



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Pyridinyl-based porphyrins are quite rare in the literature since their synthesis is cumbersome. Possible applications for these derivatives have already been reported such as photo-electrochemistry [1], light-harvesting charge separation systems [2, 3], supramolecular chemistry [4, 5, 6, 7, 8, 9, 10, 11, 12, 13], catalysis [9, 14], fast energy transfer [12, 15], molecular assembly on Au(III) [16] and dendrimer formation [17]. The full development of these pyridinylporphyrin derivatives may be restricted due to these synthetic limitations. The main issue stems from the first condensation steps with pyridinyl-based aldehyde and pyrrole, pyridinyl-based dipyrromethane with aldehydes or dipyrromethane with pyridinyl-based aldehydes, which results in low yields. Indeed, the pyridinyl fragment is able to trap protons from the Brønsted acid catalyst or deactivate the Lewis Acid catalyst in the common Adler and Longo or Lindsey condensation reactions. In our group, we have been interested in the exploitation of these pyridinyl-based porphyrins for molecular recognition of phenol derivatives [18] but also as precursors of C–N-fused cationic compounds obtained by oxidative pathway [19, 20]. For this latter purpose, mercaptopyridinylporphyrins have been successfully fused and the original physicochemical properties of the C–N-fused porphyrins have been explored in details (Previous work, Scheme 1). To extend the scope of this C–N oxidative fusion reaction, we wanted to replace the sulfur atom by a carbon atom and investigate if this reaction could work (This work, Scheme 1).
Previous work: C–N oxidative fusion with a mercaptopyridinylporphyrin; This work: planned target obtained after C–N oxidative fusion of a pyridinylmethylporphyrin precursor.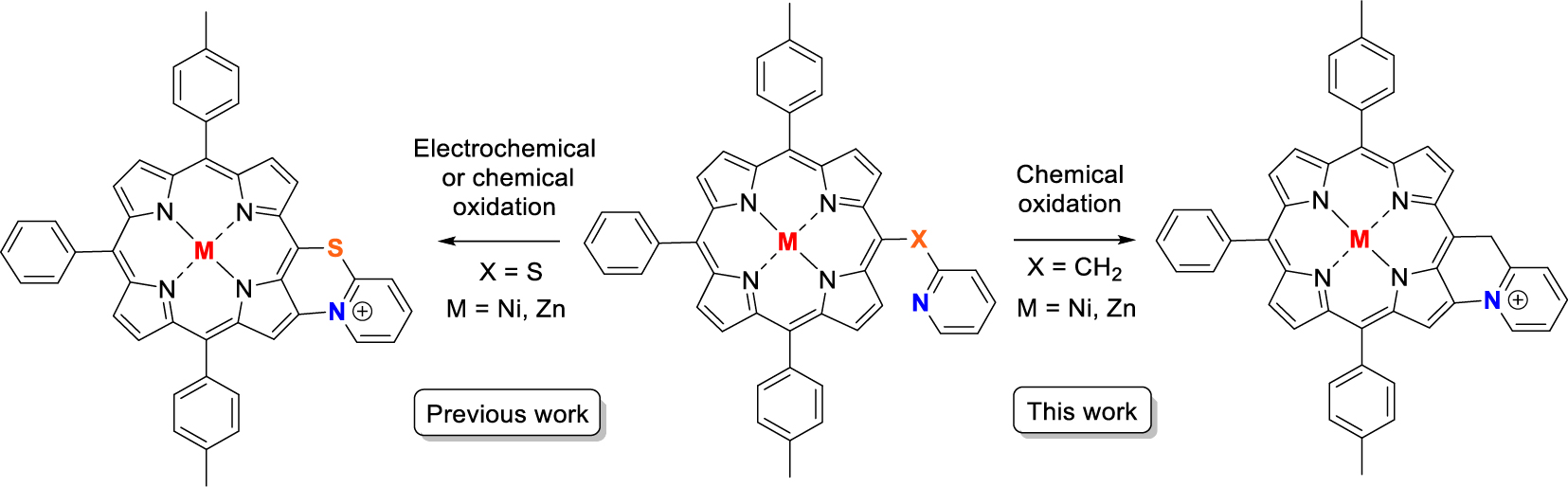
This manuscript describes the synthesis and characterization of (pyridin-2-ylmethyl)porphyrin H2-7 (Scheme 3) as well as the synthesis of its respective Zn(II) and Ni(II) complexes Zn-7 and Ni-7. Preliminary C–N oxidative fusion attempts are also described.
2. Results and discussion
2.1. Synthesis of (pyridin-2-ylmethyl)porphyrins 2, Zn-2 and Ni-2
Our initial objective was to synthesize 5,10,15,20-tetrakis(pyridin-2-ylmethyl)porphyrin H2-1 by condensation of pyrrole with aldehyde 2 produced by oxidation of commercially available 2-(2-hydroxyethyl)pyridine (3, Scheme 2).
Attempted synthesis of 5,10,15,20-tetrakis(pyridin-2-ylmethyl)porphyrin H2-1.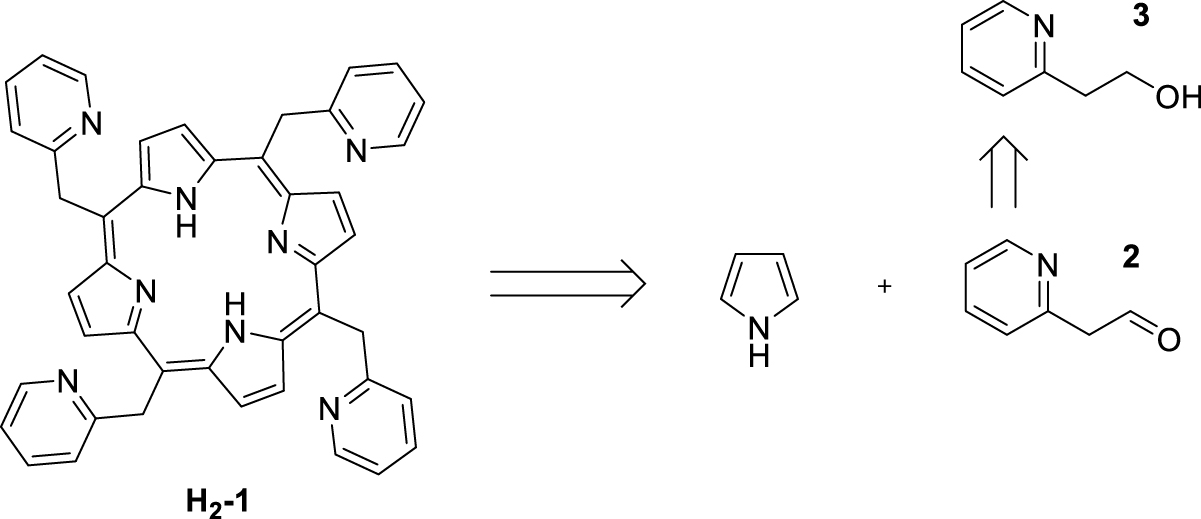
Two experimental conditions were tested for the aldehyde 2 synthesis: Swern conditions, using oxalyl chloride in DMSO and oxidation by a cerium(IV)-based oxidant [21, 22]. Despite several attempts with these two oxidants, the oxidation step of the alcohol function systematically failed. The targeted aldehyde 2 may be in equilibrium with its enol form [23], which could explain the difficulties in isolating/forming this aldehyde. Given these disappointing results, an alternative route was developed. The latter was based on the synthesis of a porphyrin monofunctionalized at the meso position with 2-methylpyridine (Scheme 3).
