

 CC-BY 4.0
CC-BY 4.0
Préambule
Ce manuscrit présente une description des transformations microscopiques, réaction chimique et transition de phase, pour laquelle l’équation de l’équilibre est établie à partir d’équations cinétiques. Le modèle élaboré constitue une remise en cause du modèle thermodynamique chimique standard :
- L’entropie calorimétrique introduite par Rudolf Clausius en 1855, appropriée pour prévoir les échanges de chaleur et travail, n’est pas jugée adéquate pour prévoir l’évolution des transformations microscopiques, en particulier et non exhaustivement celles de la réaction chimique et de la fusion.
- L’existence du potentiel chimique, postulée par Willard Gibbs en 1876, n’est pas reconnue.
Compte tenu de l’importance de la remise en question des fondements de la thermodynamique chimique, le premier chapitre est consacré à l’historique de sa construction. Il se distingue néanmoins d’une revue encyclopédique exhaustive car il ne mentionne ni les théories fondées sur l’existence du potentiel chimique, ni celles qui font usage de l’entropie calorimétrique dans les équations qui gouvernent l’équilibre chimique et l’équilibre de phases puisque ces deux notions sont remises en question.
La construction du modèle cinétique des équilibres fait l’objet du deuxième chapitre pour la réaction chimique et du quatrième chapitre pour la fusion. La description cinétique de l’équilibre n’est pas fondée sur l’existence du potentiel chimique et n’utilise pas l’entropie calorimétrique : elle est fondée sur une formulation cinétique de l’équilibre. L’entropie cinétique qui émerge de cette description se substitue à l’entropie calorimétrique dans les équations qui gouvernent l’évolution des transformations microscopiques, réaction chimique et fusion.
Les troisième et cinquième chapitres sont dédiés à la confrontation des modèles thermodynamique et cinétique des équilibres chimique et de fusion. Il est montré pour des systèmes variés, sièges d’une réaction chimique ou d’une transition de phase liquide–solide, que la description cinétique est soit aussi pertinente, soit plus appropriée, que la description thermodynamique. La description cinétique s’affranchit de l’usage de paramètres d’ajustement de nature empirique indispensables pour décrire des systèmes qualifiés de non idéaux par le modèle thermodynamique. En outre, la description cinétique rationalise des processus variés inexplicables par le modèle classique.
Le sixième et dernier chapitre est dédié au modèle cinétique de l’équilibre des transformations microscopiques dans un cadre théorique général et présente des concepts qui découlent de ce modèle.
1. Introduction historique de la thermodynamique chimique
1.1. Premier principe
Au 19ème siècle, un médecin allemand, Julius Robert von Mayer, énonce clairement le premier principe de la thermodynamique [1]. Il déduit, de l’observation physiologique de marins lors d’expéditions aux antipodes, que la chaleur de combustion de la nourriture sert à la fois à maintenir la température du corps et à fournir un effort physique [2]. Il établit en 1842 la relation entre chaleur et travail et conclut [3] :
« Ex nihilo nihil fit. »
« Rien n’est créé de rien »
Le premier principe s’exprime dans le formalisme actuel par l’existence d’une grandeur extensive conservative de tout système isolé, l’énergie [4] :
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
1.2. Second principe
Système isolé à pression constante d’une mole de deux solides A et B en contact, de même structure chimique et de températures initiales et TB différentes. La capacité calorifique molaire à pression constante et le volume des solides sont supposés indépendants de la température.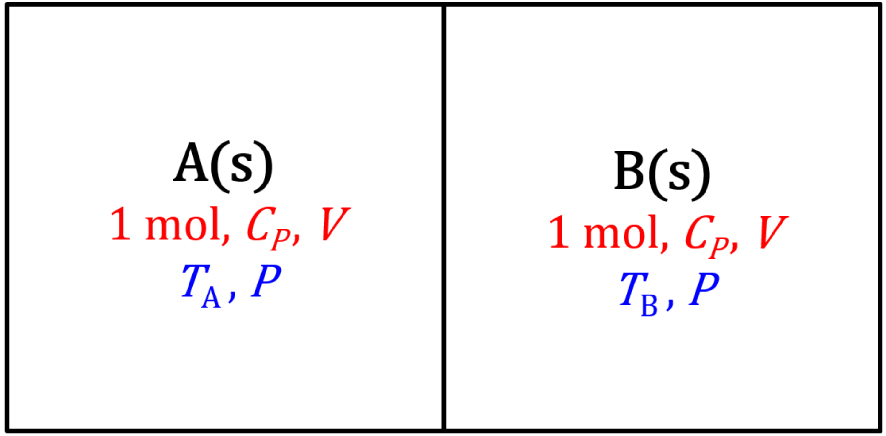
Un ingénieur français, Sadi Carnot, publie en 1824 un ouvrage à frais d’auteur intitulé « Réflexions sur la puissance motrice du feu et sur les machines propres à développer cette puissance » dans lequel il étudie la transformation par les moteurs de la chaleur de combustion en travail mécanique [6]. Il conclut à la fin de son manuscrit que le travail fourni lors de la combustion d’un kilogramme de charbon fournie par les moteurs les plus performants de son époque est très en deçà de la chaleur dégagée par la combustion de la même quantité de combustible :
| (7) |
| (8) |
| (9) |
Les deux solides forment un système isolé du milieu extérieur mais ils constituent chacun un système fermé qui échange de l’énergie avec l’autre. L’énergie du système isolé se conserve :
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
| (16) |
| (17) |
| (18) |
| (19) |
| (20) |
| (21) |
| (22) |
« Il existe une grandeur extensive et non conservative, l’entropie, qui augmente pour un système isolé avec le temps. »
Au même titre que le premier principe, le deuxième principe de la thermodynamique énoncé dans le cadre du système isolé ne permet pas l’étude de systèmes qui échangent chaleur et travail avec le milieu extérieur. Une formulation moderne du deuxième principe de la thermodynamique [5] prend en compte la possibilité de tels échanges, c’est-à-dire en considérant un système fermé :
« Il existe pour tout système fermé, délimité du milieu extérieur par une surface fermée, une fonction d’état extensive et non conservative, l’entropie. Sa variation est la somme d’une entropieSc créée à l’intérieur du système et d’une entropieSe échangée avec le milieu extérieur :
| (23) |
| (24) |
| (25) |
1.3. Energie de Gibbs
Willard Gibbs introduit en 1873 une fonction mathématique dont la variation élémentaire à température et pression constantes est nulle à l’équilibre thermodynamique [8]. La formule historique de cette fonction est :
| (26) |
| (27) |
| (28) |
| (29) |
| (30) |
1.4. La thermodynamique chimique, une science fondée sur un postulat
Gibbs postule en 1876 l’existence du potentiel chimique comme force motrice de la réaction chimique, qu’il introduit dans la différentielle de l’énergie interne [9]. La variation élémentaire d’énergie interne d’une réaction qui implique i espèces chimiques est :
| (31) |
Gibbs n’avait pas explicité dans son manuscrit l’expression du potentiel chimique, il en avait postulé l’existence comme force motrice de la réaction chimique qui vient s’additionner à la fonction qui gouverne l’équilibre d’un fluide. Le manuscrit de Gibbs traduit en français par Henry Le Chatelier inclut une note personnelle de ce dernier sur l’introduction du potentiel chimique [10] :
« C’est là en réalité une hypothèse qui revient à attribuer à chaque corps une force chimique définie, comme il possède un poids défini. Cette hypothèse très rationnelle doit être considérée comme un postulatum analogue à ceux qui servent de base à la mécanique rationnelle. »
La différentielle de l’énergie interne n’est pas, d’un point de vue pratique, appropriée pour prévoir l’évolution d’un équilibre chimique. En effet, il n’est pas aisé, lors d’une transformation, de maintenir l’entropie constante. En revanche, la différentielle de l’énergie de Gibbs dépend de celles de la température et de la pression qu’il est aisé de contrôler et s’avère de fait plus pertinente pour l’étude des équilibres chimiques :
| (32) |
| (33) |
| (34) |
| (35) |
1.5. Contenu du potentiel chimique : l’équilibre de fusion
Le premier travail des chimistes et physiciens du début du 20ème siècle, dont l’esprit avait été échauffé par l’éclosion de la thermodynamique chimique, une science nouvelle, fut de trouver l’expression du potentiel chimique dont l’existence avait été postulée par Gibbs. L’effet eutectique, l’abaissement mutuel du point de fusion des constituants d’un mélange lorsque leur proportion diminue, s’observe pour des composés qui forment une phase liquide homogène à chaud et dont les solides sont immiscibles à froid. Le diagramme de phases liquide–solide d’un mélange de deux constituants, notés A et B, est établi par l’observation expérimentale des phases stables du mélange en fonction de sa composition et de la température (Figure 2).
Topologie du diagramme de phases liquide–solide d’un système binaire à invariant eutectique. Courbes d’équilibre de fusion de la température en fonction de la fraction molaire du constituant A dans la phase liquide : constituant A (trait rouge), constituant B (trait bleu) et palier eutectique (trait noir).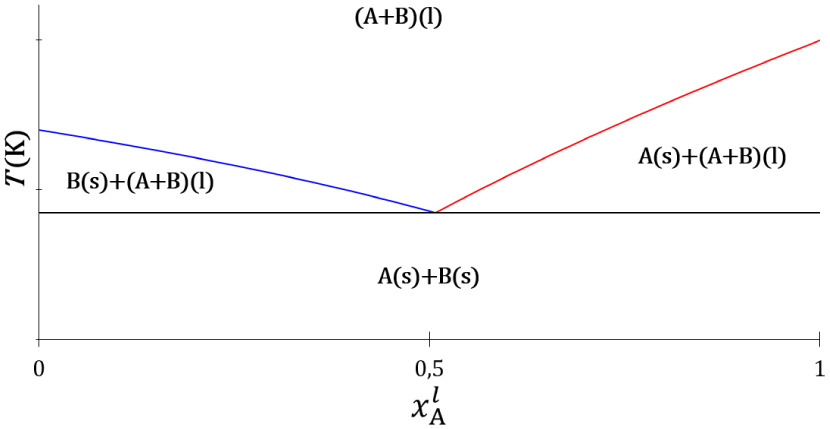
L’étude par Iw. Schröder en 1901 de l’équilibre de fusion du mélange eutectique du naphtalène avec le 1,3-dinitrobenzène est un travail précurseur et fondateur [11] (Figure 3).
Structures chimiques du naphtalène et du 1,3-dinitrobenzène.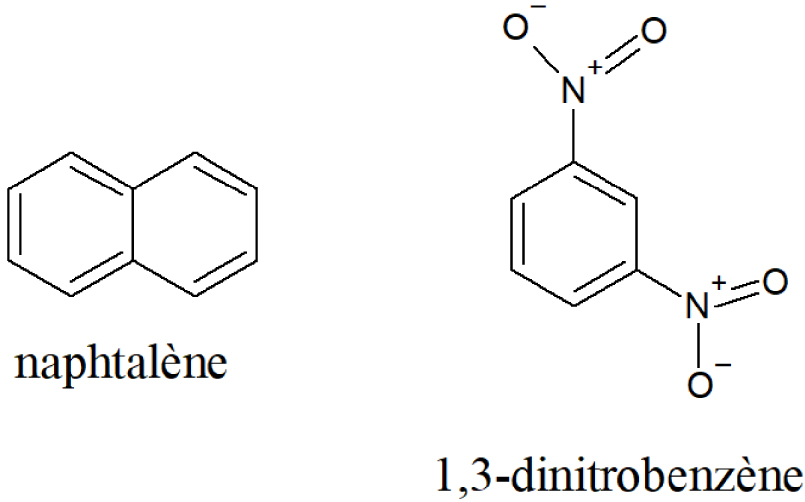
Schröder dégage de l’étude de ce mélange une équation de la courbe d’équilibre de fusion (i.e. liquidus) qui exprime la température de fusion d’un solide en fonction de sa composition dans son liquide. Cette courbe d’équilibre dépend des température et chaleur de fusion à une pression de 1 bar de l’espèce chimique pure :
| (36) |
| (37) |
| (38) |
| (39) |
1.6. Equilibre de phases liquide–solide d’un système à invariant eutectique
Une description thermodynamique de l’équilibre de fusion est présentée dans ce chapitre. Soit un constituant chimique dont le solide est en équilibre dans son mélange liquide homogène avec d’autres constituants j immiscibles dans la phase solide de i. L’équilibre de fusion est étudié à pression et température constantes :
- i. du potentiel chimique du liquide i pur ;
- ii. de sa fraction molaire dans le mélange liquide.
