



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Compounds containing heterocycles are currently among the key components of drugs. It is known that about 80% of all main active components of drugs containing cyclic systems are heterocycles with nitrogen, oxygen and sulfur [1, 2]. The pyridine- and triazine-based compounds are of particular interest since they possess a high structural diversity and are considered as agents for the treatment of diseases. The presence of various substituents on the heterocyclic core allows one to efficiently vary their biological, including antibacterial and antiviral, activities [3, 4, 5, 6]. Notably, the triazine-containing derivatives are also used in the fabrication of antimicrobial films on the surface of various materials, including implants. Such a coating is possible both due to the introduction of compounds into the polymerization mixture, as well as the ability to form their own polymeric structures and covalent interactions with the functional groups of the surface [7, 8, 9, 10]. An additional interest in the pyridine- and triazine-based derivatives as medications is currently due to potential chemical linkage with proteins. This allows their use for covalent inhibition of bacterial and viral proteins. In particular, the pyridine derivatives are known to be pronounced cysteine protease inhibitors [11, 12].
The latest research on the antiviral activity of the pyridine- and triazine-based derivatives is due to the severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) [13]. The newest agents for coronavirus disease 2019 (COVID-19) therapy are also constructed from the pyridine and/or triazine rings [14]. Furthermore, triazine is a precursor of another well-known drug, remdesivir [15, 16]. Some of its derivatives are also known as efficient antiviral agents [17]. At the same time, analysts predict an intensive growth of the triazine market due to its high demand for medicine [18].
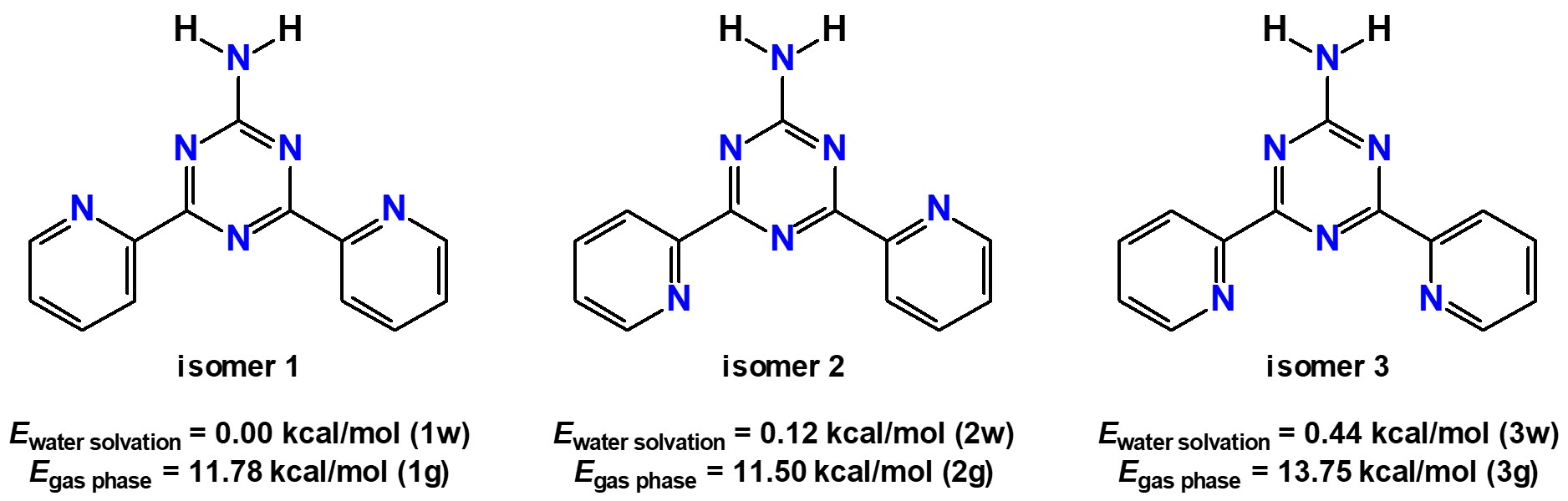
We have also been interested in the chemistry of nitrogen-containing six-membered rings [19, 20, 21, 22, 23, 24, 25, 26] and in computational studies of potential biological activity of different compounds [27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47]. In the present work, I have focused on 2-amino-4,6-bis(2-pyridyl)-1,3,5-triazine (NH2Py2T), which is constructed from the triazine and two 2-pyridine rings (Figure 1). Furthermore, this molecule also contains a triazine-linked primary amine group, which is of potential interest for further possible functionalizations of NH2Py2T through, e.g., reactions of condensation and addition.
Diagrams of the three discussed NH2Py2T isomers together with their corresponding energies under water solvation and in gas phase.
The Cambridge Structural Database (CSD) [48] did not contain crystal structures for NH2Py2T. Notably, although NH2Py2T is of great interest for the coordination chemistry due to the 2,2′-bipyridine- (bpy) or 2,2′;6′,2′ ′-terpyridine (terpy)-like coordination pockets, the CSD contains only 19 hits for crystal structures of complexes with a series of lanthanide(III) cations [49], one crystal structure with Cd2+ [50] and one with Ru2+ [51]. Interestingly, the CSD also contains a crystal structure of the dihydrochloride salt of NH2Py2T with two protonated pyridine rings [52]. Thus, it becomes important to study structural features of NH2Py2T. As such, I have performed density functional theory (DFT)-based calculations to establish and study the most favourable NH2Py2T isomers, including its electronic and reactivity properties. Furthermore, by means of a molecular docking approach, I have examined the binding of NH2Py2T to a series of SARS-CoV-2 proteins. Stability of the resulting protein–NH2Py2T complexes was assessed with molecular dynamics studies.
2. Methods
2.1. DFT calculations
The gas phase and aqueous structures of the NH2Py2T isomers were optimized with GaussView 6.0 molecular visualization [53] and Gaussian 09, Revision D.01 [54] using the DFT/B3LYP/6-311++G(d,p) method [55, 56, 57]. The calculation in water were performed using the integral equation formalism polarizable continuum model (IEFPCM).
2.2. Molecular docking
AutoDock Vina was used for molecular docking simulations (Lamarckian Genetic Algorithm (LGA) scoring function with GA runs = 100, population size = 500, maximum number of evaluations = 25000000) [58, 59]. The RCSB Protein Data Bank (RCSB PDB) was used as a source to download the structures of proteins [60], from which water molecules were removed, and hydrogen atoms and missing residues and charges were inserted. The grid box (30 × 30 × 30 Å with 0.375 Å grid spacing) was defined by AutoDock Tools (v. 1.5.7). The docking procedure was applied to flexible ligands and rigid proteins. The conformers of the ligands with the best binding scores were retrieved from the 10 top-ranked poses. The results of molecular docking were visualized in BIOVIA Discovery Studio 2020 [61].
2.3. In silico drug-likeness analysis
The absorption, distribution, metabolism, excretion and toxicity (ADMET) properties of NH2Py2T were predicted using the SwissADME [62], which also includes the brain or intestinal estimated permeation method (BOILED-Egg) [63], and ProTox-II [64, 65] web tools.