Synthesis of (pyridin-2-ylmethyl)porphyrin H2-7 and the corresponding Zn(II) and Ni(II) complexes Zn-7 and Ni-7.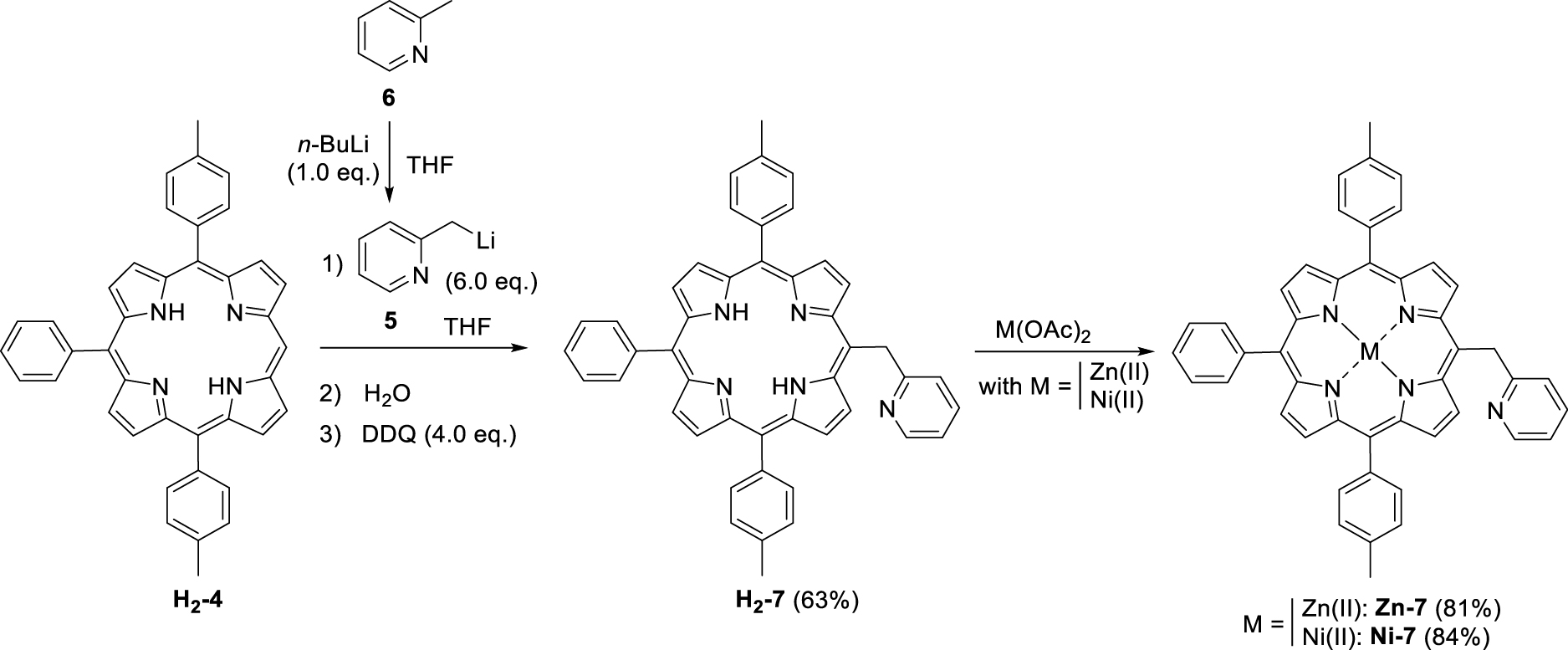
The first step consisted in the preparation of the (pyridin-2-ylmethyl)lithium (5) reagent by addition of n-butyllithium on 2-picoline (6). This orange solution was then added in excess to the tri-meso-arylporphyrin H2-4 following a protocol similar to the one previously described by Senge and colleagues for the phenylation of porphyrins [24]. Under these conditions, the original (pyridin-2-ylmethyl)porphyrin H2-7 was isolated in 63% yield (Scheme 3). The corresponding Zn(II) and Ni(II) complexes (Zn-7 and Ni-7, respectively) were also synthesized in good yields by standard metalation procedure (see experimental part) using the respective acetate salts [25]. It should be noted that another report from Barbe and coworkers describes the synthesis of a pyridin-2-ylmethylporphyrin (Zn-14, Scheme 4) [26]. This synthesis was based on the Suzuki cross-coupling of 5-bromo-2-chloromethyl-pyridine (13) (synthesized in two steps from bromopicolinaldehyde (11), Scheme 4) with zinc(II) meso-substituted dioxoborolaneporphyrin Zn-10 (synthesized in three steps from the 5,10,15-trimesitylporphyrin). According to these authors and contrary to their expectations, only the chlorinated carbon atom of 13 was coupled with the porphyrin and not the brominated one.
Synthesis of pyridin-2-ylporphyrin Zn-14 as described by Barbe and coworkers [26].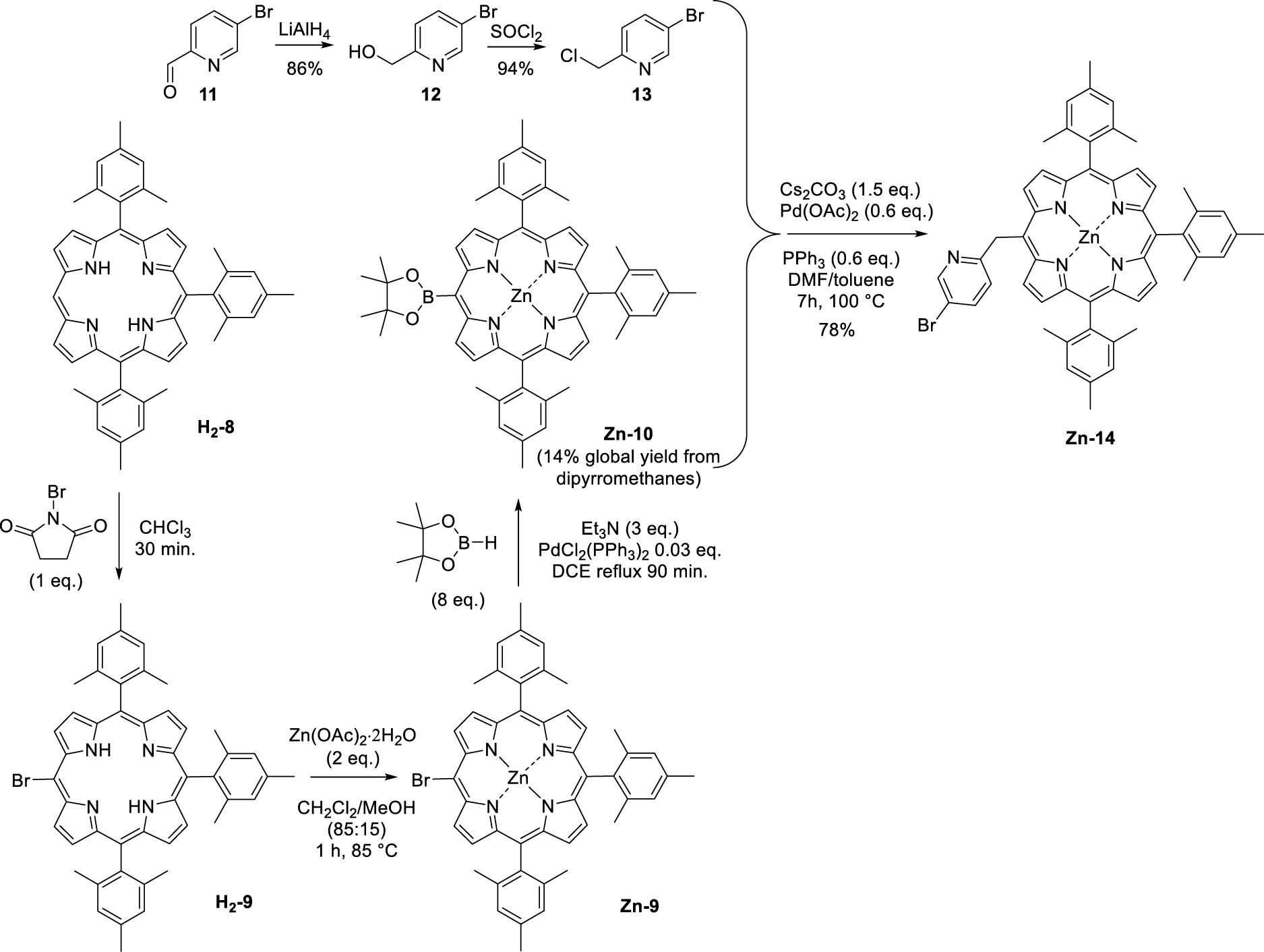
Thus our proposed synthesis is more straightforward since it involves only two steps for the synthesis of H2-7 starting from the tri-meso-arylporphyrin H2-4.