Soit :
| (40) |
| (41) |
| (42) |
| (43) |
| (44) |
| (45) |
L’équation de l’équilibre de fusion est usuellement exprimée avec le formalisme des grandeurs molaires :
| (46) |
| (47) |
| (48) |
| (49) |
| (50) |
Le comportement de certains mélanges à invariant eutectique est en accord avec ce modèle, l’équation de l’équilibre de fusion déduite de mesures calorimétriques est en accord avec celle du diagramme de phases expérimental :
| (51) |
| (52) |
| (53) |
| (54) |
| (55) |
| (56) |
1.7. Equilibre chimique dans une phase homogène gazeuse ou liquide
Marcellin Berthelot et Louis–Denis Péan de Saint–Gilles établissent en 1863 que diverses réactions d’estérification donnent lieu à l’établissement d’un équilibre, caractérisé par l’invariance de la concentration de chaque espèce réactive engagée. Ils étudièrent en particulier la réaction d’estérification de l’acide éthanoïque par l’éthanol [13] :
| (57) |
| (58) |
| (59) |
| (60) |
| (61) |
| (62) |
| (63) |
| (64) |
| (65) |
Evolution de ln(K) en fonction de l’inverse de la température d’une réaction chimique.
La concordance numérique entre la constante d’équilibre déterminée par le dosage des réactifs et produits d’une réaction à l’équilibre et celle déduite des propriétés calorimétriques est vérifiée pour certains équilibres chimiques, qualifiés d’idéaux :
| (66) |
| (67) |
| (68) |
| (69) |
| (70) |
| (71) |
| (72) |
| (73) |
1.8. Le coefficient d’activité, paramètre empirique de la thermodynamique chimique
Le coefficient d’activité a été introduit dans les équations thermodynamiques de l’équilibre de fusion et de l’équilibre chimique pour décrire certains systèmes dont le comportement s’écartait de celui prévu par le modèle thermodynamique chimique. Le coefficient d’activité ne présente pas dans sa définition de restriction sur les valeurs numériques qu’il peut prendre, il sera donc toujours possible de l’ajuster de manière à corriger n’importe quel écart observé entre le comportement expérimental et celui prévu par le modèle :
| (74) |
| (75) |
1.9. Description microscopique du gaz parfait
L’équation d’état du gaz parfait, établie par l’expérience et formulée par Emile Clapeyron en 1833 dans son mémoire sur la puissance motrice de la chaleur, est une équation phénoménologique pour laquelle la température est exprimée en degrés Celsius [20] :
| (76) |
| (77) |
| (78) |
| (79) |
| (80) |
| (81) |
| (82) |
| (83) |
1.10. Synthèse
La thermodynamique chimique constitue un modèle prédictif puissant et de large portée de l’évolution des transformations microscopiques. Elle est utilisée avec succès depuis plus d’un siècle dans des domaines variés. Elle présente néanmoins des faiblesses et incohérences :
- i. la thermodynamique chimique est fondée sur le postulat de Gibbs, l’existence du potentiel chimique;
- ii. la thermodynamique chimique est fondée sur l’hypothèse selon laquelle l’entropie d’un équilibre chimique ou de phases s’identifie à l’entropie calorimétrique issue du second principe qui gouverne l’évolution de systèmes qui échangent chaleur et travail ;
- iii. la description de systèmes non idéaux ne peut s’affranchir de l’utilisation de coefficients d’activité de nature empirique. Le caractère ajustable du coefficient d’activité ne confère pas à la thermodynamique chimique le statut de théorie scientifique définie en termes de réfutabilité [26] ;
- iv. les équations qui permettent le calcul des coefficients d’activité sont de portées restreintes à des systèmes particuliers ;
- v. les fonctions thermodynamiques qui gouvernent les équilibres, comme la fonction de Gibbs, s’affranchissent de toute considération géométrique, alors qu’elle joue un rôle primordial dans les modèles thermodynamiques prédictifs les plus performants.
Comme la thermodynamique chimique est perfectible, la question scientifique adressée par cette étude est la suivante :
« Est-il possible de décrire l’équilibre des transformations microscopiques, réaction chimique et transition de phase, sans utiliser l’entropie calorimétrique et le potentiel chimique ? »
2. Théorie cinétique de l’équilibre chimique
2.1. Cadre de l’analyse
Le cadre de la théorie cinétique de l’équilibre chimique est celui du premier principe et celui du second principe restreint aux échanges de travail et chaleur. Ainsi, le potentiel chimique et l’entropie calorimétrique issue du second principe ne seront pas utilisés pour établir la fonction qui gouverne l’équilibre.
2.2. Définition de l’équilibre cinétique d’une réaction chimique
L’équilibre cinétique d’une réaction chimique est défini ainsi :
« L’équilibre chimique d’un système fermé est atteint lorsque la vitesse de formation de chaque constituant chimique est compensée par sa vitesse de disparition. »
Soit une réaction chimique qui se produit dans une phase homogène liquide ou gazeuse (note 3 ) :
| (84) |
2.3. Loi de vitesse d’une réaction chimique
Soit une réaction élémentaire entre deux espèces chimiques A et B, dans une phase homogène liquide ou gazeuse, à température et pression P de 1 bar constantes :
| (85) |
| (86) |
| (87) |
| (88) |
Diagramme du col d’enthalpie standard de transition d’une réaction chimique.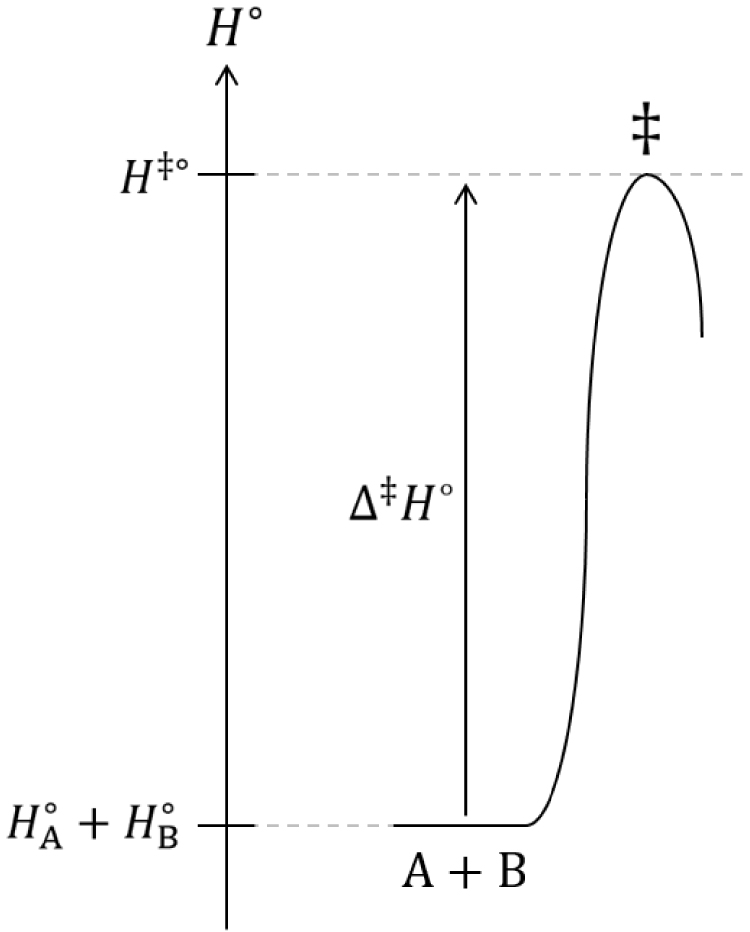
s’apparente au facteur stérique 𝜌 introduit par Cyril Norman Hinshelwood mais il ne s’identifie pas exactement à ce dernier : contrairement au facteur stérique 𝜌, cette probabilité est inférieure à 1 :
| (89) |
| (90) |
2.4. Equation cinétique de l’équilibre chimique
Les grandeurs z±°, p±° et 𝛥‡ H±° utilisées dans cette analyse seront supposées indépendantes de la température et de l’avancement de la réaction. Soit la réaction chimique en phase gazeuse renversable ci-après à température et pression égale à 1 bar constantes :
Diagramme du col d’enthalpie standard de transition d’une réaction chimique.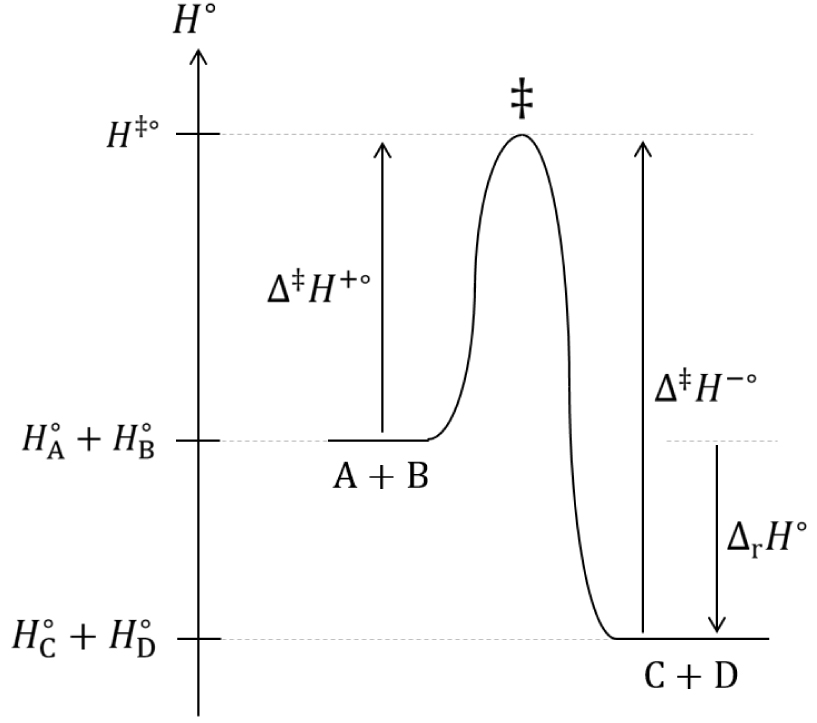
L’enthalpie standard de réaction est :
| (91) |
| (92) |
| (93) |
| (94) |
| (95) |
| (96) |
| (97) |
| (98) |
Evolution du système et de l’enthalpie libre cinétique de réaction d’un état initial arbitraire à l’état d’équilibre.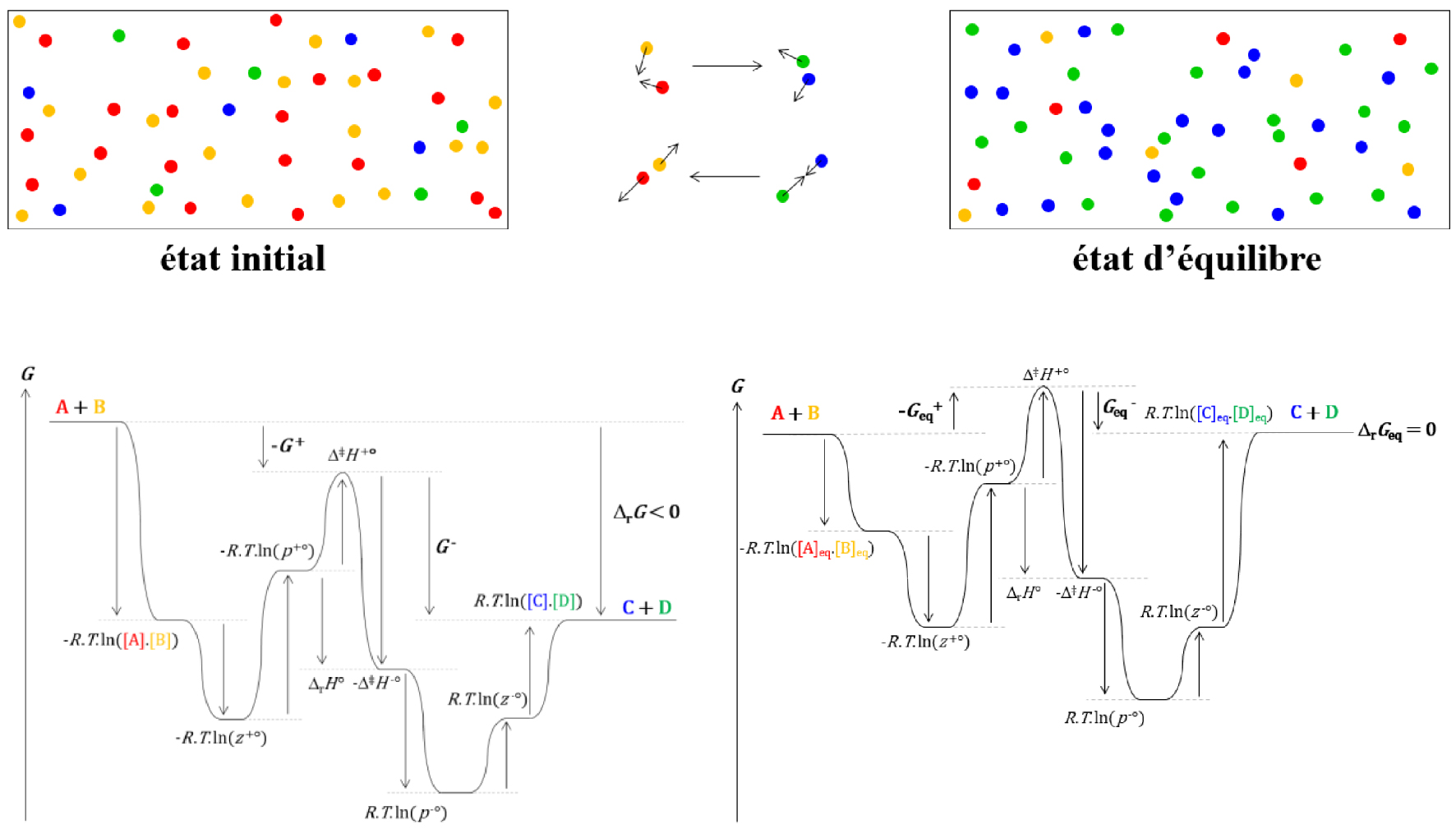
L’enthalpie libre cinétique de réaction 𝛥r G peut se décomposer en deux termes, G+ et G−, qui reflètent respectivement les vitesses de passage des réactifs aux produits et des produits aux réactifs.