2.4. Molecular dynamics simulation
The WebGRO online service was used for molecular dynamics simulations [66]. Parameters such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), and intermolecular hydrogen bonds were assessed. GROMOS96 54a7 forcefield was applied for complexes, equilibrated using the isothermal–isobaric (NPT) and canonical (NVT) ensembles. Topology of the ligand was generated with the PRODRG tool [67]. Simple point-charge (SPC) was used as solvent model (triclinic water box with size 50 × 75 × 70 Å) [68]. The steepest-descent algorithm (5000 steps) was applied to minimize the energy of the system. 0.15 M NaCl was used during the simulation at 310 K and 1.0 bar with 1000 frames per simulation. Three independent simulation runs with a duration of 50 ns were performed for each protein–ligand complexes and identical results were obtained.
3. Results and discussion
The structure of NH2Py2T both in gas phase and in water was examined using DFT (Table S1 in the Supplementary Material). Conformational analysis (Figure S1 in the Supplementary Material) showed that three different isomers, depending on the orientation of the pyridine rings, are the most favorable in both gas phase and water (Figure 1). In particular, under water solvation, isomer 1, namely 1w, was found to be the most energetically favorable, while isomers 2 and 3, namely 2w and 3w, respectively, were only 0.12 and 0.44 kcal/mol less favorable (Figure 1). In gas phase, isomer 2, namely 2g, was the most favorable, while isomers 1 and 3, namely 1g and 3g, respectively, were 0.28 and 2.25 kcal/mol less favorable (Figure 1). Furthermore, under water solvation conditions the corresponding isomers were 11.78, 11.38 and 13.31 kcal/mol more favorable in comparison to gas phase (Figure 1). Thus, the formation of all three isomers is almost equally possible, especially in water. The same conformational analysis results (Figure S1 in the Supplementary Material) showed that the energy barriers for conversion of 1w to 2w, 1w to 3w and 2w to 3w were 8.87, 9.10 and 17.92 kcal/mol, respectively. In gas phase, the energy barriers for conversion of 1g to 2g, 1g to 3g and 2g to 3g were 9.27, 18.76 and 10.65 kcal/mol, respectively. The computed energy barriers are quite high for both gas phase and under water solvation. Thus, each isomer in both phases does not exist in equilibrium with the other two isomers.
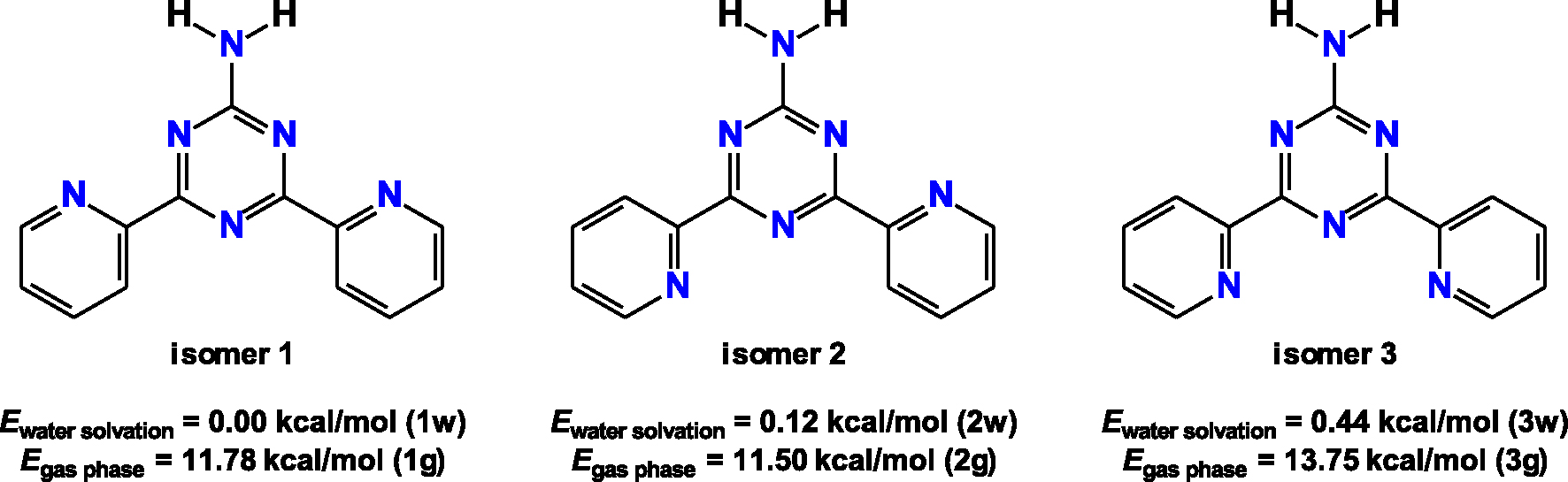
The calculated C–C and C–N bond lengths within the aromatic rings are 1.328–1.401 Å (Table S2 in the Supplementary Material). The C1–N6 bond is 1.345 Å in the structures of the isomers obtained under water solvation. This value is 0.006–0.012 Å longer for the isomers in gas phase (Table S2 in the Supplementary Material). The C–C bonds between the triazine and pyridine carbon atoms are about 1.5 Å in all the discussed isomers (Table S2 in the Supplementary Material). The bond angles formed by the non-hydrogen atoms are about 115–125° (Table S2 in the Supplementary Material). The structures of 3w and 1g, followed by 3g and 1w, are the most deviated from planarity (Figure 2), as evidenced from the corresponding dihedral angles (Table S2 in the Supplementary Material).
The DFT-optimized structures of the NH2Py2T isomers.
Each isomer forms a pair of weak C–H⋯N interactions yielding two five-membered pseudo-aromatic rings (Figure 2, Table 1). These rings are characterized by a conjugation effect with partial covalency [69, 70]. According to the Harmonic Oscillator Model for Heterocycles with π-electrons and/or n-electron delocalization (HOMHED) [70], the calculated aromaticity for these non-covalent rings varies from 0.665 to 0.678, which is similar to those of (is)oxazoles and furans [70]. The same index for the triazine and pyridine rings was found to be 0.994–0.999.