2.2. Characterization of (pyridin-2-ylmethyl)porphyrins H2-7, Zn-7 and Ni-7
2.2.1. NMR characterization of (pyridin-2-ylmethyl)porphyrins H2-7, Zn-7 and Ni-7
A focus on the aromatic part of the 1H NMR spectra of compounds 6, H2-7, Zn-7 and Ni-7 is presented in Figure 1. Noteworthy, for Zn-7, CDCl3 had to be replaced by (CD3)2CO due to solubility problems. In addition, one drop of pyridine-d5 was used to prevent the formation of coordination polymers upon coordination of the pyridinyl nitrogen atom onto the metallic center of another Zn-7 porphyrin. 2-Picoline 6 is characterized by a singlet at 2.33 ppm for the methyl group and four multiplets between 6.84 ppm and 8.28 ppm. The 1H NMR spectra of H2-7, Zn-7 and Ni-7 show similar chemical shifts for the pyridinyl moiety (6.73 < 𝛿 < 7.32 ppm) except for the 𝛼 proton doublet which appears between 8.59 and 8.75 ppm. For comparison purpose, the 𝛼 proton doublet of 6 appears at a slightly lower chemical shift (8.28 ppm). Thus the porphyrin ring has little influence on the chemical shifts of the pyridinyl moiety contrary to what is observed for the –CH2– spacer for which the methylene singlets of H2-7, Zn-7 and Ni-7 are observed between 6.21 and 6.70 ppm, a much higher value as compared to 2.33 ppm for 6. The same range of chemical shift was observed by Barbe and coworkers with the free-base analogue of Zn-14 [26], where the methylene bridge appeared at 6.59 ppm. This behavior is typically observed for protons that are lying outside the shielding cone of the porphyrin. The β-pyrrolicprotons of H2-7, Zn-7 and Ni-7 are located between 8.70 and 9.74 ppm. The more deshielded β-pyrrolicdoublet appears between 9.32 and 9.74 ppm and is attributed to the protons that are closest to the methylpyridinyl substituent .
Partial 1H NMR spectra of 6, H2-7, Zn-7 and Ni-7 (CDCl3 ((CD3)2CO + one drop of pyridine-d5 for Zn-7), 500 MHz, 298 K). The asterisk ∗ indicates not fully deuterated pyridine signals in the 1NMR spectrum of Zn-7. Small signals observed on the 1NMR spectrum of 6 correspond to unknown impurities that are present in the commercial reagent.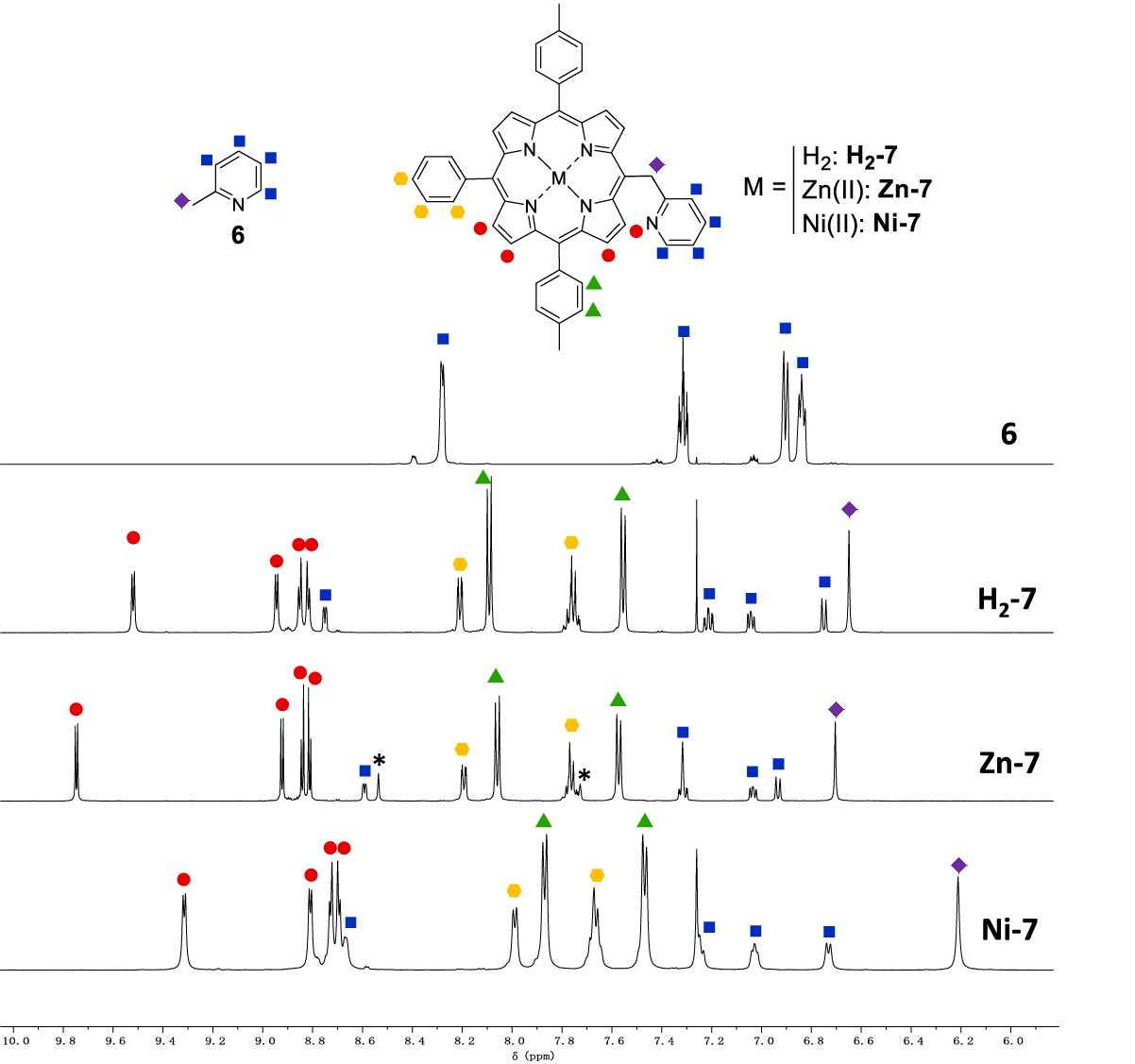
2.2.2. UV–visible absorption spectroscopy of H2-7, Zn-7 and Ni-7
UV–visible absorption spectra of H2-7, Zn-7 and Ni-7 are presented in Figure 2. Porphyrin H2-7 exhibits a Soret band at 419 nm and four less intense Q bands at 516, 552, 592 and 648 nm, as expected for a free base porphyrin. The corresponding nickel(II) complex Ni-7 shows a 3-nm hypsochromic shift of the Soret band as compared to H2-7. Indeed, while the metallic cation accepts the lone-pair electrons of the nitrogen atoms of the porphyrin ring, its electrons are donated to the macrocycle. The resulting flow of electrons of the delocalized π-system increases the energy available for electron transition, leading to the blueshift observed for the Soret band of Ni-7 [27]. The number of Q bands for this derivative is reduced to one as shown by comparison with the UV–visible absorption spectrum of H2-7, in agreement with the enhanced symmetry of the porphyrin induced by the metalation.
UV–visible absorption spectra of H2-7 (black curve), Zn-7 (red-curve) and Ni-7 (blue curve) in CH2Cl2 or for Zn-7 in DMF.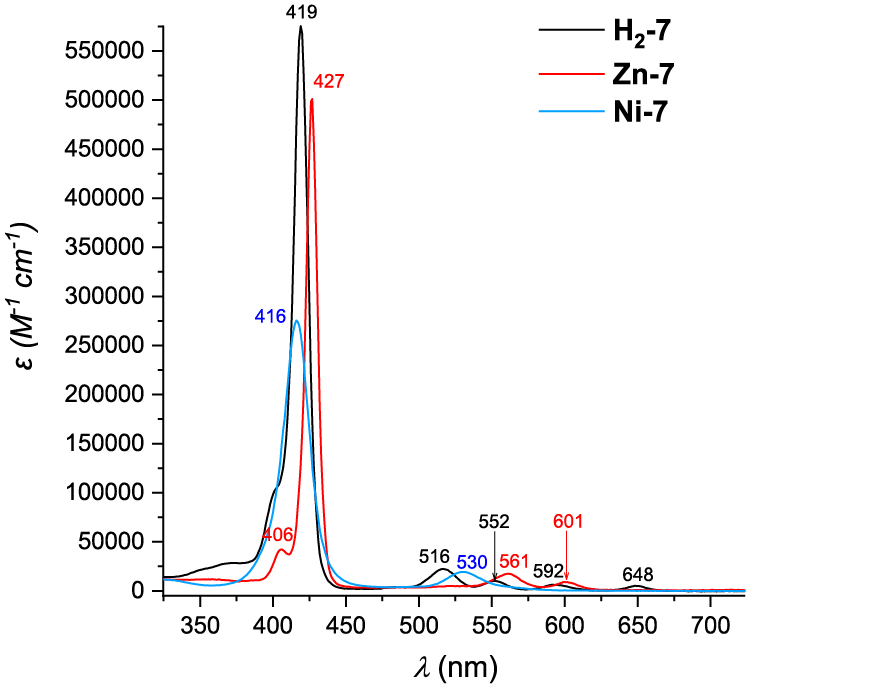
In the specific case of Zn-7, the CH2Cl2 solvent had to be replaced by DMF to improve the solubility of the porphyrin by preventing the formation of the coordination polymers. The Soret band is observed at 427 nm and only two Q bands are seen at 561 and 601 nm, for symmetry reasons.