| (99) |
| (100) |
| (101) |
| (102) |
| (103) |
| (104) |
| (105) |
| (106) |
| (107) |
| (108) |
| (109) |
| (110) |
| (111) |
| (112) |
2.5. Synthèse
L’équation de l’équilibre a été établie en utilisant une loi cinétique issue de mesures expérimentales de vitesse de réaction. L’entropie calorimétrique et le potentiel chimique n’ont pas été utilisés. Les équations de l’énergie de Gibbs de réaction et de l’enthalpie cinétique de réaction sont formellement identiques :
| (113) |
| (114) |
| (115) |
| (116) |
| (117) |
| (118) |
| (119) |
3. Etude thermodynamique et cinétique d’équilibres chimiques
3.1. Cadre de l’analyse
Ce chapitre confronte les modèles thermodynamique et cinétique aux observations expérimentales d’équilibres chimiques dans les conditions standards. Les enthalpies et entropies standards cinétique et thermodynamique seront considérées indépendantes de la température.
3.2. Formation d’un complexe d’association intermoléculaire
Soit l’équilibre d’association de la -cyclodextrine avec l’acide salicylique en solution aqueuse (Figure 8).
Equilibre d’association de la β-cyclodextrine (A) avec l’acide salicylique (B).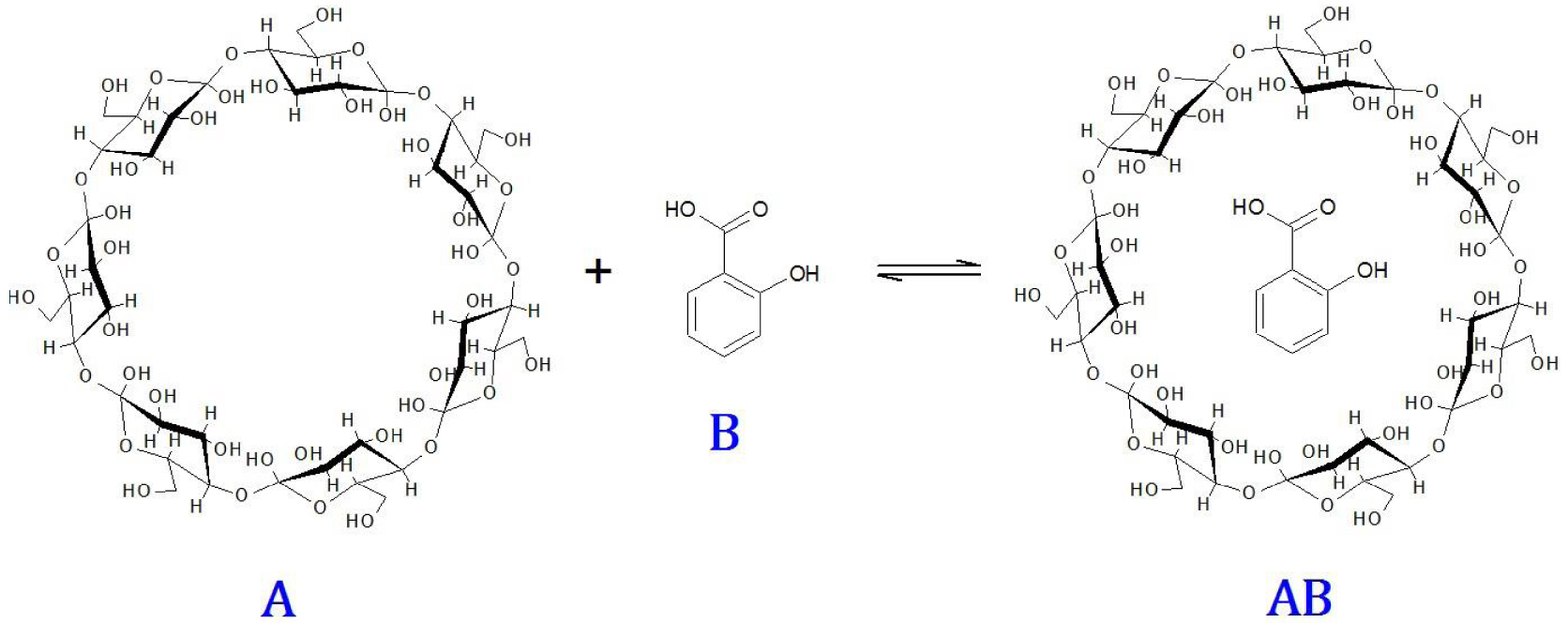
Cette réaction d’association se produit dans le sens direct suite à une collision entre A et B. Dans le sens opposé, le complexe d’association AB est dissocié lors de sa collision avec n’importe quelle molécule X du milieu : A, B, AB ou H2O. L’équation chimique de l’équilibre s’écrit, en indiquant entre crochets les espèces qui entrent en collision :
| (120) |
| (121) |
| (122) |
| (123) |
| (124) |
| (125) |
| (126) |
| (127) |
| (128) |
| (129) |
Evolution de la constante d’équilibre cinétique (bleu) et calorimétrique (rouge) avec l’inverse de la température de l’association entre la -cyclodextrine et l’acide salicylique.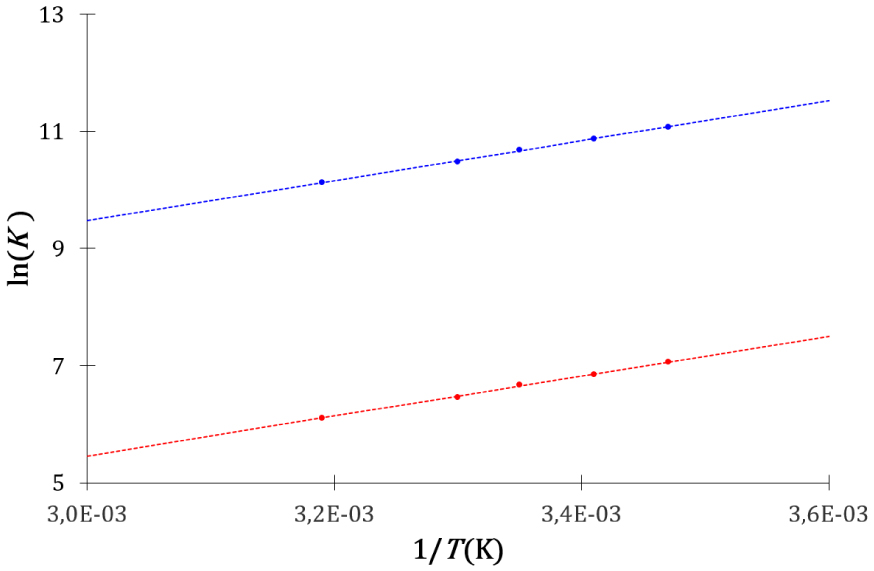
La manière différente de définir les constantes d’équilibre cinétique et thermodynamique est arbitraire, elle ne constitue pas un critère pour comparer leurs performances prédictives. Les descriptions cinétique et thermodynamique conduisent à des équations concordantes avec la dépendance en température de la constante d’équilibre établie par l’expérience.
3.3. Estérification de Fischer
Soit l’équilibre de l’estérification de Fischer entre un acide carboxylique, l’acide lévulinique, et le n-butanol, un alcool d’alkyle primaire (Figure 10).
Equation de l’équilibre de l’estérification de Fischer entre l’acide lévulinique et le n-butanol.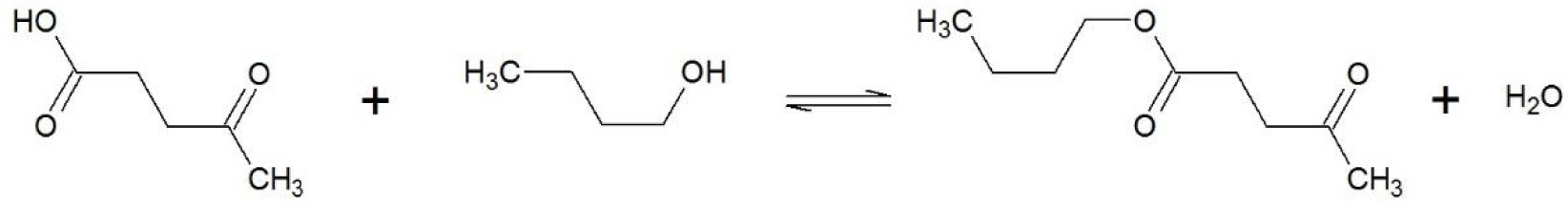
Cette réaction est catalysée par un acide de Bronsted qui est soit un acide ajouté au milieu réactionnel, soit présent dans le milieu. En effet, cette réaction est connue pour être catalysée par un de ses réactifs, l’acide carboxylique. Le mécanisme de l’estérification de Fischer se décompose en cinq étapes élémentaires (Figure 11). Les espèces qui entrent en collision préalablement à chaque réaction sont indiquées entre crochets. L’espèce chimique X se rapporte à n’importe quelle espèce chimique présente dans le milieu réactionnel : réactif, produit ou solvant.