Lengths (Å) and angles (°) of the C–H⋯N interactions, and aromaticity indexes of the triazine, pyridine and five-membered non-covalent rings in the optimized structures of the NH2Py2T isomers
| C–H⋯N | d(C–H) | d(H⋯N) | d(C⋯N) | ∠(CHA) | Aromaticity index | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Triazine | PyN4 | PyN5 | R1 | R2 | ||||||
| 1w | C5–H3⋯N3 | 1.080 | 2.447 | 2.800 | 97.44 | 0.994 | 0.996 | 0.996 | 0.667 | 0.667 |
| C10–H7⋯N3 | 1.081 | 2.447 | 2.800 | 97.73 | ||||||
| 2w | C5–H3⋯N1 | 1.081 | 2.448 | 2.799 | 97.3 | 0.995 | 0.996 | 0.996 | 0.667 | 0.667 |
| C10–H7⋯N3 | 1.080 | 2.442 | 2798 | 97.55 | ||||||
| 3w | C5–H3⋯N1 | 1.081 | 2.465 | 2.805 | 96.71 | 0.997 | 0.996 | 0.996 | 0.666 | 0.665 |
| C10–H7⋯N2 | 1.081 | 2.465 | 2.805 | 96.71 | ||||||
| 1g | C5–H3⋯N3 | 1.081 | 2.461 | 2.811 | 97.31 | 0.995 | 0.996 | 0.996 | 0.672 | 0.672 |
| C10–H7⋯N3 | 1.081 | 2.461 | 2.811 | 97.31 | ||||||
| 2g | C5–H3⋯N1 | 1.081 | 2.442 | 2.800 | 97.70 | 0.997 | 0.996 | 0.995 | 0.672 | 0.678 |
| C10–H7⋯N3 | 1.081 | 2.408 | 2.779 | 98.35 | ||||||
| 3g | C5–H3⋯N1 | 1.081 | 2.444 | 2.798 | 97.43 | 0.999 | 0.995 | 0.995 | 0.672 | 0.672 |
| C10–H7⋯N2 | 1.081 | 2.445 | 2.798 | 97.41 | ||||||
According to atomic charges in the discussed NH2Py2T isomers, all the H atoms carry a positive charge with the highest values for NH2 hydrogens (Figure S2 in the Supplementary Material). The C(NH2) carbon atom, and the C6 and C11 carbons carry the largest and lowest charges, respectively, within the non-hydrogen atoms (Figure S2 in the Supplementary Material). The other positively charged atoms are C5 and C10 carbons, followed by the pyridine and triazine N3 nitrogen atoms in 1g–3g (Figure S2 in the Supplementary Material). The latter nitrogen atoms in 1w–3w are either negatively charged or close to zero (Figure S2 in the Supplementary Material). The C4 and C9 pyridine carbon atoms, followed by the amine nitrogen atom, are also among the most negatively charged atoms in the discussed isomers (Figure S2 in the Supplementary Material).
According to vibrational analysis, no negative frequencies were obtained (Figure 3, Table S3 in the Supplementary Material), testifying to correct optimized structures of all the NH2Py2T isomers. Notably, the calculated spectra are pairwise very similar for the corresponding isomers under water solvation and in gas phase (Figure 3, Table S3 in the Supplementary Material). However, some discrepancies clearly appear between spectra for 1w–3w and 1g–3g. In particular, bands for the NH groups were found at 3604 and 3733 cm−1 in the water solvation spectra, while the same bands are shifted by about 8–11 and 11–18 cm−1 to higher frequencies in the gas phase spectra (Table S3 in the Supplementary Material). Furthermore, a similar shift was observed for the band of the H1–N6–H2 bending and C1–N6 stretching modes from ∼1640 cm−1 to ∼1628 cm−1 (Table S3 in the Supplementary Material).
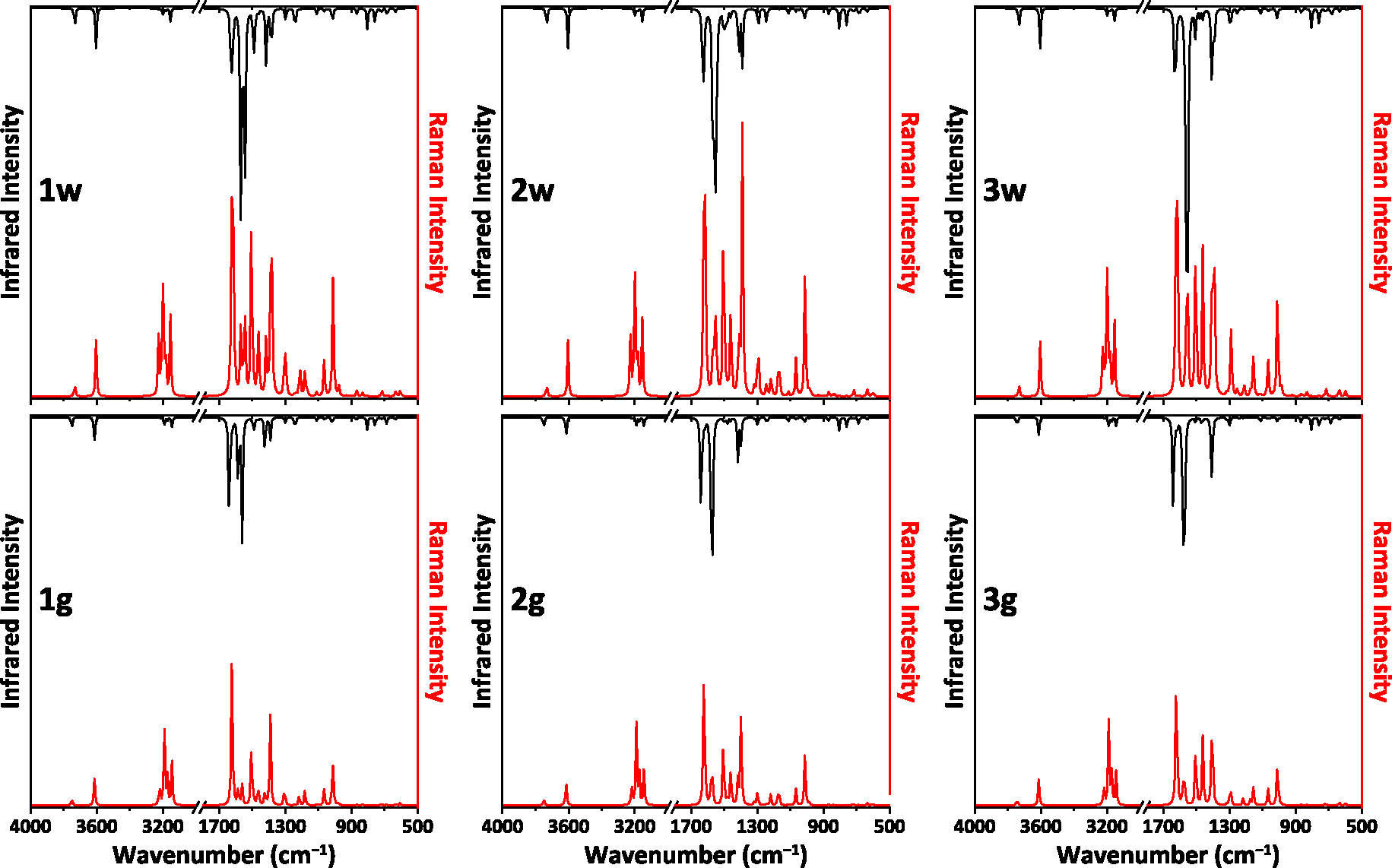
The calculated IR (black) and Raman (red) spectra of the NH2Py2T isomers. Intensities of the spectra are scaled to the same factor.
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) for all NH2Py2T isomers range from −7.242 to −6.764 and from −2.483 to −2.193 eV, respectively (Table 2). Notably, the HOMO decreases and the LUMO increases from 1w through 2w to 3w, while the reverse is observed for the HOMO of 1g–3g and the LUMO is almost the same for the latter isomers (Table 2). All this leads to a gradual increase of the HOMO–LUMO energy gap for 1w–3w and to a gradual decrease for 1g–3g, with the highest values obtained for 3w and 1g (Table 2).