2.2.3. Electrochemical characterization of H2-7, Zn-7 and Ni-7
All potential values reported in this manuscript are vs. the saturated KCl calomel electrode (SCE), unless otherwise mentioned. The cyclic voltammogram of Ni-7 is presented in Figure 3 (top black curve). The first fully irreversible oxidation takes place at Epa(O1) = 0.85 V which is less positive than the first oxidation potentials of Ni(II) porphyrins functionalized with 2-mercaptopyridine (Epa ≈ 1.10 V) [19]. This behavior agrees with the donor inductive effect of the –CH2– group, compared to the sulfur atom involved in a thioether function. This irreversibility which follows the porphyrin cation radical formation might also be a good indication that the C–N fusion reaction takes place. Two other reversible oxidation peaks are observed at Epa(O2) = 0.98 V and Epa(O3) = 1.29 V. Only one poorly reversible reduction peak is seen at Epc(R1) = −1.56 V.
Cyclic voltammograms of Ni-7 (top, black curve) and Zn-7 (bottom, blue curve). Conditions: C = 10−3 M, CH2Cl2 0.1 M TBAPF6, WE = Pt Ø = 2 mm, v = 100 mV⋅s−1. For solubility reason, 1.0 equiv of pyridine has been added to the solution containing Zn-7.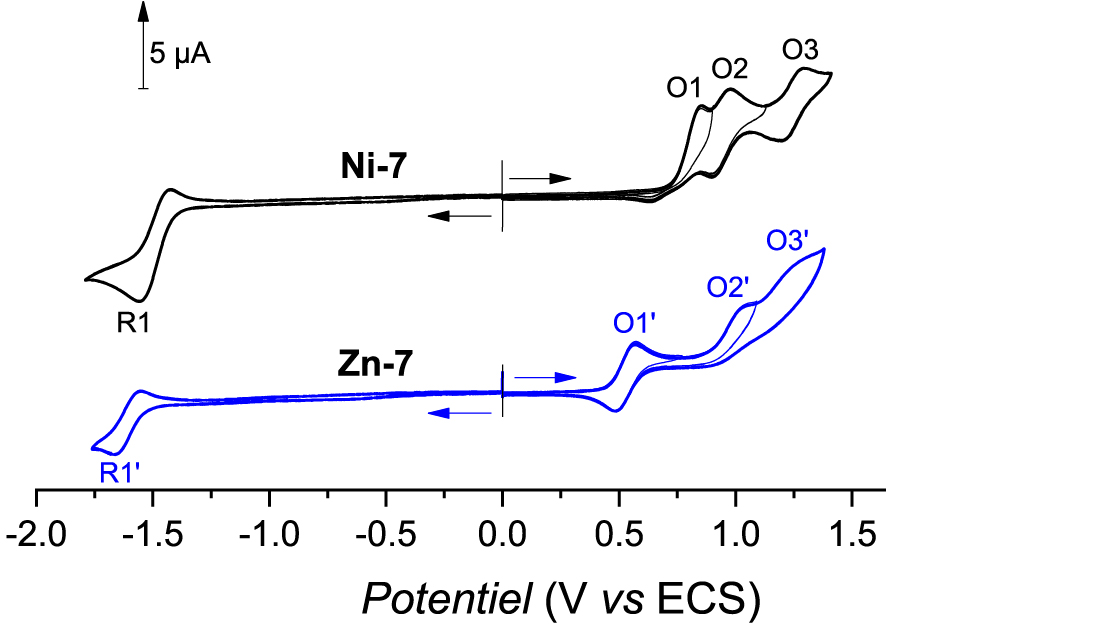
The zinc(II) complex Zn-7 exhibits very low solubility in CH2Cl2. The latter probably arises from coordination polymers that may be formed by coordination of the pyridinyl moiety onto the zinc(II) atom of another porphyrin [3, 10, 11, 17]. This behavior has been already experienced by us with the zinc(II) complexes substituted by one or two mercaptopyridinyl fragment(s) [20]. The addition of 1.0 equiv of pyridine to a solution of Zn-7 in CH2Cl2 was supposed to break the coordination polymers. Indeed, all the porphyrin became soluble which supported our initial hypothesis. The first oxidation wave is much more reversible than for Ni-7 (Epa(O1′) = 0.57 V, E1∕2 = 0.53 V, 𝛥Ep = 80 mV). Two other irreversible oxidation peaks are observed at ca. Epa(O2′) = 1.05 V and (Epa(O3′) = 1.27 V. The reduction peak R1′ is almost reversible and is observed at Epc(R1′) = −1.66 V.
2.3. Oxidative C–N fusion attempts
Following our previous work on the oxidative C–N fusion of pyri(mi)dine-based porphyrins (see Scheme 1 and Refs. [19, 20, 28]), fusion tests have been performed in CH2Cl2 on Ni-7 and Zn-7 with PIFA ((bis(trifluoroacetoxy)iodo)benzene), a two-electron acceptor. In principle, if the C–N oxidative fusion reaction of 2-picoline on the porphyrin nucleus occurs, the use of a base to neutralize the protons released by the C–N coupling reaction is necessary. Indeed, unlike 2-mercaptopyridine whose pKa is very low (−1.07) [29, 30], 2-picoline has a pKa of 5.96 [31] which is higher than that of trifluoroacetate ions (pKa = 0.23). Thus, the trifluoroacetate anions released during the reduction of PIFA are no longer sufficiently basic to capture the protons released during the formation of the C–N bond. A first reaction test between Ni-7 and PIFA was carried out in CH2Cl2 in the presence of potassium carbonate as a base. After reacting for 1 h with 1.0 equiv of PIFA, a brown product, more polar than compound Ni-7, was observed on a TLC plate (SiO2, CH2Cl2). Then 0.2 and 0.8 equiv of PIFA were added at one-hour intervals. One hour after the last addition of oxidant, the stirring was stopped because no further development was observed. 2,6-Lutidine, whose pKa is 6.72 [31], is perfectly miscible under our experimental conditions, unlike potassium carbonate which mainly remains as a precipitate. This organic base was therefore chosen for a new C–N fusion test of 2-picoline on the porphyrin core. Thus, the reaction between Ni-7 and PIFA, in the presence of 2.0 equiv of 2,6-lutidine, in dichloromethane, was monitored by TLC (SiO2, CH2Cl2), in order to determine the amount of oxidant necessary for the consumption of the starting product. After 23 h of reaction at room temperature and addition of 2.0 equiv of PIFA, the crude reaction no longer seemed to evolve. The latter contained Ni-7 and a more polar brown product which did not migrate onto the TLC plate. MALDI-TOF mass spectrometry analysis of the crude reaction shows a peak at m∕z = 712.1, attributed to the C–N fusion product (m∕z = 712.2026), a peak at m∕z = 713.1 corresponding to Ni-7 and a more intense peak at m∕z = 726.1 corresponding to the exact mass of compound Ni-7 to which 13 mass units are added. This mass may be related to the fusion of 2-picoline with porphyrin and formation of a carbonyl on the CH2 bridge (Figure 4).