Mécanisme réactionnel de l’estérification de Fischer, les mouvements électroniques ont été omis par souci de clarté.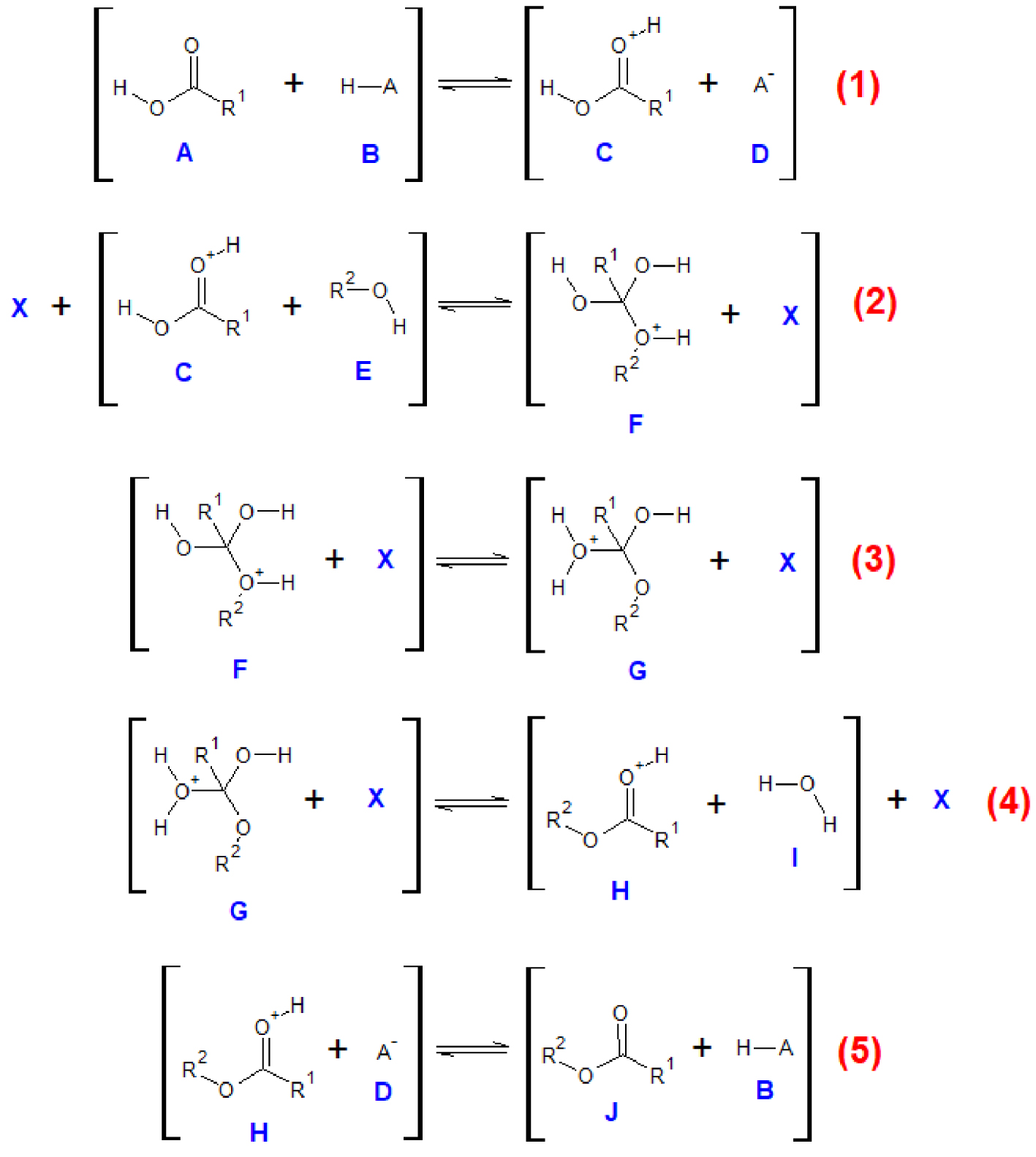
Les équations de l’équilibre cinétique sont :
| (130) |
| (131) |
| (132) |
| (133) |
| (134) |
| (135) |
| (136) |
| (137) |
| (138) |
| (139) |
| (140) |
| (141) |
| (142) |
| (143) |
| (144) |
Evolution du logarithme népérien de la constante d’équilibre avec l’inverse de la température de la réaction d’estérification de l’acide lévulinique par le n-butanol.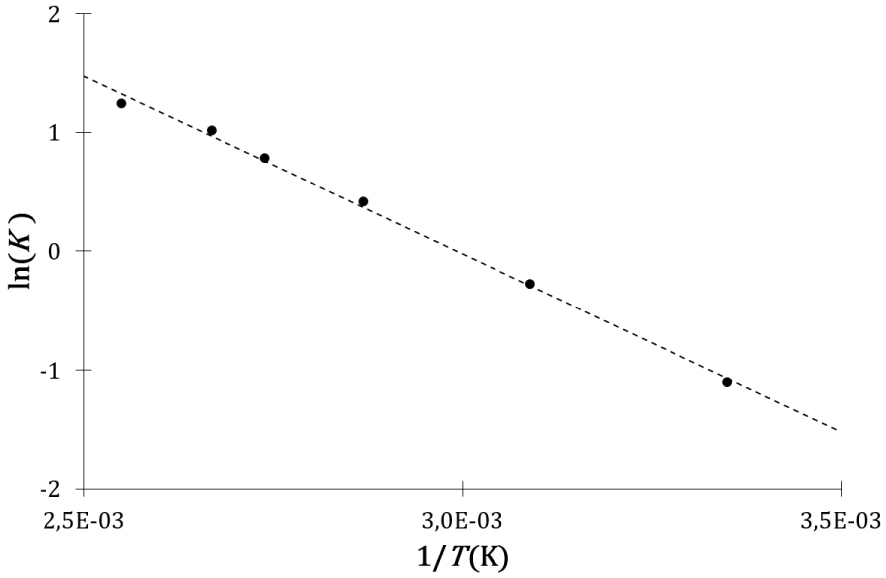
Une fois encore, même si l’interprétation de l’entropie des descriptions cinétique et calorimétrique est différente, les expressions mathématiques des constantes d’équilibre cinétique et thermodynamique sont formellement identiques et en accord avec l’expérience, c’est-à-dire avec une évolution linéaire du logarithme népérien de la constante d’équilibre avec l’inverse de la température.
3.4. Catalyse de l’estérification de Fischer par un acide fort
Soit l’équilibre d’estérification de l’acide éthanoïque par l’éthanol :
Constantes et temps d’établissement de l’équilibre d’estérification de l’acide éthanoïque par l’éthanol
| T | K | Catalyseur | teq | Référence |
|---|---|---|---|---|
| 323 K | 3,4 | Aucun | 22 jours | Calvar et al. [32] |
| 323 K | 4,6 | RSO3H 0,2 M | 3 heures | He et al. [33 ] |
L’ajout d’une faible quantité de l’hydrogénosulfate de 1-sulfobutyl-3-méthylimidazolium (RSO3H) ne modifie pas significativement la constante de l’équilibre mais accélère dans des proportions considérables la vitesse avec laquelle il s’établit. Les expressions de la constante d’équilibre et de l’entropie standard de réaction cinétiques de l’estérification de Fischer, établies dans le chapitre précédent, sont respectivement :
| (145) |
| (146) |
| (147) |
| (148) |
| (149) |
Diagrammes réactionnels cinétique (gauche) et thermodynamique (droite) de l’estérification de Fischer.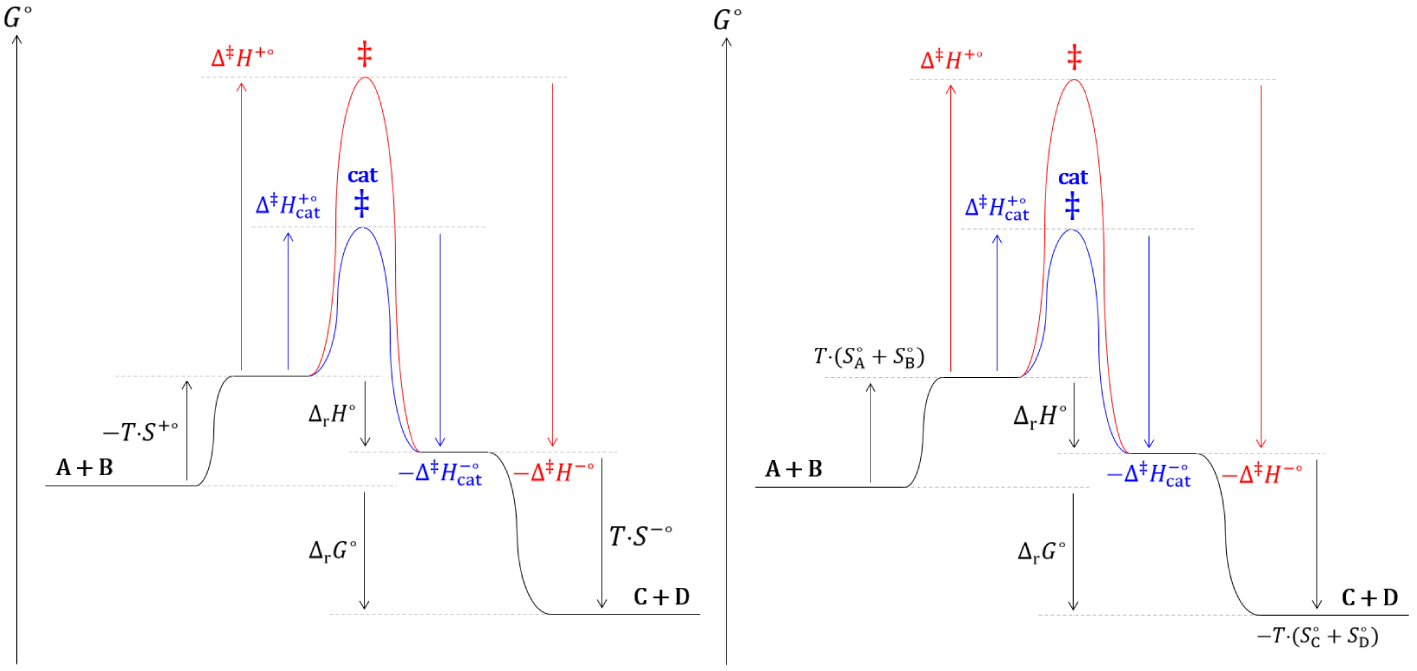
Les analyses thermodynamique et cinétique d’une réaction chimique catalysée prévoient un comportement identique et en accord avec les observations expérimentales.
3.5. Catalyse enzymatique d’une transestérification
Les énantiomères réactifs engagés dans certaines réactions catalysées par une molécule chirale sont synthétisés en produits énantiomères dans des proportions différentes. L’équilibre de la transestérification du butanoate de 2,2,2-trifluoroéthyle par les énantiomères du 3-fluoro-1-phénoxypropan-2-ol (Figure 14), dont la transformation est catalysée par l’enzyme Candida Antartica [34], présente ce caractère.