HOMO and LUMO, gap value, descriptors and charge transfer parameters for the NH2Py2T isomers
| Parameter | 1w | 2w | 3w | 1g | 2g | 3g |
|---|---|---|---|---|---|---|
| EHOMO (eV) | −7.204 | −7.230 | −7.242 | −6.972 | −6.878 | −6.764 |
| ELUMO (eV) | −2.483 | −2.474 | −2.448 | −2.193 | −2.206 | −2.201 |
| ΔELUMO−HOMO = ELUMO − EHOMO (eV) | 4.721 | 4.756 | 4.795 | 4.779 | 4.672 | 4.563 |
| Ionization energy, I = −EHOMO (eV) | 7.204 | 7.230 | 7.242 | 6.972 | 6.878 | 6.764 |
| Electron affinity, A = −ELUMO (eV) | 2.483 | 2.474 | 2.448 | 2.193 | 2.206 | 2.201 |
| Electronegativity, 𝜒 = (I + A)∕2 (eV) | 4.843 | 4.852 | 4.845 | 4.582 | 4.542 | 4.482 |
| Chemical potential, 𝜇 = −𝜒(eV) | −4.843 | −4.852 | −4.845 | −4.582 | −4.542 | −4.482 |
| Global chemical hardness, 𝜂 = (I − A)∕2 (eV) | 2.361 | 2.378 | 2.397 | 2.389 | 2.336 | 2.282 |
| Global chemical softness, S = 1∕(2𝜂) (eV−1) | 0.212 | 0.210 | 0.209 | 0.209 | 0.214 | 0.219 |
| Global electrophilicity index, 𝜔 =𝜇 2∕(2𝜂) (eV) | 4.969 | 4.950 | 4.896 | 4.394 | 4.416 | 4.403 |
| Maximum additional electric charge, ΔNmax = −𝜇∕𝜂 | 2.052 | 2.040 | 2.021 | 1.918 | 1.944 | 1.965 |
| Molecule-to-metal electron charge transfer, ΔN1 = (𝛷 − 𝜒)∕𝜂: | ||||||
| Ti | −0.22 | −0.22 | −0.21 | −0.11 | −0.09 | −0.07 |
| Fe | −0.15 | −0.15 | −0.14 | −0.03 | −0.02 | 0.01 |
| Zr | −0.34 | −0.34 | −0.33 | −0.22 | −0.21 | −0.19 |
| Co | 0.07 | 0.06 | 0.06 | 0.17 | 0.20 | 0.23 |
| Cu | −0.08 | −0.08 | −0.08 | 0.03 | 0.05 | 0.07 |
| Cr | −0.15 | −0.15 | −0.14 | −0.03 | −0.02 | 0.01 |
| Ni | 0.13 | 0.13 | 0.13 | 0.24 | 0.26 | 0.29 |
| Mn | −0.31 | −0.32 | −0.31 | −0.20 | −0.19 | −0.17 |
| Mo | −0.10 | −0.11 | −0.10 | 0.01 | 0.02 | 0.05 |
| Zn | −0.22 | −0.22 | −0.21 | −0.11 | −0.09 | −0.07 |
| Al | −0.24 | −0.24 | −0.24 | −0.13 | −0.11 | −0.09 |
| W | −0.12 | −0.13 | −0.12 | −0.01 | 0.00 | 0.03 |
| Ag | −0.25 | −0.25 | −0.24 | −0.13 | −0.12 | −0.10 |
| Au | 0.11 | 0.10 | 0.11 | 0.22 | 0.24 | 0.27 |
| Total negative charge, TNC (e) | −3.36553 | −3.62912 | −3.60288 | −3.62619 | −3.10979 | −3.37761 |
The overall distribution of HOMO and LUMO is very similar for all isomers (Figure 4). In particular, the LUMO is localized over the whole molecule except for the CNH2, N3, C8–H6 and C13–H10 fragments (Figure 4). In isomers 1 and 3, the HOMO is also localized over almost the whole molecule except for the N4, N5, C6–H4 and C11–H8 fragments in 1w and 1g, and C6–H4 and C11–H8 fragments in 3w and 3g, respectively (Figure 4). In isomers 2, the HOMO is located over the whole triazine–NH2 fragment, one of the pyridine rings, and the C11–H8 group and C4–N4 fragments of the second pyridine ring (Figure 4). The density-of-states (DOS) plots of the discussed NH2Py2T isomers are shown in Figure S3 in the Supplementary Material.
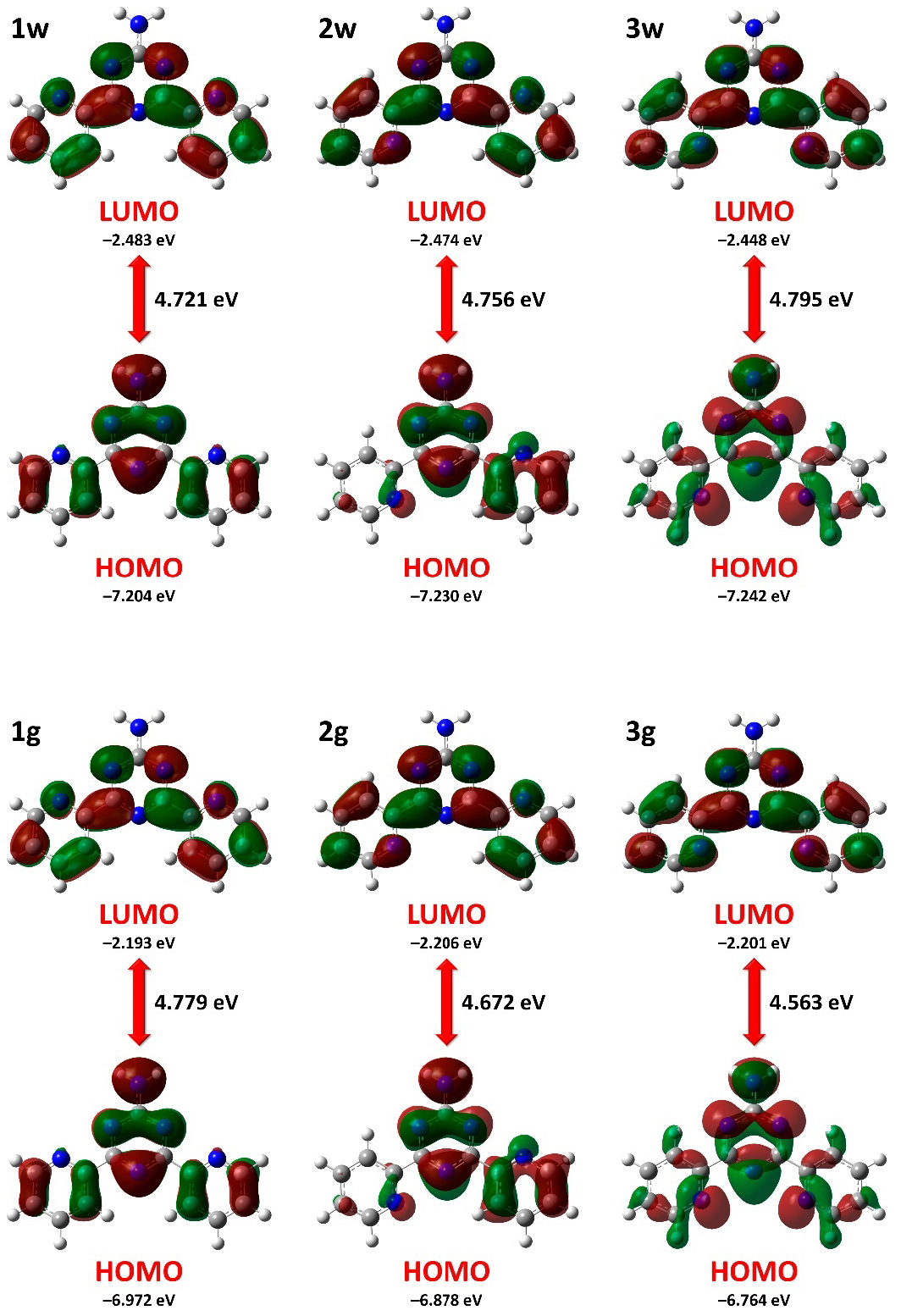
The HOMO and LUMO of the NH2Py2T isomers. Results under 0.02 a.u. isovalue.