MALDI-TOF spectrum (global and zoom views) obtained from the crude solution resulting from the addition of 2.0 equiv of 2,6-dimethylpyridine and 2.0 equiv of PIFA on Ni-7, after stirring for 23 h in CH2Cl2.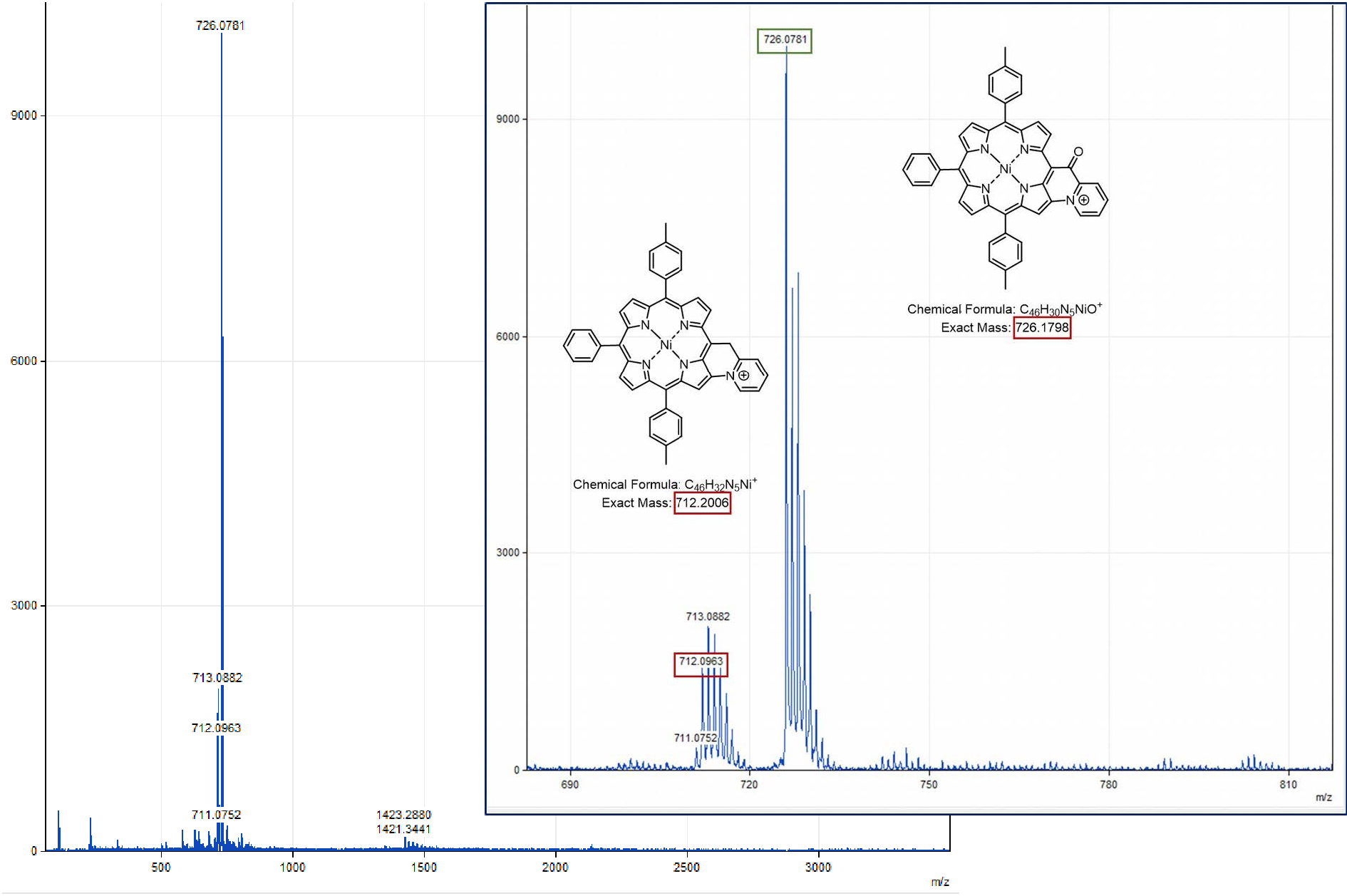
Two chemical fusion tests were also carried out on Zn-7 with PIFA at room temperature in dichloromethane, one in the presence of 2.0 equiv of potassium carbonate, the other in the presence of 2.0 equiv of 2,6-lutidine. When the reaction is carried out in the presence of potassium carbonate as base, 1.8 equiv (first 1.0 equiv, then 1.5 h of stirring, then 0.8 additional equiv added in 4 times at one-hour intervals) of PIFA are necessary to consume all the precursor. Under these conditions, two products are formed, one being more polar (purple/pink) and the other less polar (dark purple) than the product Zn-7 (purple). The 1H NMR analysis of the raw reaction product carried out in acetone-d6 in the presence of a drop of pyridine-d5 did not give any information (poorly resolved signals). An ESI mass analysis was performed to obtain more information, but none of the observed peaks could be assigned. The oxidation of compound Zn-7 in the presence of 2,6-lutidine only leads to the formation of a single dark purple product, more polar than the precursor to be fused. In this case, the use of a total of 2.2 equiv of PIFA did not allow all the starting material to be consumed, even after 23 h of reaction. MALDI-TOF analysis of the crude reaction (Figure 5) shows a peak at an m∕z value of 719.1 corresponding to the initial product (m∕z theoretical = 719.2027) but also at m∕z = 718.1 (C–N coupling product, theoretical m∕z = 718.1944) and 717.1 (loss of an additional proton), all three minor in terms of intensity compared to a peak at 732.1, whose m∕z ratio corresponds to the mass of Zn-7 to which 13 units of mass are added. The latter may, as in the case of the oxidation of derivative Ni-7, correspond to the fusion product with the formation of a carbonyl function on the bridging carbon atom. Finally, an isotopic cluster between m∕z = 1435 and m∕z = 1445 is also observed. It could be assigned to the formation of a dimer of unknown structure.
MALDI-TOF spectrum (global and zoom views) obtained from the crude solution resulting from the addition of 2.0 equiv of 2,6-dimethylpyridine and 2.0 equiv of PIFA on Zn-7, after stirring for 23 h in CH2Cl2.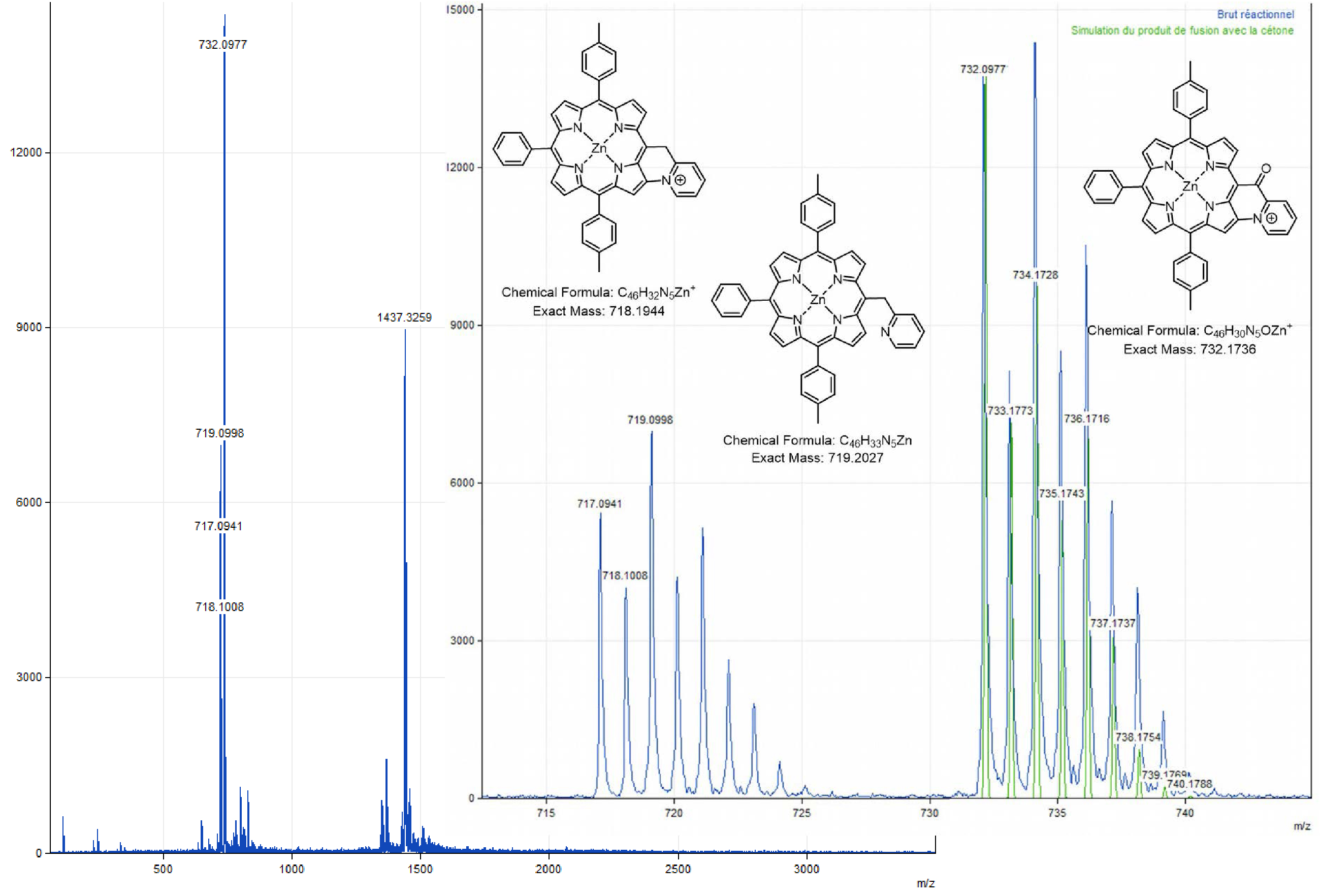
3. Experimental section
3.1. Reagents and instrumentation
Unless otherwise noted, all reactions were carried out without protection from air. CH2Cl2, MeCN, THF were dried over alumina cartridges using a solvent purification system PureSolv PS-MD-5 model from Innovative Technology and kept under argon. The following reagents were used: n-BuLi(1.6 M in hexane, Acros Organics), 2-picoline (98%, Acros Organics), 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ, 98%, Sigma-Aldrich), [bis(trifluoroacetoxy)iodo]benzene ((PIFA), 98%, Acros Organics), 2,6-lutidine (99%, Sigma-Aldrich), pyridine (99%, Acros Organics), K2CO3 (99%, anhydrous, Alfa Aesar), Zn(OAc)2⋅2H2O (99.5%, Acros Organics), Ni(OAc)2⋅4H2O (99%, Alfa Aesar), 10-phenyl-5,15-di-p-tolylporphyrin H2-4 was synthesized as reported in [32]. Our data (1H NMR, 13C NMR, UV–Visible absorption, and MALDI-TOF mass spectrum) were consistent with those described in Ref. [32]. Tetra-n-butylammonium hexafluorophosphate was synthesized by mixing stoichiometric amounts of HPF6 (60% in water, Acros Organics) and tetra-n-butylammonium hydroxide (40% in water, Alfa Aesar). The resulting solution was filtrated on a Büchner filter under vacuum and the white crystals were collected. These crystals were recrystallized twice in EtOH and then dried in a stove at 110 °C for 48 h.
1H, 13C NMR spectra were recorded either on a Bruker Avance 400, on a Bruker Avance 500 or on a Bruker Avance 600 III HD spectrometer. Chemical shifts are expressed in parts per million (ppm) from the residual non-deuterated solvent signal [33]. J values are expressed in Hz.
High-resolution mass spectra (HRMS) were recorded either on a Thermo LTQ Orbitrap XL apparatus equipped with an ESI source or on a Bruker UltraflexII LRF 2000 MALDI-TOF mass spectrometer (matrix: dithranol).