Equilibres de transestérification du butanoate de 2,2,2-trifluoroéthyle par les énantiomères et S du 3-fluoro-1-phénoxypropan-2-ol.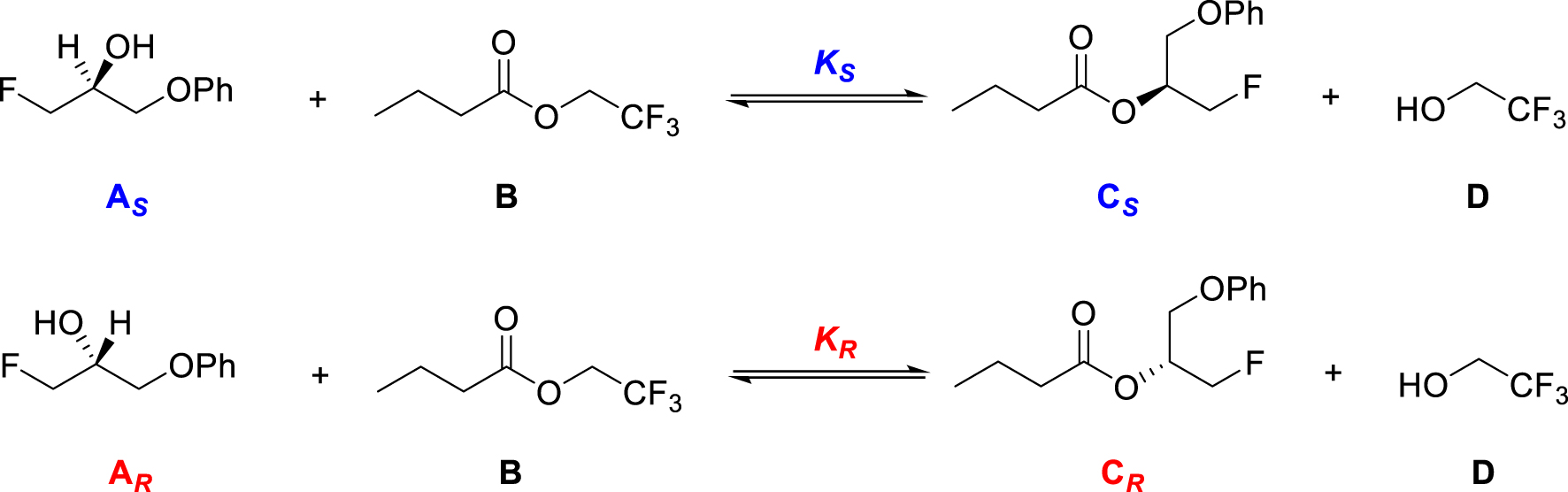
A l’équilibre, le facteur d’énantiosélectivité de cette transestérification, de 490, est supérieur à 1, ce qui signifie que la constante d’équilibre de la réaction avec l’énantiomère S est supérieure à celle de la réaction avec l’énantiomère R :
| (150) |
| (151) |
Le mécanisme de transestérification résulte d’une succession de réactions élémentaires consécutives, que la réaction soit catalysée ou non (cf. estérification de Fischer), il est donc possible d’exprimer les constantes d’équilibre cinétique ainsi :
| (152) |
| (153) |
| (154) |
| (155) |
Les enthalpies de transestérification et R sont égales car les enthalpies de formation de deux énantiomères sont égales.
| (156) |
| (157) |
La séquence peptidique du domaine catalytique de l’enzyme Candida Antartica (Trp104, His224, Ser105) permet d’établir avec l’énantiomère et l’ester B achiral un réseau de liaisons hydrogènes et d’interactions de Van der Waals qui augmente la vitesse de formation de l’énantiomère S, ce qui se traduit par une valeur plus élevée du rapport des probabilités de la transestérification S :
| (158) |
| (159) |
| (160) |
| (161) |
Diagramme réactionnel cinétique des transestérifications du butanoate de 2,2,2-trifluoroéthyle par les énantiomères et S du 3-fluoro-1-phénoxypropan-2-ol.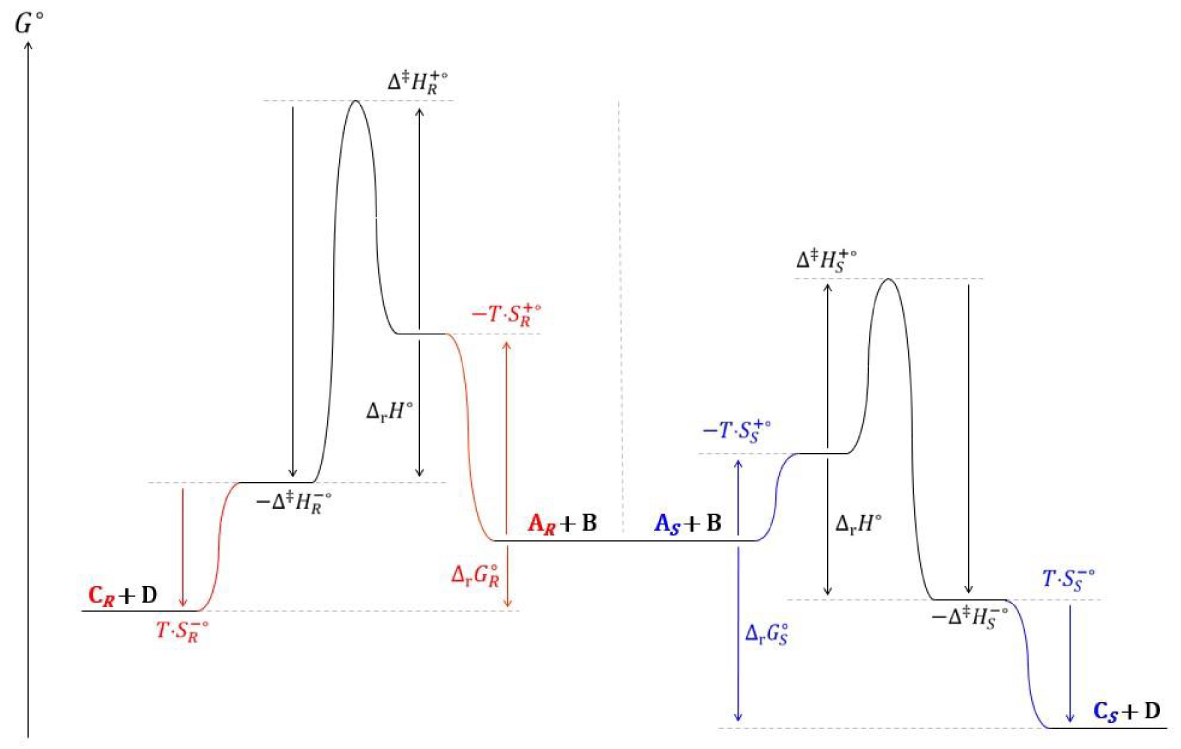
La description cinétique de cet équilibre est satisfaisante. Pour le modèle thermodynamique, les enthalpies et capacités calorifiques standards de deux énantiomères sont égales :
| (162) |
| (163) |
Ces deux égalités induisent l’égalité entre les enthalpies de transestérifications des énantiomères d’une part et de leurs entropies d’autre part, soient respectivement :
| (164) |
| (165) |
| (166) |
| (167) |
Diagramme réactionnel thermodynamique des transestérifications du butanoate de 2,2,2-trifluoroéthyle par les énantiomères et S du 3-fluoro-1-phénoxypropan-2-ol.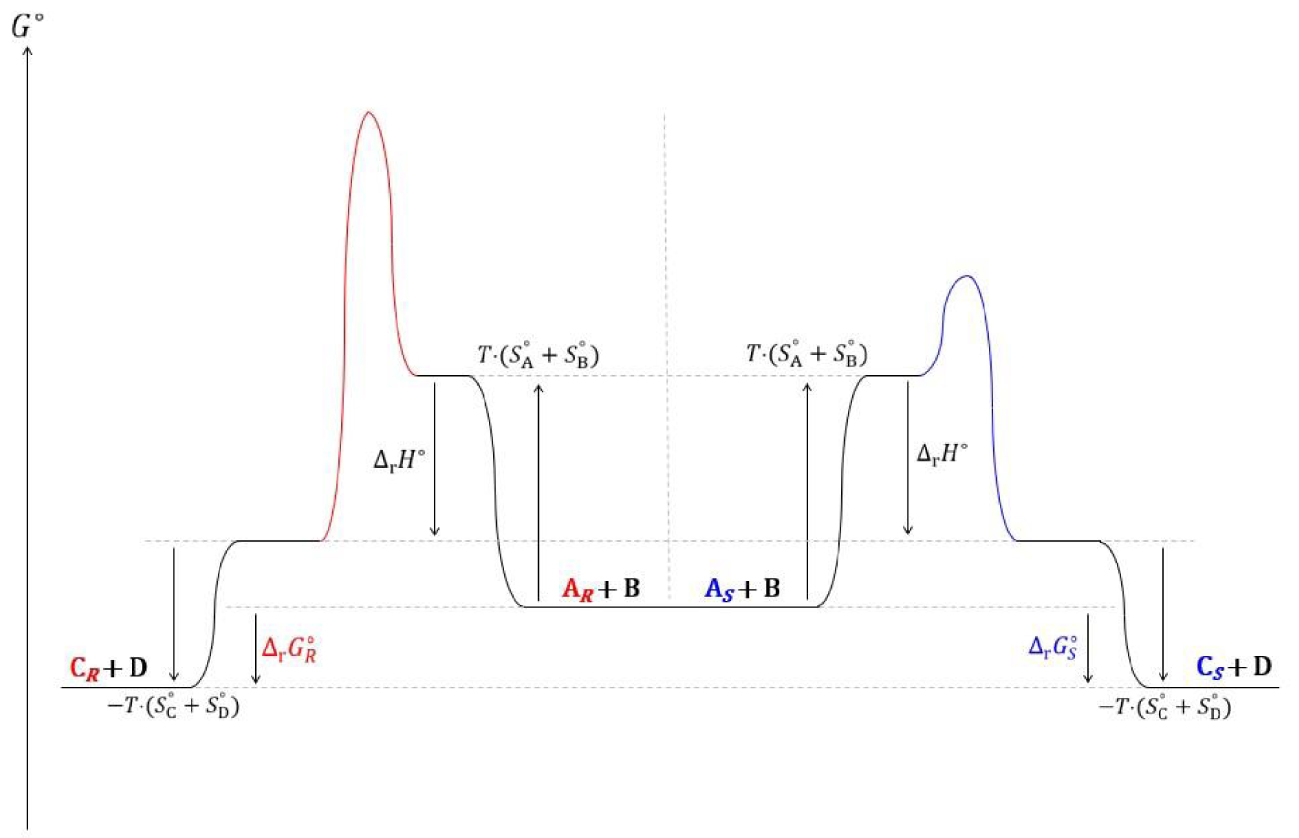
Le modèle cinétique de l’équilibre offre une description plus cohérente de l’équilibre de transestérification enzymatique que le modèle thermodynamique. Il conviendrait à la lumière de cette description de nuancer deux types de catalyse. La première, qui ne modifie pas la valeur de la constante d’équilibre (i.e. estérification de Fischer), est de nature enthalpique, la deuxième, qui peut drastiquement l’affecter (i.e. transestérification énantiosélective), est de nature entropique.
3.6. Dissociation de mono- et di-acides faibles
Les mono- et di-acides carboxyliques, solubles dans l’eau à basse concentration, présentent un caractère acide (Figure 17).
Equilibres de dissociation de première acidité de mono- et di-acides carboxyliques linéaires en solution aqueuse.
La constante d’acidité est définie par le produit de la constante d’équilibre d’une réaction acide-base avec la concentration de l’eau, supposée constante en solution diluée :
| (168) |
| (169) |
Constantes de première acidité exprimées en échelle logarithmique de réactions de dissociation de mono- et di-acides carboxyliques issues de la littérature
| Monoacide (M) | p | Référence | Diacide (D) | pKa | Référence | |
|---|---|---|---|---|---|---|
| HCO2H | 3,76 | Kim et al. [35 ] | HO2CCO2H | 1,32 | Mateus et al. [36 ] | |
| Dasgupta et al. [37] | 1,27 | Lugo et al. [38] | ||||
| H(CH3)1CO2H | 4,74 | Espinoza et al. [39 ] | HO2C(CH2)1CO2H | 2,83 | Khalil et al. [40 ] | |
| H(CH3)2CO2H | 4,876 | Lomas et al. [41 ] | 2,86 | Gelles et al. [42 ] | ||
| Körtum et al. [43] | HO2C(CH2)2CO2H | 4,19 | Khalil et al. [40 ] | |||
| Mansfield et al. [44] | HO2C(CH2)3CO2H | 4,34 | German et al. [45 ] | |||
| 4,81 | Chen et al. [46] | HO2C(CH2)4CO2H | 4,46 | Radalla et al. [47 ] | ||
| H(CH3)3CO2H | 4,86 | Headley et al. [48 ] | Jeffery et al. [49 ] | |||
| Gane et al. [50] | ||||||
| HO2C(CH2)5CO2H | 4,51 | Radalla et al. [47 ] | ||||
| Jeffery et al. [49] | ||||||
| Gane et al. [50] | ||||||
| HO2C(CH2)6CO2H | 4,52 | Jeffery et al. [49 ] | ||||
Le pKa de première acidité des mono- et di-acides carboxyliques augmente avec la longueur de la chaîne hydrocarbonée, il atteint une valeur limite pour des longueurs élevées de la chaîne hydrocarbonée (Figure 18).