Corrosion inhibition properties of a compound can be inferred from the HOMO and LUMO values [71, 72, 73, 74, 75]. Corrosion inhibition is of importance considering metallic implants [76, 77, 78]. In this regard, we have investigated the ability of the NH2Py2T isomers to inhibit corrosion in a set of metals, which are actively used in biomedical implants (Table 2) [78], using the work function (Φ) [79]. Of all the metals studied, it was established that the isomers are active toward Ni, Au and Co (Table 2).
Molecular electrostatic potential (MEP) was calculated for all NH2Py2T isomers to establish nucleophilic and electrophilic regions of the molecule. The MEP surface revealed the triazine and pyridine nitrogen atoms, which form either a bpy- or terpy-like pocket, as the most pronounced nucleophilic centers, and the amine hydrogen atoms as the most electrophilic region (Figure S4 in the Supplementary Material).
The absorption spectra calculated for the NH2Py2T isomers each contain bands exclusively in the UV region up to ∼350 nm (Figure 5). In particular, the spectra exhibit an intense narrow band at ∼185 nm and a broad band of about half intensity centered at ∼250–260 nm (Figure 5). The main transitions responsible for the observed bands are given in Table S4 in the Supplementary Material.
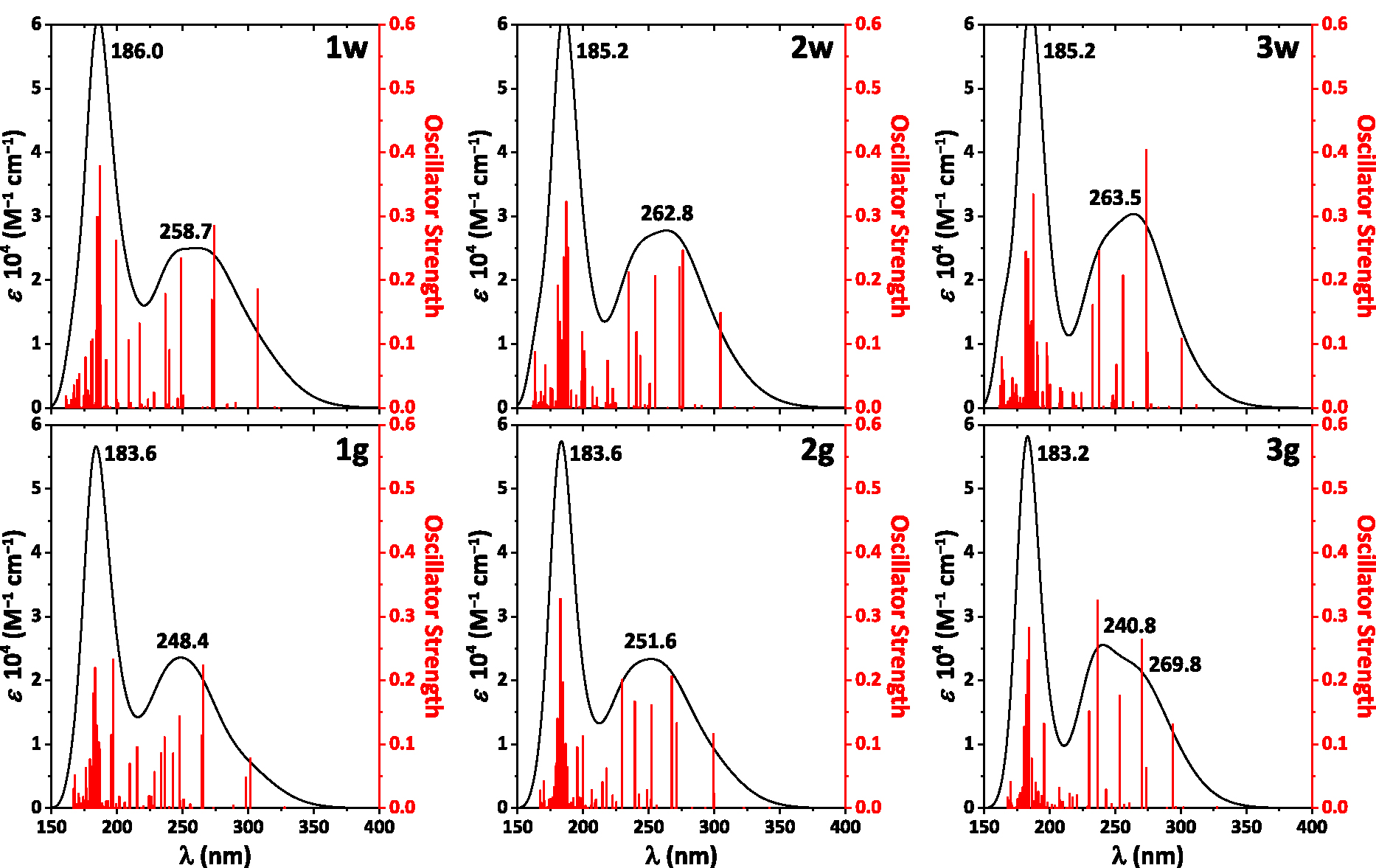
The calculated absorption spectra of the NH2Py2T isomers.
The values of I and A (Table 2) [80] for the NH2Py2T isomers indicate their electron-acceptor nature, which is also supported by the corresponding chemical potentials (𝜇) (Table 2). A low 𝜂 value together with a relatively high S value indicate that the isomers tend to exchange their electron cloud with the surrounding environment (Table 2) [80]. The value of 𝜔 is about 4.90–4.97 eV and 4.39–4.42 eV for 1w–3w and 1g–3g, respectively (Table 2), indicating their strong electrophilic nature [81]. The ΔNmax values show that the isomers can accept ∼2 electrons (Table 2).
In the calculated 1H NMR spectra of the reported NH2Py2T isomers, signals for the amine hydrogen atoms are observed at 5.36–5.43 ppm for 1w–3w, and, as expected, upfield-shifted up to 4.66–5.01 ppm in the spectra of 1g–3g (Table S5 in the Supplementary Material). The signals for protons H4 and H8, or H5 and H9 are shown at 7.68–8.17 or 7.29–7.84 ppm in the spectra of 1w–3w or 1g–3g, respectively (Table S5 in the Supplementary Material). The signals for the remaining protons, viz. H3, H6, H7, and H10, are significantly downfield-shifted and found at 8.94–9.55 ppm (Table S5 in the Supplementary Material). For H6 and H10, this shift is explained by the influence of the neighbouring electronegative nitrogen atom, thus leading to deshielding, while the H3 and H7 signals are downfield-shifted due to participation in the C–H⋯N hydrogen bonds described above. Notably, the calculated 1H NMR spectra of 1w and 3w are in good agreement with the experimental 1H NMR spectrum of NH2Py2T recorded in DMSO-d6 except for the signal of the NH2 protons [49], which is obviously due to their acidic nature.