UV–visible absorption spectra were recorded on a VARIAN Cary 50 UV–Visible spectrophotometer using quartz cells.
All electrochemical manipulations were performed using Schlenk techniques in an atmosphere of dry oxygen-free argon at room temperature (T = 20 °C ± 3 °C). The supporting electrolyte was degassed under vacuum before use and then dissolved to a concentration of 0.1 mol⋅L−1. Voltammetric analyses were carried out in a standard three-electrode cell, with an Autolab PGSTAT 302 N potentiostat, connected to an interfaced computer that employed Electrochemistry Nova software or on a Biologic SP-300 potentiostat, connected to an interfaced computer that employed EC-Lab (v. 11.25) software. The reference electrode was a saturated KCl calomel electrode (SCE) separated from the analysed solution by a sintered glass disk filled with the background solution. The auxiliary electrode was a platinum wire separated from the analysed solution by a sintered glass disk filled with the background solution. For all voltammetric measurements, the working electrode was a platinum disk electrode (Ø = 1.6 mm). In these conditions, when operating in CH2Cl2 (0.1 M TBAPF6), the formal potential for the ferrocene (+∕0) couple was +0.46 V.
3.2. Synthesis of 5-(Pyridin-2-methyl)-10,20-bis(p-tolyl)-15-phenylporphyrin (H2-7)
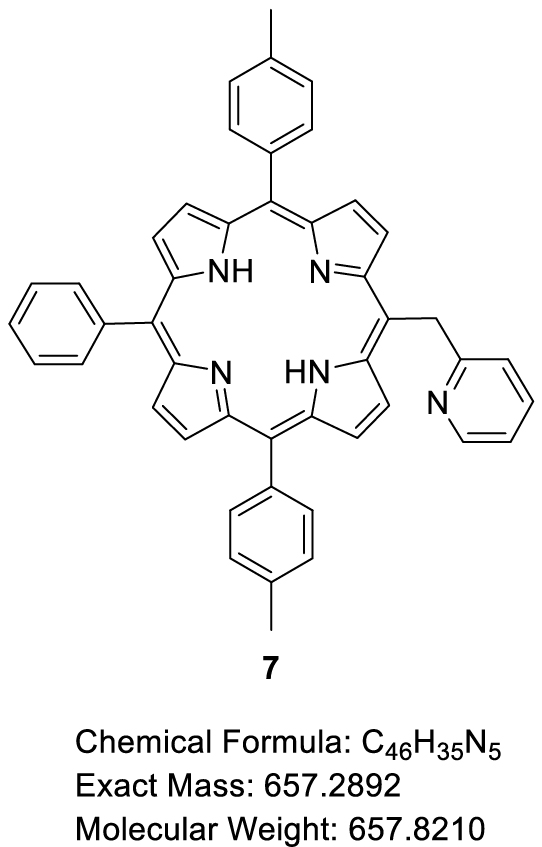
This protocol was inspired by the one described by Senge and coworkers for aryl and alkyl functionalization of porphyrins [24]. A 20 mL Schlenk flask was evacuated and flushed with argon three times. 2-Picoline (197.5 μL, 2.0 mmol, 1.0 equiv) was then added to the Schlenk flask followed by dry THF (0.88 mL). The solution was cooled to −10 °C then a commercial solution of n-BuLi (1.6 M in n-hexane) was added dropwise (1.25 mL, 2.0 mmol, 1.0 equiv). The resulting orange solution was stirred at room temperature for 1 h. H2-4 (100.00 mg, 1.76 × 10−1 mmol, 1.0 equiv) was kept under vacuum for 2 h in a second Schlenk flask and then flushed with argon. Dry THF (45 mL) was then introduced into the Schlenk flask containing the porphyrin and the solution was cooled to −30 °C. 1.2 mL (1.032 mmol, 6.0 equiv) of the orange solution obtained in the first Schlenk flask were added dropwise to the solution of porphyrin. The resulting solution was stirred at room temperature for 20 min, then a solution of H2O (0.84 mL) and dry THF (8.4 mL) previously degassed by bubbling argon was added dropwise to the reaction mixture. The green solution was stirred for a further 10 min at room temperature and DDQ (163.72 mg, 7.07 × 10−1 mmol, 4.0 equiv) was added to the Schlenk flask. The resulting red solution was stirred at room temperature for 1 h and then filtered on a pad of alumina, eluting with CH2Cl2. The crude product was purified by column chromatography (SiO2, CH2Cl2/CH3OH (99.5:0.5, v/v)). Three fractions were collected containing an unknown impurity (Fraction 1), H2-7 (Fraction 2) and traces of a product which could be unreacted H2-4 (Fraction 3). Fraction 2 was recrystallized in a CH2Cl2/CH3OH mixture. The obtained precipitate was filtered under vacuum, washed with CH3OH and dried at 110 °C overnight to give H2-7 in 63% yield (74.28 mg, 1.13 × 10−1 mmol). 1H NMR (CDCl3, 500 MHz, 298 K): 𝛿 (ppm) 9.52 (d, 3 JH–H = 4.8 Hz, 2H), 8.94 (d, 3 JH–H = 4.8 Hz, 2H), 8.85 (d, 3 JH–H = 4.8 Hz, 2H), 8.82 (d, 3 JH–H = 4.7 Hz, 2H), 8.75 (ddd, 3 JH–H = 5.0 Hz, 4 JH–H = 1.9 Hz, 4 JH–H = 0.9 Hz, 1H), 8.21, (dd, 3 JH–H = 7.7 Hz, 4 JH–H = 1.6 Hz, 2H), 8.09 (d, 3 JH–H = 7.9 Hz, 4H), 7.82 − 7.70 (m, 3H), 7.55 (d, 3 JH–H = 7.6 Hz, 4H), 7.21 (td, 3 JH–H = 7.7 Hz, 4 JH–H = 1.9 Hz, 1H), 7.04 (ddd, 3 JH–H = 7.3 Hz, 4 JH–H = 5.0 Hz, 4 JH–H = 1.2 Hz, 1H), 6.75 (d, 3 JH–H = 8.1 Hz, 1H), 6.65 (s, 2H), 2.72 (s, 6H), −2.64 (s, 2H); 13C{1H} NMR (CDCl3, 126 MHz, 298 K): 𝛿 (ppm) 164.1, 149.1, 142.2, 139.4, 137.5, 136.6, 134.6, 134.6, 127.8, 127.5, 126.8, 124.5, 121.1, 120.2, 120.1, 114.7, 43.7, 21.7; Rf 0.07 (SiO2, CH2Cl2); 𝜆max (CH2Cl2)/nm (log 𝜀): 419 (5.76), 516 (4.35), 552 (4.10), 592 (3.81), 648 (3.71); HRMS (ESI+): m∕z cald for C46H35N5 [M + H]+ 658.29652, found 658.29638.