Evolution du pKa de première acidité de mono- et di-acides carboxyliques avec la longueur de la chaîne hydrocarbonée.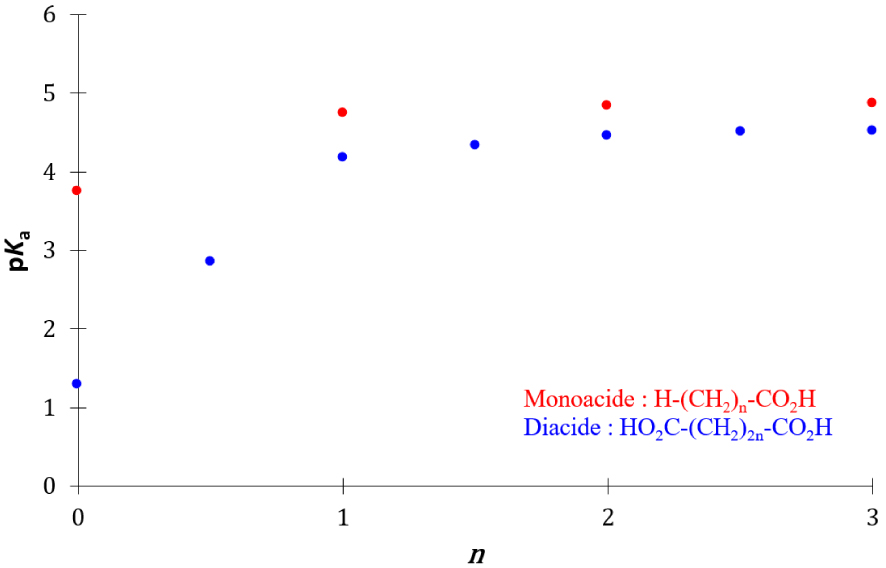
Cet effet d’atténuation du pKa est en accord avec celui de l’atténuation des effets inductifs avec la longueur de la chaîne aliphatique. Les vitesses des réactions directe et inverse des monoacides carboxyliques sont respectivement :
| (170) |
| (171) |
| (172) |
| (173) |
| (174) |
| (175) |
| (176) |
| (177) |
| (178) |
| (179) |
| (180) |
Diagramme réactionnel cinétique de la dissociation d’acides mono- et di-carboxyliques.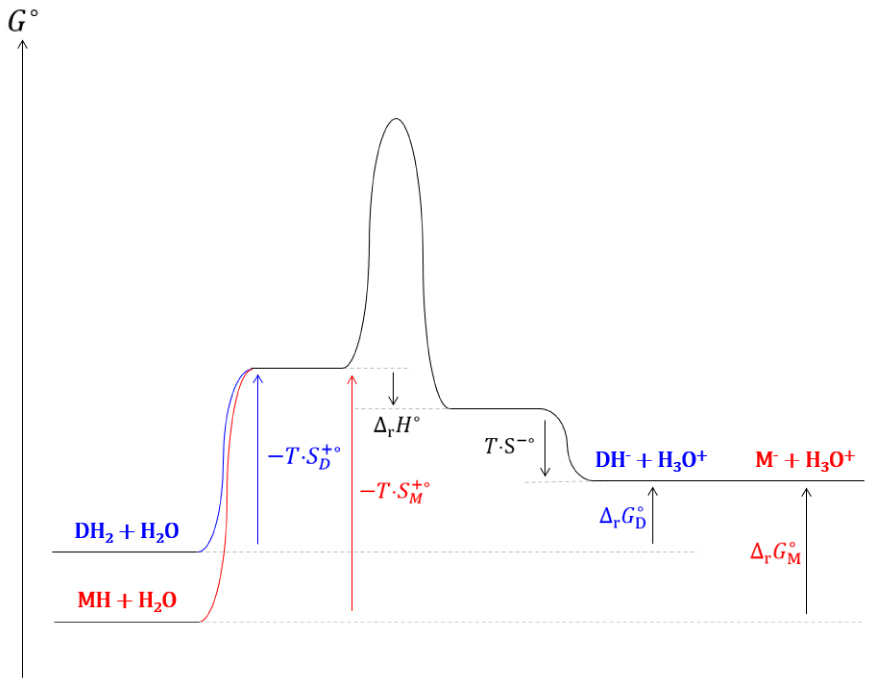
3.7. Synthèse
Les modèles thermodynamique et cinétique de l’équilibre chimique prévoient une évolution linéaire du logarithme népérien de la constante d’équilibre avec l’inverse de la température, en accord avec le comportement de nombreuses réactions. Les deux approches donnent une explication satisfaisante de la catalyse de certaines réactions. En revanche, le modèle cinétique est plus approprié que le modèle thermodynamique pour comprendre un équilibre chimique où la géométrie des réactifs lors de leur collision favorise sélectivement une des réactions possibles, la transestérification enzymatique. L’analyse cinétique de l’équilibre permet de distinguer deux types de catalyse : la catalyse enthalpique pour laquelle l’enthalpie de l’état de transition diminue et la catalyse entropique pour laquelle la probabilité de réaction suite à une collision augmente indépendamment de l’enthalpie. Le modèle cinétique de l’équilibre permet également de quantifier l’écart entre les constantes d’acidité de mono- et di-acides carboxyliques linéaires sur la base du mécanisme collisionnel en amont de la réaction chimique.
4. Théorie cinétique de l’équilibre de fusion
4.1. Cadre de l’analyse
Le cadre de la théorie cinétique de l’équilibre de fusion est le même que celui de la théorie cinétique de l’équilibre chimique présentée dans le troisième chapitre, à savoir celui du premier principe et celui du second principe restreint aux échanges de travail et chaleur. Ainsi, le potentiel chimique et l’entropie calorimétrique issue du second principe ne seront pas utilisés pour établir la fonction qui gouverne l’équilibre de fusion.
4.2. Equilibre cinétique d’une fusion
L’équilibre cinétique d’une fusion est défini de manière analogue à l’équilibre cinétique d’une réaction chimique (note 4 ) :
« L’équilibre de fusion d’un constituant chimique d’un système fermé est atteint lorsque la vitesse de sa fusion est compensée par sa vitesse de solidification. »
Soit l’équilibre de fusion de l’espèce chimique A pure ou dans un mélange liquide homogène avec d’autres espèces chimiques immiscibles dans la phase solide de A :
| (181) |
4.3. Lois de vitesse d’une fusion et d’une solidification
La fusion est la transformation d’une espèce chimique A (atome, molécule, paire d’ions, complexe d’association) de l’état solide à l’état liquide. Réciproquement, la solidification est la transformation d’une espèce chimique A de l’état liquide à l’état solide. L’interface () de l’équilibre de fusion est constituée du domaine de contact entre le solide et le liquide où les propriétés de l’espèce chimique A sont différentes du cœur du solide (s) et du cœur du liquide (l) (Figure 20).
Schéma de l’équilibre de fusion.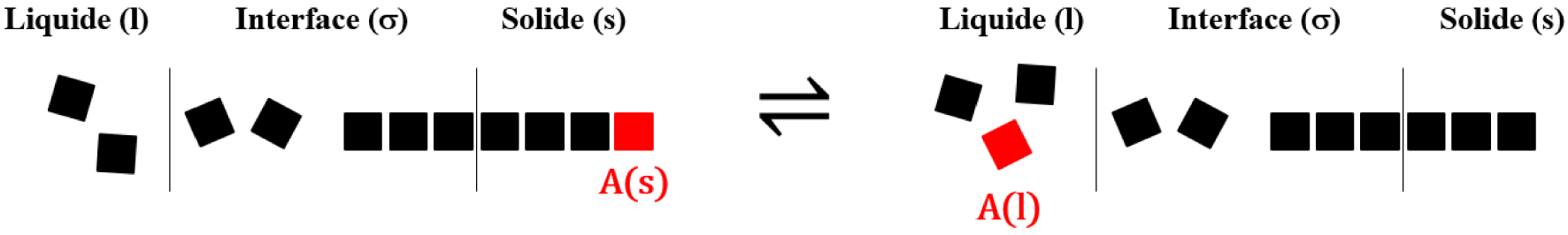
Il est important de préciser afin d’éviter toute confusion que les segments verticaux entre (l) et (σ) d’une part, et entre (σ) et (s) d’autre part, ne constituent pas des interfaces. Ces segments verticaux sont les lignes de démarcation de l’interface (σ) avec le liquide (l) et le solide (s). Dans cette description, l’interface (σ) est un volume. Un état de transition, de même nature que celui existant pour les réactions chimiques en phase homogène, est franchi lors de la fusion et de la solidification (Figure 21).