We have also examined the potential nonlinear optical properties of 1w–3w and 1g–3g, (Table S6 in the Supplementary Material). Regarding the calculated dipole moment (μ), the isomers ranked as follows: 3 > 1 > 2. Notably, for isomers 1 and 3, the μy component is the main contributor to the overall dipole moment, while it is μx for isomers 2 (Table S6 in the Supplementary Material). Such an increase in dipole moment from isomers 2 through isomers 1 to isomers 3 is due to increasing charge disparity on the corresponding MEP surfaces (Figure S4 in the Supplementary Material). The calculated polarizability (α) and first-order hyperpolarizability (β) for the NH2Py2T isomers are about 7.9–10.8, except for 1g (Table S6 in the Supplementary Material). These values are higher to those of urea (Table S6 in the Supplementary Material), which is a common reference [82]. For 1g, the first-order hyperpolarizability parameter is about 10 times lower than that of urea (Table S6 in the Supplementary Material).
According to ProTox-II [64, 65], NH2Py2T was predicted to belong to toxicity class 4 (Figure 6), and could target the following classes: kinase, oxidoreductase, family C G protein-coupled receptor, ligand-gated ion channel, and other cytosolic protein with probabilities 46.7%, 33.3%, 6.7%, 6.7% and 6.7%, respectively (Figure 6). NH2Py2T was also predicted to be hepatotoxic, and active towards Aryl hydrocarbon Receptor (AhR) and Estrogen Receptor Alpha (ER) (Figure 6).
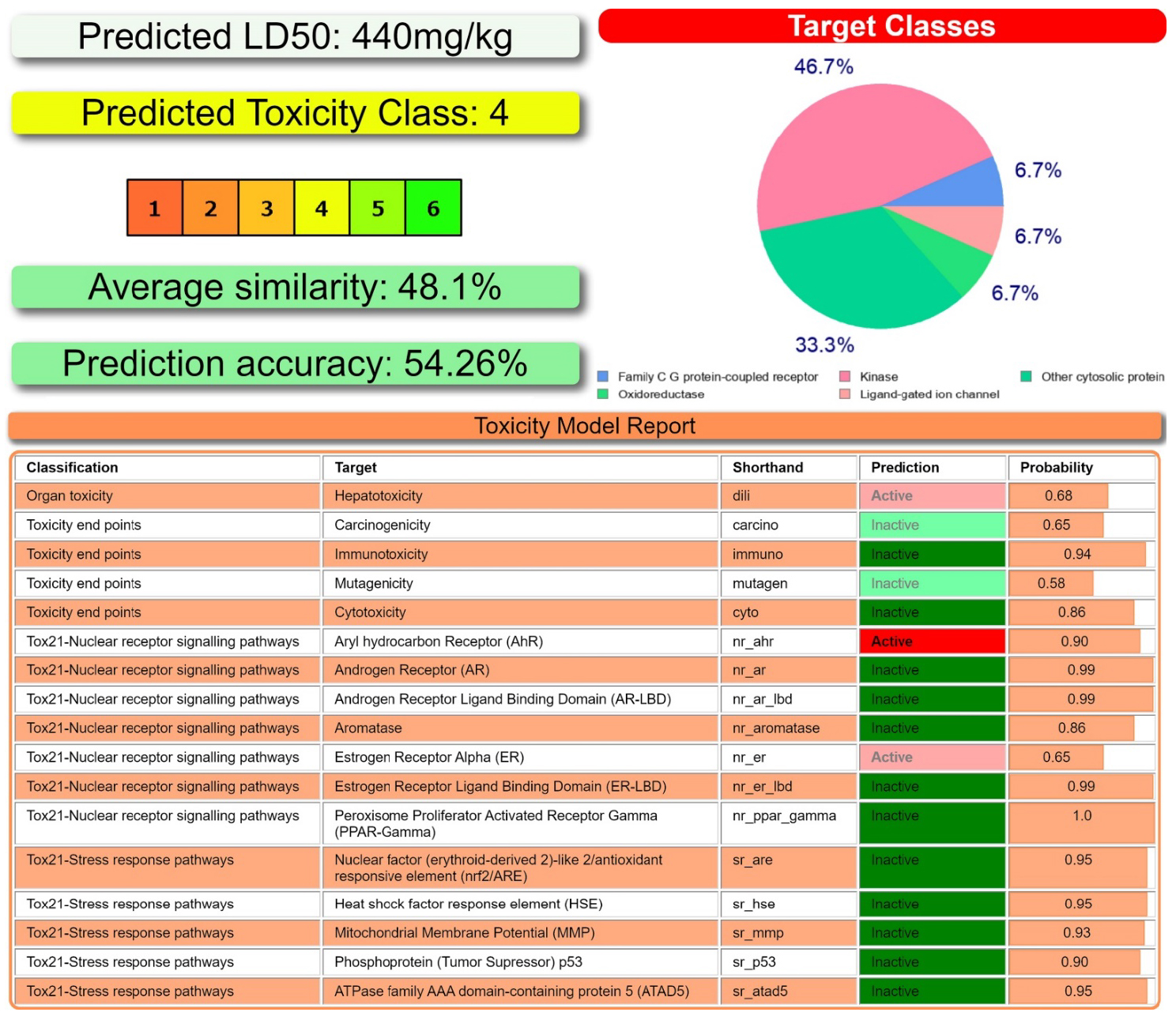
(Top left and bottom) Toxicity results of NH2Py2T calculated by ProTox-II. (Top right) Druggability predictions of NH2Py2T calculated by SwissADME.
The SwissADME bioavailability radar [62] indicates that NH2Py2T is favored in five parameters but has a poor result in the insaturation parameter (Figure 7), which is obviously due to the absence of the sp3-hybridized carbons. The BOILED-Egg method is powerful to predict the human blood–brain barrier (BBB) penetration and gastrointestinal absorption (GA) [63]. The dot position for NH2Py2T indicates a low absorption probability for BBB and high for GA with blue indicating a possible substrate of the P-glycoprotein (Figure 7).
