolerance to match peaks was set at 0.04 Da, and cosine scores were only considered when a minimum of two peaks were matched between 2 spectra. The number of iterations, perplexity, learning speed, and early exaggeration were set to 5000, 25, 10, and 12, respectively, for the t-SNE representation. Figures were generated using the MetGem export function and ChemDraw Professional 16.0 (PerkinElmer). The NMR spectra were processed and analyzed using TopSpin 3.6.2 (Bruker, Rheinstetten, Germany).
4.5. Biological essay
The crude extracts and pure isolated compounds were tested on human pathogenic methicillin-resistant Staphylococcus aureus (ATCC 33591). The test was performed in conformance with the reference protocols of the European Committee on Antimicrobial Susceptibility Testing [44]. The MIC was obtained after 24 h. Vancomycin was used as a positive control.
For cytotoxic assays, the crude extracts and isolated compounds were tested in triplicate at concentrations of 10 μg∕mL and 1 μg∕mL in the MRC5 cell line (ATCC CCL-171, Human Lung Fibroblast Cells), following the procedure described by Tempête et al. [45].
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Acknowledgments
The authors are grateful to N. Pollet, Université Paris-Saclay, CNRS, IRD, Évolution Génomes Comportement & Écologie, 91198 Gif-Sur-Yvette, France for their assistance in strain identification.