3.3. Synthesis of [5-(Pyridin-2-methyl)-10,20-bis(p-tolyl)-15-phenylporphyrinato]zinc(II) (Zn-7)
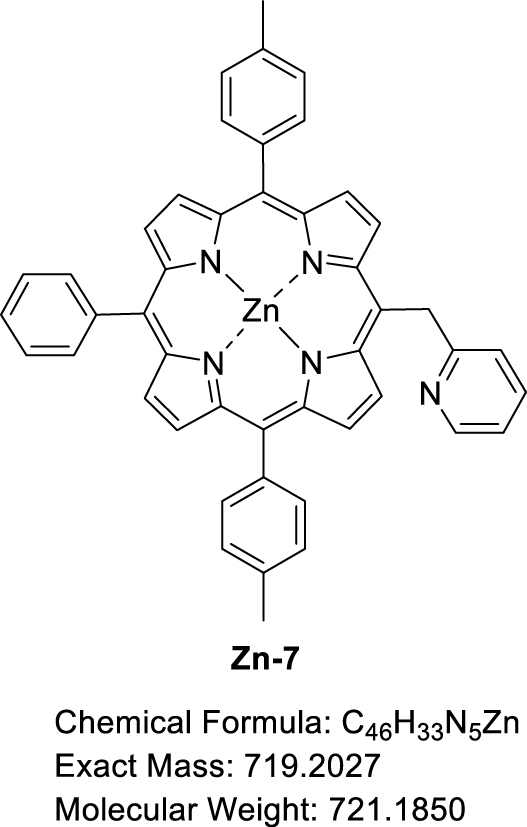
A solution of H2-7 (30.05 mg, 4.57 × 10−2 mmol, 1.0 equiv) and Zn(OAc)2⋅2H2O (10.20 mg, 9.16 × 10−2 mmol, 2.0 equiv) in a mixture of CHCl3 (2.7 mL) and CH3OH (1.0 mL) was stirred at 60 °C for 1.5 h, monitoring the progress of the reaction by TLC (SiO2, CH2Cl2). The solvent was then removed by rotary evaporation and the crude product was recrystallized in a CH2Cl2/CH3OH mixture. The precipitate was filtered under vacuum, washed with a small amount of CH3OH and dried at 110 °C overnight to give Zn-7 in 81% yield (26.72 mg, 3.71 × 10−2 mmol). 1H NMR ((CD3)2CO + one drop pyridine-d5, 500 MHz, 298 K): 𝛿 (ppm) 9.75 (d, 3 JH–H = 4.7 Hz, 2H), 8.92, (d, 3 JH–H = 4.7 Hz, 2H), 8.84 (d, 3 JH–H = 4.6 Hz, 2H), 8.81 (d, 3 JH–H = 4.6 Hz, 2H), 8.59 (ddd, 3 JH–H = 5.0 Hz, 4 JH–H = 1.9 Hz, 4 JH–H = 0.9 Hz, 1H), 8.19 (dd, 3 JH–H = 7.6 Hz, 4 JH–H = 1.7 Hz, 2H), 8.06 (d, 3 JH–H = 7.8 Hz, 4H), 7.80–7.70 (m, 3H), 7.57 (d, 3 JH–H = 7.5 Hz, 4H), 7.34–7.29 (m, 1H), 7.03 (ddd, 3 JH–H = 7.3 Hz, 4 JH–H = 4.9 Hz, 4 JH–H = 1.2 Hz, 1H), 6.93 (d, 3 JH–H = 8.1 Hz, 1H), 6.70 (s, 2H), 2.68 (s, 3H); 13C{1H} NMR ((CD3)2CO + one drop Pyridine-d5, 126 MHz, 298 K): 𝛿 (ppm) 151.8, 150.9, 150.7, 150.4, 150.2, 150.0, 149.7, 141.4, 137.7, 137.7, 136.8, 136.8, 136.3, 136.1, 135.9, 135.2, 135.2, 133.0, 132.4, 132.2, 130.7, 128.1, 127.9, 127.3, 124.6, 124.2, 124.0, 123.8, 121.5, 121.2, 117.0, 21.5; Rf 0.47 (SiO2, CH2Cl2); 𝜆max (DMF)/nm (log 𝜀): 406 (4.55), 427 (5.70), 561 (4.05), 600 (3.44); HRMS (ESI+): m∕z cald for C46H33N5Zn [M + H]+ 720.21002, found 720.21151.
3.4. Synthesis of [5-(Pyridin-2-methyl)-10,20-bis(p-tolyl)-15-phenylporphyrinato]nickel(II) (Ni-7)
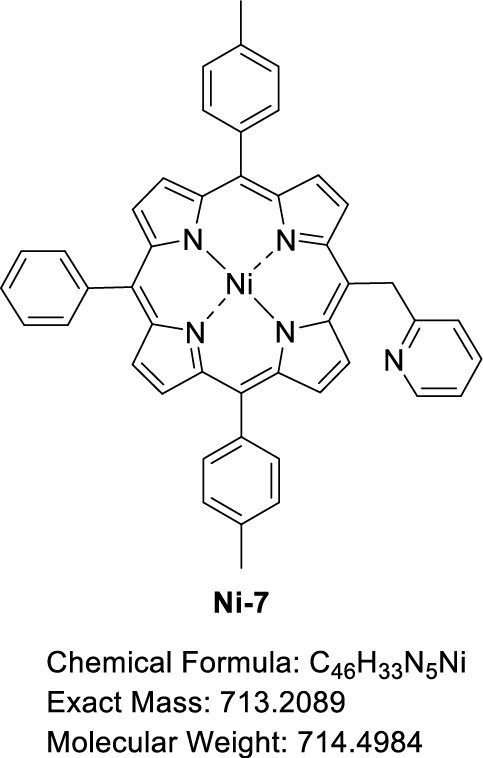
A Schlenk flask containing H2-7 (40.02 mg, 6.08 × 10−2 mmol, 1.0 equiv) and Ni(OAc)2⋅4H2O (30.31 mg, 1.22 × 10−1 mmol, 2.0 equiv) was evacuated and flushed with argon three times. Dry DMF (3 mL) was then added and the solution was stirred at 160 °C under argon for 1.5 h, monitoring the progress of the reaction by TLC (SiO2, CH2Cl2). The red solution obtained was allowed to cool to room temperature, then water was added (10 mL) to precipitate the product. The precipitate obtained was filtered under vacuum, washed with water and dried at 150 °C under vacuum for 4.5 h to give Ni-7 in 84% yield (36.61 mg, 5.12 × 10−2 mmol). 1H NMR (CDCl3, 500 MHz, 298 K): 𝛿 (ppm) 9.31 (d, 3 JH–H = 5.0 Hz, 2H), 8.81 (d, 3 JH–H = 4.9 Hz, 2H), 8.73 (d, 3 JH–H = 4.9 Hz, 2H), 8.69 (d, 3 JH–H = 4.9 Hz, 2H), 8.67 (d, 3 JH–H = 5.0 Hz, 1H), 7.99 (d, 3 JH–H = 6.4 Hz, 2H), 7.87 (d, 3 JH–H = 7.5 Hz, 4H), 7.74 − 7.61 (m, 3H), 7.47 (d, 3 JH–H = 7.6 Hz, 4H), 7.24 (d, 3 JH–H = 9.5 Hz, 1H), 7.03 (t, 3 JH–H = 6.2 Hz, 1H), 6.73 (d, 3 JH–H = 8.1 Hz, 1H), 6.21 (s, 2H), 2.64 (s, 2H); 13C{1H} NMR (CDCl3, 126 MHz, 298 K): 𝛿 (ppm) 149.1, 143.2, 142.7, 142.5, 141.0, 138.0, 137.6, 136.6, 133.8, 133.7, 133.2, 132.4, 132.2, 130.2, 127.8, 127.7, 127.0, 124.1, 121.1, 118.9, 113.3, 21.6; Rf 0.17 (SiO2, CH2Cl2); 𝜆max (CH2Cl2)/nm (log 𝜀): 416 (5.44), 530 (4.29); HRMS (ESI+): m∕z cald for C46H33N5Ni [M + H]+ 714.21622, found 714.21738.