Diagramme du col d’enthalpie standard de transition d’une fusion.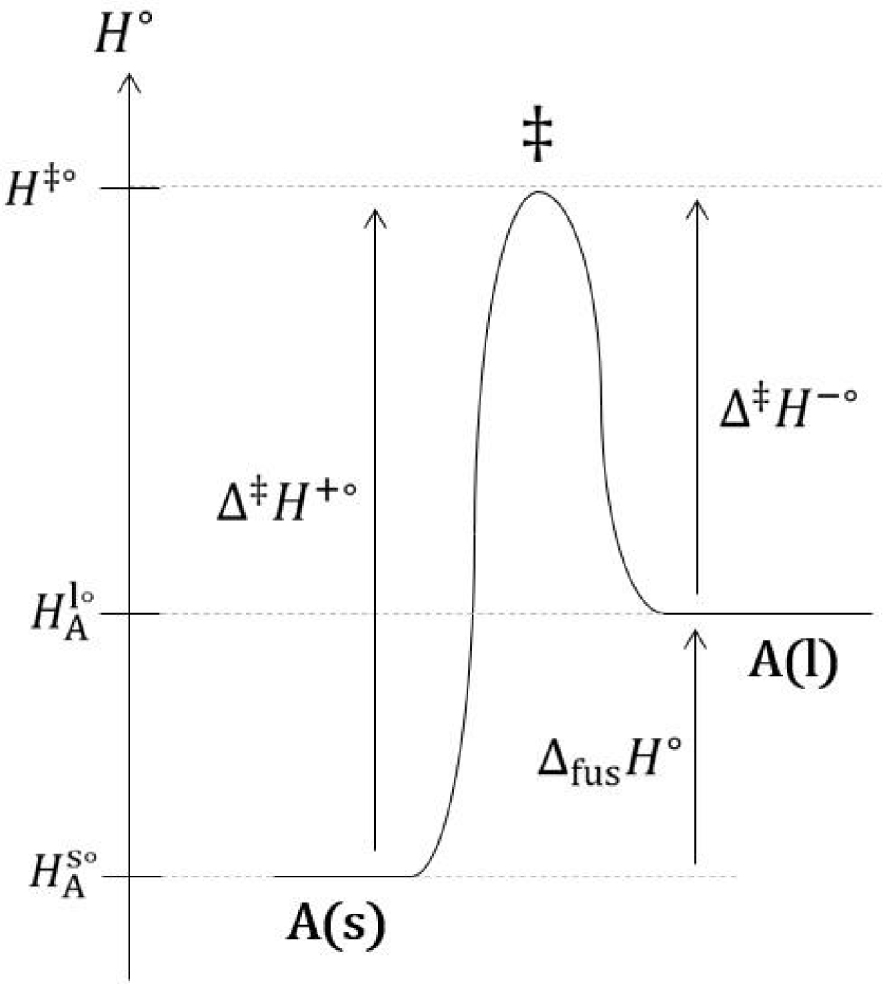
L’expression de l’enthalpie standard de fusion est :
| (182) |
| (183) |
| (184) |
4.4. Equation cinétique d’évolution d’une fusion
L’équilibre de fusion s’exprime par l’égalité entre les vitesses de fusion et solidification :
| (185) |
| (186) |
| (187) |
| (188) |
| (189) |
| (190) |
L’enthalpie libre cinétique standard de fusion déduite de l’équation de l’équilibre est nulle :
| (191) |
| (192) |
Diagramme réactionnel cinétique de l’équilibre de fusion d’un corps pur soumis à une pression de 1 bar.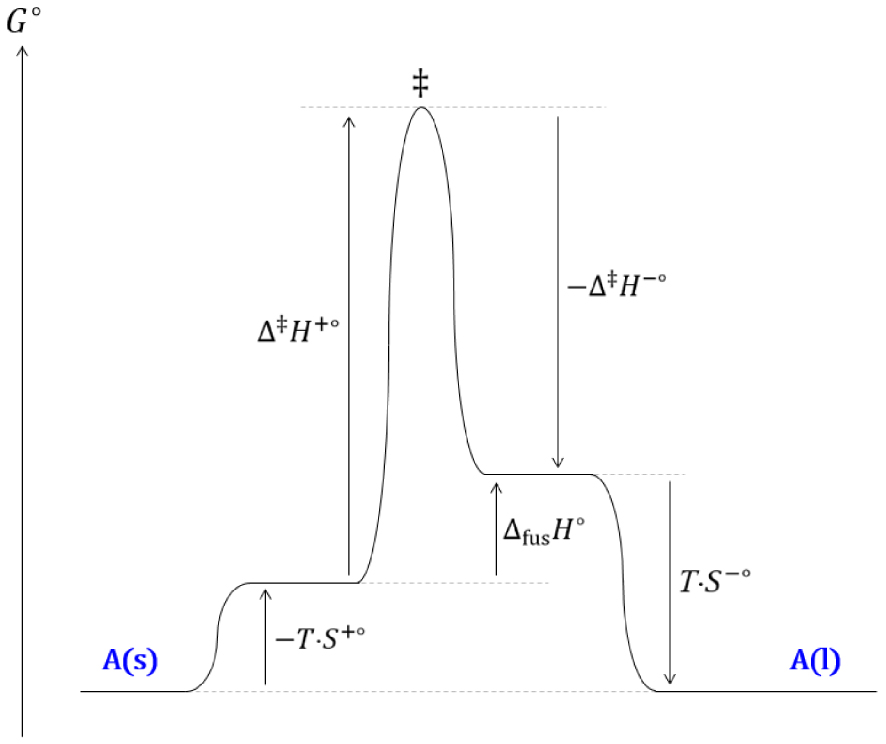
Dans le cas d’un mélange de A avec d’autres espèces qui forment une phase liquide homogène avec la phase liquide de A mais qui sont immiscibles dans la phase solide de A, les vitesses de fusion et solidification sont respectivement :
| (193) |
| (194) |
L’équilibre de fusion s’exprime par l’égalité entre les vitesses de fusion et solidification :
| (195) |
| (196) |
| (197) |
| (198) |
| (199) |
4.5. Synthèse
L’équation de l’équilibre de fusion a été établie en utilisant des lois cinétiques. De manière analogue à l’étude cinétique de l’équilibre chimique, l’entropie calorimétrique et le potentiel chimique n’ont pas été utilisés pour établir l’équation de l’équilibre de fusion. Les équations thermodynamique et cinétique de l’équilibre de fusion sont formellement très proches, respectivement :
| (200) |
| (201) |
Les enthalpies de fusion thermodynamique et cinétique sont, en vertu du premier principe de la thermodynamique, strictement égales :
| (202) |
| (203) |
| (204) |
5. Etude thermodynamique et cinétique d’équilibres de fusion
5.1. Cadre de l’analyse
Ce chapitre confronte les modèles thermodynamique et cinétique aux observations expérimentales d’équilibres de fusion à la pression standard. Les enthalpies et entropies standards cinétique et thermodynamique seront considérées indépendantes de la température. Pour le modèle cinétique, la dilution d’une espèce chimique dans sa phase liquide est quantifiée par le rapport entre sa concentration dans le mélange liquide et sa concentration [A]° dans sa phase liquide lorsqu’il est pur :
| (205) |
| (206) |
5.2. Mélange binaire idéal de sels ioniques
Soient les équilibres de fusion du système binaire à invariant eutectique du mélange entre le chlorure de sodium et le chlorure de rubidium :
Les équations cinétiques de ces équilibres de fusion sont respectivement :
| (207) |
| (208) |
Avec :
| (209) |
| (210) |
| (211) |
Les propriétés physiques issues de la littérature du chlorure de sodium, du chlorure de rubidium et de leur mélange, sont renseignées dans les Tableaux 3 et 4.
Propriétés physiques du mélange NaCl/RbCl
| 823 | 1,69 | 2,39 |
| Pelton et al. [53] | Janz [51] | Janz [51] |
| Nasch et al. [52] | Nasch et al. [52] | |
Le diagramme de phases expérimental du mélange entre le chlorure de sodium et le chlorure de rubidium [53] et les diagrammes de phases théoriques issus des modèles thermodynamique (Annexe 2) et cinétique (Annexe 3), calculés avec les valeurs physiques des Tableaux 3 et 4, sont tracés sur la Figure 23.
Diagrammes de phases liquide–solide composition-température du mélange NaCl–RbCl : expérimental (disques noirs), théorique thermodynamique (traits rouges) et théorique cinétique (traits bleus).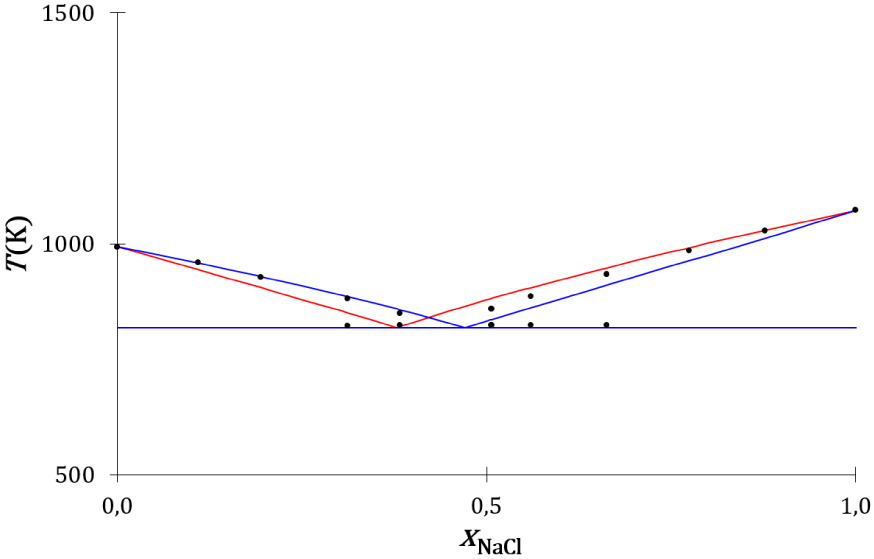
Les liquidus du chlorure de sodium et du chlorure de rubidium déduits du modèle thermodynamique, sont :
| (212) |
| (213) |
| (214) |
Les équations cinétique et thermodynamique de l’équilibre de fusion sont proches. Elles ne se différencient que par la variable de composition du mélange : la fraction de concentration pour le modèle cinétique et la fraction molaire pour le modèle thermodynamique. Les liquidus thermodynamiques sont présentés sur la Figure 23, ils sont proches des liquidus cinétiques et proches des liquidus expérimentaux.
5.3. Mélange binaire non idéal de sels ioniques
Soient les équilibres de fusion du mélange du chlorure de sodium avec l’eau :
Les équations cinétiques de l’équilibre de fusion du chlorure de sodium et de l’eau dans le mélange sont :
| (215) |
| (216) |
Avec :
| (217) |
| (218) |
| (219) |
Les liquidus thermodynamique (Annexe 4) et cinétique (Annexe 5), déterminés avec les propriétés des corps purs renseignées dans le Tableau 5, sont comparés aux liquidus expérimentaux [54] (Figure 24) : le diagramme de phases expérimental présenté ne prend pas en compte la formation de l’hydrate du chlorure de sodium qui se forme à basse température, il utilise le point de fusion eutectique expérimental métastable du mélange entre NaCl et H2O reporté dans la littérature (Tableau 6).
Propriétés physiques de NaCl et H2O
| Composé | 𝛥fus H° (kJ∕mol) | |||
|---|---|---|---|---|
| NaCl | 58,4 | 1,56 | 1072 | 28,0 |
| IUPAC [61] | Janz [51] | Pelton et al. [53] | Kubaschewski et al. [54] | |
| Nasch et al. [52] | ||||
| H2O | 18,0 | 1,00 | 273,2 | 6,0 |
| IUPAC [61] | Wagner et al. [57] | Giauque et al. [58] | ||
Propriétés physiques du mélange eutectique NaCl–H2O
| Teut (K) | ||
|---|---|---|
| 249 | 2,00 | 1,00 |
| Li et al. [56] | Janz [51] | |
| Nasch et al. [52] | ||
Diagrammes de phases liquide–solide composition-température du mélange NaCl–H2O expérimental (disques noirs) et calculés avec les modèles thermodynamique (traits rouges), cinétique (traits bleus) et cinétique avec prise en compte de la formation de la première sphère de solvatation de la paire d’ions Na+, Cl- (courbes vertes).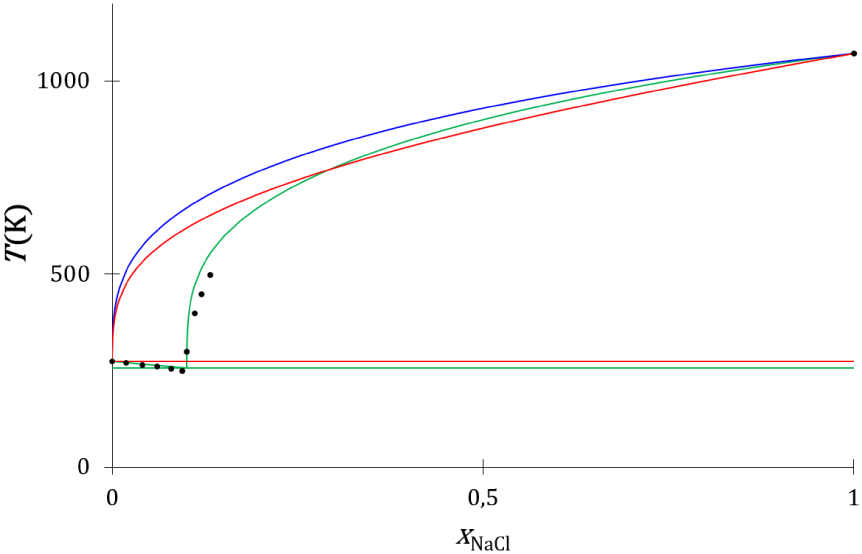
Les liquidus du chlorure de sodium déterminés par les modèles thermodynamique (courbe rouge) et cinétique (courbe bleu) s’écartent fortement du liquidus expérimental (disques noirs). En effet, à la composition eutectique, la température de fusion expérimentale est de 249,5 K, celle du liquidus thermodynamique est de 607 K, et celle du liquidus cinétique de 658 K.
Le chlorure de sodium en solution dans l’eau à haute concentration est une paire d’ions solvatée par des molécules d’eau. Le nombre de molécules d’eau de la première sphère de solvatation déterminée par des modèles théoriques est compris entre 8 et 10 (Tableau 7).