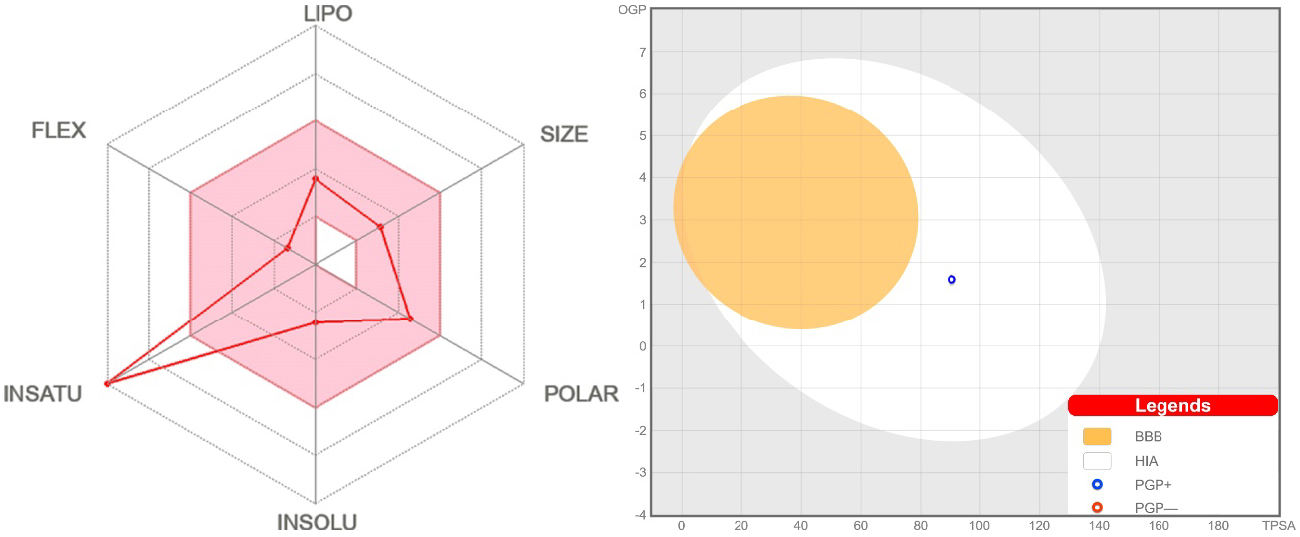
(Left) Bioavailability radar for NH2Py2T (colored zone is the suitable physicochemical space for oral bioavailability). (Right) BOILED-Egg model of NH2Py2T.
We have also studied potential inhibition properties of NH2Py2T toward a series of SARS-CoV-2 proteins using a molecular docking approach [83, 84, 85]. Notably, the structures optimized under water solvation (1w–3w) were investigated as potential inibitors of the herein-selected series of proteins and similar docking results were obtained. It was found that NH2Py2T is active against all the tested SARS-CoV-2 proteins and the best binding score was found for Nonstructural protein 3 (Nsp3-MES) (Table 3). Furthermore, according to our results, NH2Py2T shows significantly higher absolute values of binding energy compared to Favipiravir and Aspirin, but inferior to Remdesivir and all the redocked initial ligands except for the complex of Nsp3-MES with the corresponding initial ligand (Table 3) [31]. Interactions responsible for the binding of NH2Py2T with the studied proteins are shown in Figure 8 and collected in Table S7 in the Supplementary Material.
The best poses of NH2Py2T inside the binding sites of the listed proteins
| Protein | PDB code | Initial liganda | NH2Py2T | Favipiravir | Aspirin | Remdesivir |
|---|---|---|---|---|---|---|
| Main protease (Mpro) | 6LU7 | −8.00 | −6.9(0) | −4.70 | −5.40 | −8.10 |
| Papain-like protease (PLpro) | 6WUU | −9.70 | −6.8(0) | −5.30 | −5.30 | −8.70 |
| Nonstructural protein 3 (Nsp3-AMP) | 6W6Y | −7.50 | −6.8(1) | −4.60 | −5.60 | −7.50 |
| Nonstructural protein 3 (Nsp3-MES) | 6W6Y | −5.80 | −7.4(0) | −5.60 | −6.20 | −8.50 |
a(From top to bottom) Initial ligand = N-[(5-methylisoxazol-3-yl)carbonyl]alanyl-L-valyl-N ∼1∼-((1R,2Z)-4-(benzyloxy)-4-oxo-1-{[(3R)-2-oxopyrrolidin-3-yl]methyl}but-2-enyl)-L-leucinamide, methyl 4-[2-[[(2∼{S})-2-[[(2∼{S})-2-acetamido-4-(1,3-benzothiazol-2-yl)butanoyl]amino]-3-azanyl-propanoyl]amino]ethanoylamino]butanoate, [(2R,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-bis(oxidanyl)oxolan-2-yl]methyl dihydrogen phosphate, 2-morpholin-4-ium-4-ylethanesulfonate.
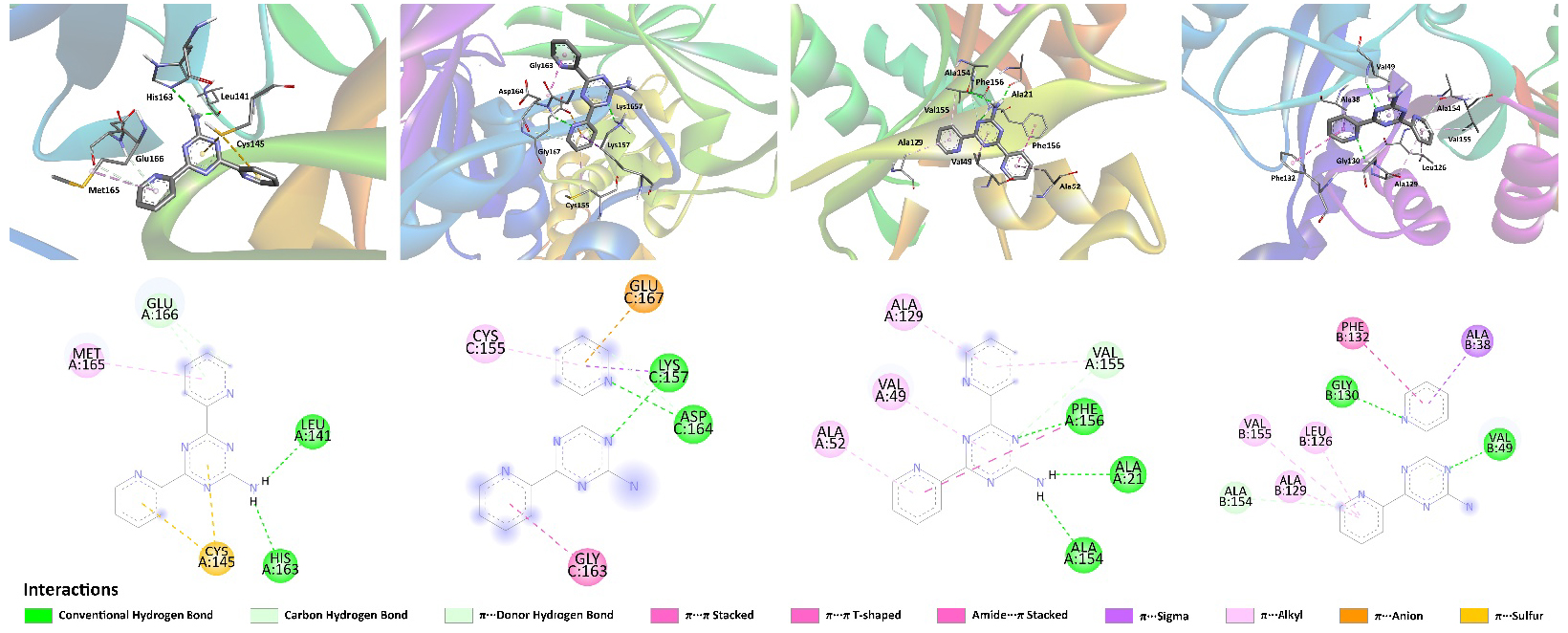
Views on the interaction of NH2Py2T with (from left to right) Mpro, PLpro, Nsp3-AMP and Nsp3-MES.