3.5. Attempted synthesis of fused [5-(Pyridin-2-methyl)-10,20-bis(p-tolyl)-15-phenylporphyrinato] nickel(II) (Ni-7+,PF6−)
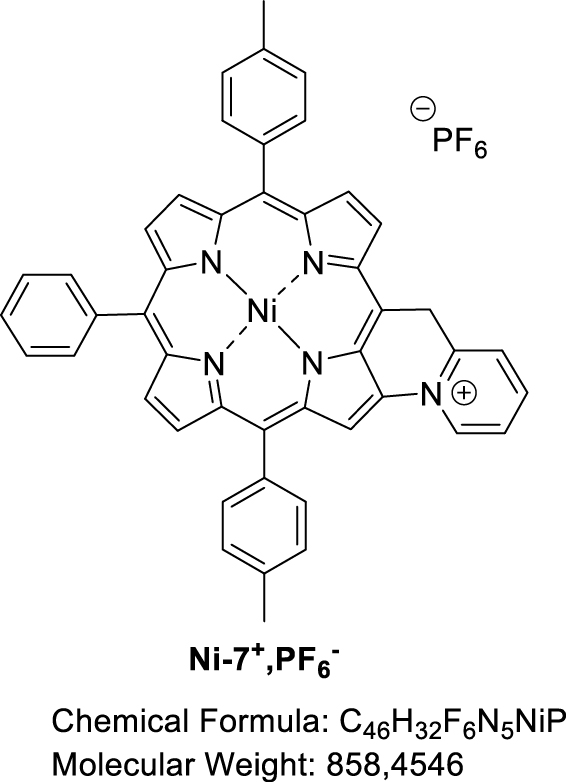
Nickel(II) porphyrin Ni-7 (1.0 equiv), 2,6-lutidine (2.0 equiv) and PIFA (1.0 equiv) were dissolved in dry CH2Cl2 (0.05 M). The reaction mixture was then stirred at room temperature for 1 h, monitoring the progress of the reaction by TLC (SiO2, CH2Cl2). At that time, an additional amount of PIFA (0.2 equiv) was added. The mixture was stirred for 1.5 h and a further amount of PIFA was again added (1.0 equiv). After a total of 23 h of reaction, the solvent was evaporated. MALDI-TOF: m∕z cald for C46H32N5Ni+ [M-PF6−] 712.2026, found 712.2006 (assigned to the fused compound); 713.0963 (assigned to Ni-7) and 726.1798 ([M-2H+-PF6−+O] assigned to the fused compound with a carbonyl function on the bridging carbon atom).
3.6. Attempted synthesis of fused [5-(Pyridin-2-methyl)-10,20-bis(p-tolyl)-15-phenylporphyrinato] zinc(II) (Zn-7+,PF6−)
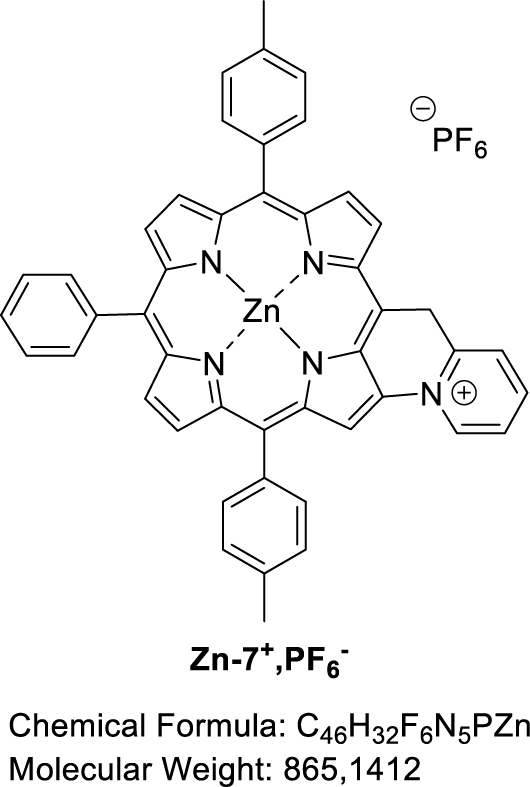
Zinc(II) porphyrin Zn-7 (1.0 equiv), 2,6-lutidine (2.0 equiv) and PIFA (1.0 equiv) were dissolved in dry CH2Cl2 (0.05 M). The reaction mixture was then stirred at room temperature for 1 h, monitoring the progress of the reaction by TLC (SiO2, CH2Cl2). At that time, an additional amount of PIFA (0.2 equiv) was added. The mixture was stirred for 1.5 h and a further amount of PIFA was again added (1.0 equiv). After a total of 23 h of reaction, the solvent was evaporated. MALDI-TOF: m∕z cald for C46H32N5Zn+ [M-PF6−]718.1944, found 717.0941 (assigned to the fused compound with the loss of one proton), 718.1008 (assigned to the fused compound); 719.0998 (assigned to Zn-7) and 732.0977 ([M-2H+-PF6−+O] assigned to the fused compound with a carbonyl function on the bridging carbon atom).
4. Conclusion
In summary, a new straightforward route for the synthesis of meso-(pyridin-2-ylmethyl)porphyrins was developed. This original pathway involves the nucleophilic attack of (pyridin-2-ylmethyl)lithium reagent on 10-phenyl-5,15-di-p-tolylporphyrin bearing one free meso position. The obtained free base porphyrin was then metalated providing the zinc(II) and nickel(II) complexes. These products were characterized by common analytical techniques (NMR, UV–vis., mass spectrometry and electrochemistry). Preliminary attempts to chemically generate the corresponding C–N-fused compounds (new bond between the nitrogen of the pyridinyl substituent and the β-pyrrolic position of the porphyrin) by oxidation with PIFA have been explored. Unfortunately, none of these attempts have lead to the formation and isolation of the desired fused porphyrins. Although the analyses performed on the crude mixtures suggest the formation of the fused porphyrins with formation of a carbonyl on the CH2 bridge, the isolation of these compounds to confirm their structure was not successful.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
This work was supported by the Université de Bourgogne, Université de Bourgogne Franche-Comté, CNRS (Centre National de la Recherche Scientifique), Conseil Régional de Bourgogne through the “Plan d’Actions Régional pour l’Innovation (PARI)” and the “Fonds Européen de Développement Régional (FEDER)” programs). MB and CHD thank the Agence Nationale de la Recherche for funding (ANR-15-CE28-0018-01). CHD acknowledges the Conseil Régional de Bourgogne Franche-Comté for a PhD grant (PORFELEC project) and the French “Investissement d’Avenir” program, project ISITE-BFC (contract ANR-15-IDEX-03) for funding.
Acknowledgments
The authors are thankful to Sophie Fournier for technical support, to the “Plateforme d’Analyse Chimique et de Synthèse Moléculaire de l’Université de Bourgogne” (PACSMUB, http://www.wpcm.fr) for ESI-HRMS analyses.