Using molecular dynamics simulations, we have evaluated interactions in complexes of NH2Py2T with the tested SARS-CoV-2 proteins. In particular, complex with Mpro showed an RMSD around 0.3 nm with an average value of 0.299 nm (Figure 9). However, complexes of NH2Py2T with Nsp3-AMP and Nsp3-MES showed a higher RMSD around 0.4 nm with average values of 0.392 and 0.364 nm, respectively (Figure 9). Notably, complex of NH2Py2T with PLpro showed a much higher RMSD over the simulation time reaching approximately 2.8 nm with an average value of 1.566 nm (Figure 9). Thus, the latter complex is unstable. The RMSF values for complexes of NH2Py2T with Mpro, Nsp3-AMP and Nsp3-MES were below 0.511, 0.389 and 0.534 nm, respectively (Figure 9). The strongest fluctuations of amino acid residues for each complex are listed in Table S8 in the Supplementary Information. Rg values for complexes of NH2Py2T with Mpro, Nsp3-AMP and Nsp3-MES form relatively stable profiles (Figure 9), with values in ranges 2.173–2.287, 2.319–2.475 and 2.317–2.489 nm, respectively. The SASA profiles were obtained to assess the interaction between complexes and solvents. As a result, the binding of NH2Py2T to Mpro, Nsp3-AMP and Nsp3-MES did not impair the stability of the proteins and interaction of the proteins with the solvent molecule (Figure 9). The average SASA was found as 148.44, 163.15 and 163.43 nm2 for complexes of NH2Py2T with Mpro, Nsp3-AMP and Nsp3-MES, respectively. Complexes of NH2Py2T with Mpro, Nsp3-AMP and Nsp3-MES, mainly, form one intermolecular hydrogen bond during the simulation time and two with a much lower frequency (Figure 9).
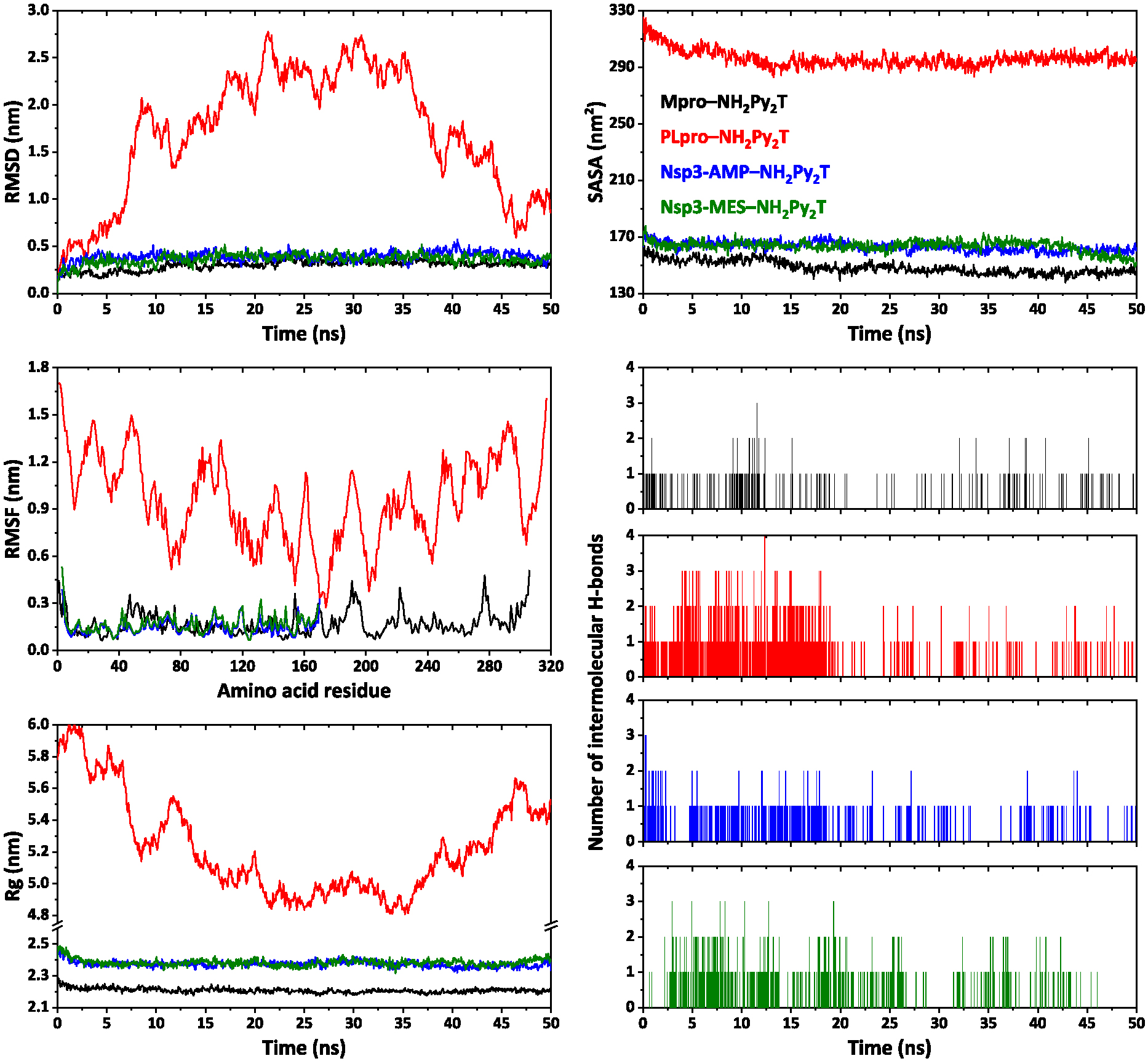
RMSD, RMSF, Rg, SASA and intermolecular hydrogen bonds analysis profiles of complexes of NH2Py2T with Mpro, PLpro, Nsp3-AMP and Nsp3-MES.
4. Conclusions
In this work, we have performed in silico analyses of 2-amino-4,6-bis(2-pyridyl)-1,3,5-triazine (NH2Py2T) using DFT. According to the conformational analysis, it was found that three different isomers, depending on the orientation of the pyridine rings, are the most favorable in both gas phase and water. Under water solvation, 1w was found to be the most energetically favorable, while 2w and 3w are 0.12 and 0.44 kcal/mol less favorable. In gas phase, 2g was found most favorable, while 1g and 3g are 0.28 and 2.25 kcal/mol less favorable. It was also established that under water solvation conditions the corresponding isomers are about 11–13 kcal/mol more favorable in comparison to gas phase. The global chemical reactivity descriptors of the studied NH2Py2T isomers allowed to assess their electron-accepting and -donating features. The calculated polarizability and first-order hyperpolarizability parameters of all the isomers, except for 1g, are higher in comparison to those of urea, which is a commonly used reference.
ADMET properties were predicted using SwissADME, BOILED-Egg and ProTox-II. It was found that human blood–brain barrier penetration is unlikely and gastrointestinal absorption likely with NH2Py2T predicted to be substrate of the P-glycoprotein. NH2Py2T was predicted to be hepatotoxic and active towards Aryl hydrocarbon Receptor (AhR) and Estrogen Receptor Alpha (ER).
According to in silico molecular docking results, NH2Py2T is active toward all the studied SARS-CoV-2 proteins. The best binding of NH2Py2T was found with Nsp3-MES. Molecular dynamics simulation showed that NH2Py2T forms stable complexes with Mpro, Nsp3-AMP and Nsp3-MES, while its complex with PLpro is unstable.
Declaration of interests
The author does not work for, advise, own shares in, or receives funds from any organization that could benefit from this article, and has declared no affiliations other than their research organizations.