Nombre de molécules d’eau de la première sphère de solvatation du chlorure de sodium déterminée par modélisation
| NH 2O | Modèle | Référence |
|---|---|---|
| 8 | Dynamique moléculaire (SHAKE) | Belch et al. [59] |
| 10 | Ab initio (Hartree–Fock) | Petersen et al. [60] |
Tableau d’avancement de formation de la première sphère de solvatation du chlorure de sodium dans son mélange avec l’eau
| Espèce | Na+Cl− (aq) | 9H2O(l) | Na+Cl−(H2O)9(aq) |
|---|---|---|---|
| Etat initial | xNaCl | 1 − xNaCl | 0 |
| Etat final | 0 | ||
Soit la réaction de formation de la première sphère de solvatation de la paire d’ions du chlorure de sodium, pour un nombre de molécules d’eau de 9, intermédiaire entre les deux valeurs issues de la littérature :
La collision d’une paire d’ions du chlorure de sodium solvatée avec la surface d’un cristal de glace provoque la solidification (haut) tandis que la collision d’une paire d’ions avec la surface d’un cristal de chlorure de sodium non (bas).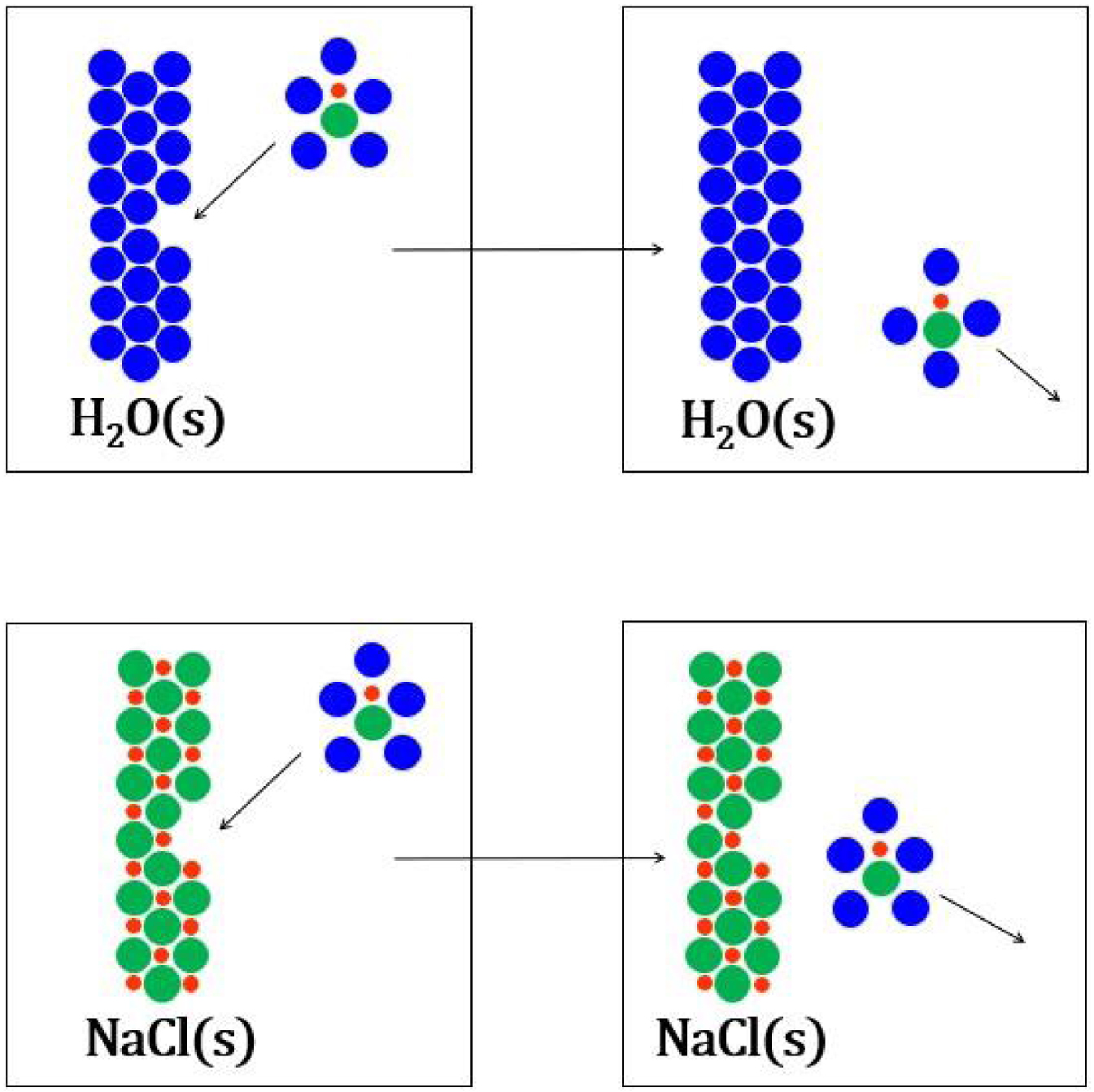
La situation est différente pour la solidification de l’eau car les molécules d’eau de la première sphère de solvatation sont au contact de la surface des cristaux de glace lors d’une collision (Figure 25). D’après ce qui précède, la solidification de l’eau devrait être faiblement affectée dans le mélange, l’équation cinétique du liquidus de l’eau n’est donc pas modifiée :
| (220) |
Le liquidus cinétique du chlorure de sodium calculé en prenant en compte la composition statistique de paires d’ions libres, c’est-à-dire non solvatées (Annexe 6) est :
| (221) |
| (222) |
| (223) |
Le diagramme de phases construit avec les liquidus cinétiques de l’eau et du chlorure de sodium qui prennent en considération la formation de la première sphère de solvatation est proche du diagramme de phases expérimental (Figure 24, traits verts). Cette description cinétique de l’équilibre ne fait pas appel à un coefficient d’activité de nature empirique, elle utilise une caractéristique du système, le nombre de molécules d’eau par paire d’ions solvatée issu de mesure expérimentales, et prend en compte la dynamique collisionnelle de la solidification. Cet effet entropique de nature cinétique est à l’origine de la grande différence de température de fusion eutectique des mélanges du chlorure de sodium avec le chlorure de rubidium et l’eau en particulier, et des sels ioniques en solution aqueuse en général. La description thermodynamique du système à invariant eutectique chlorure de sodium-eau ne peut pas s’affranchir de l’utilisation d’un coefficient d’activité de nature empirique [12].
5.4. Mélange binaire idéal de molécules organiques
Soient les équilibres de fusion du système binaire de l’acide laurique avec l’acide oléique (Figure 26) :
Structures de l’acide laurique (LA) et de l’acide oléique (OL).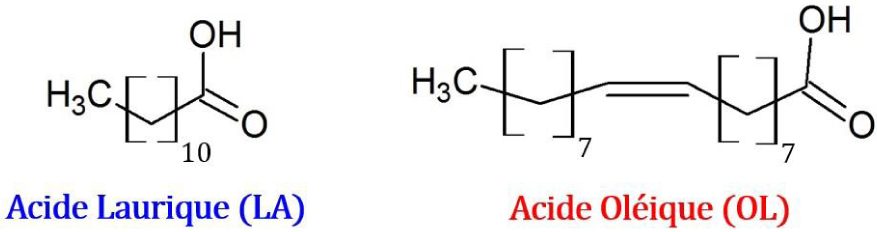
Les équations cinétiques de ces équilibres de fusion sont respectivement :
| (224) |
| (225) |
Avec :
| (226) |
| (227) |
| (228) |
Les masses molaires, densités, températures et enthalpies de fusion de l’acide laurique et de l’acide oléique issues de la littérature sont renseignées dans le Tableau 9.
Propriétés physiques de l’acide laurique et de l’acide oléique
| Composé | 𝛥fus H° (kJ∕mol) | |||
|---|---|---|---|---|
| Acide laurique | 200,3 | 0,87 | 317,4 | 36,6 |
| IUPAC [61] | Hossein et al. [62] | Tohru et al. [63] | Tohru et al. [63] | |
| Acide oléique | 282,5 | 0,89 | 286,5 | 39,6 |
| IUPAC [61] | Hossein et al. [62] | Tohru et al. [63] | Tohru et al. [63] | |
Le diagramme de phases expérimental du mélange entre l’acide laurique et l’acide oléique [63] et les diagrammes de phases théoriques thermodynamique (Annexe 7) et cinétique (Annexe 8) calculés avec les valeurs du Tableau 9 sont présentés sur la Figure 27.
Diagrammes de phases liquide–solide composition-température du mélange acide laurique-acide oléique expérimental (disques noirs), thermodynamique (traits rouges) et cinétique (traits bleus).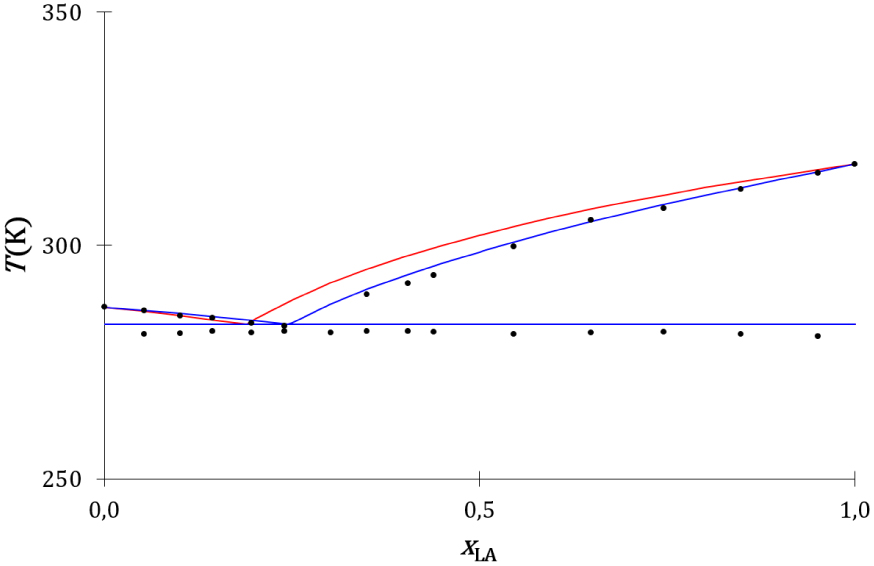
Les liquidus cinétique et thermodynamique sont proches l’un de l’autre et proches des liquidus expérimentaux. Leurs équations ne se différencient que faiblement : la fraction de concentration est utilisée pour le modèle cinétique et la fraction molaire pour le modèle thermodynamique. Aucune différence significative n’est observée entre les descriptions cinétique et thermodynamique de l’équilibre de fusion du mélange binaire à invariant eutectique entre l’acide laurique et l’acide oléique.
Propriétés physiques de l’acide laurique et de la lidocaïne
| Composé | M (g∕mol) | 𝛥fus H° (kJ∕mol) | ||
|---|---|---|---|---|
| Acide laurique | 200,3 | 0,87 | 317,4 | 36,6 |
| IUPAC [61] | Hossein et al. [62] | Tohru et al. [63] | Tohru et al. [63] | |
| Lidocaïne | 234,3 | 1,04 | 340,7 | 16,4 |
| IUPAC [61] | Lazerges et al. [65] | Lazerges et al. [66] | Lazerges et al. [66] | |
5.5. Mélange binaire non idéal de molécules organiques
Soient les équilibres de fusion du mélange de l’acide laurique (LA) avec la lidocaïne (LC) :
Les équations du liquidus de l’acide laurique et de la lidocaïne issues de la description cinétique, obtenues par une analyse analogue à celle du mélange de l’acide laurique avec l’acide oléique, sont :
| (229) |
| (230) |
Avec :
| (231) |
| (232) |
| (233) |
Les liquidus thermodynamique (Annexe 9) et cinétique (Annexe 10) sont calculés avec les propriétés des corps purs renseignées dans le Tableau 10 afin d’être comparés aux liquidus expérimentaux [64].
Le diagramme de phases expérimental et les diagrammes de phases théoriques du mélange acide laurique-lidocaïne sont présentés sur la Figure 28 : les liquidus thermodynamique et cinétique de l’acide laurique s’écartent fortement du liquidus expérimental. L’acide laurique, à l’état solide comme à l’état liquide, est sous forme de dimère associé par une double liaison hydrogène [67], mais ce dimère peut se dissocier dans le mélange liquide de l’acide laurique avec la lidocaïne [64] (Figure 29) :
Une double liaison hydrogène se forme entre la fonction acide carboxylique de l’acide laurique et la fonction amide de la lidocaïne. La lidocaïne à l’état solide n’établit pas avec elle-même de double liaison hydrogène [68]. La solidification de la lidocaïne se produit lors de la collision du complexe d’association avec la surface d’un cristal de lidocaïne (Figure 30).
Diagrammes de phases liquide–solide composition-température du mélange lidocaïne-acide laurique expérimental (disques noirs), thermodynamique (traits rouges) et cinétiques sans (traits bleus) et avec prise en compte de la formation d’un complexe d’association entre l’acide laurique et la lidocaïne (traits verts).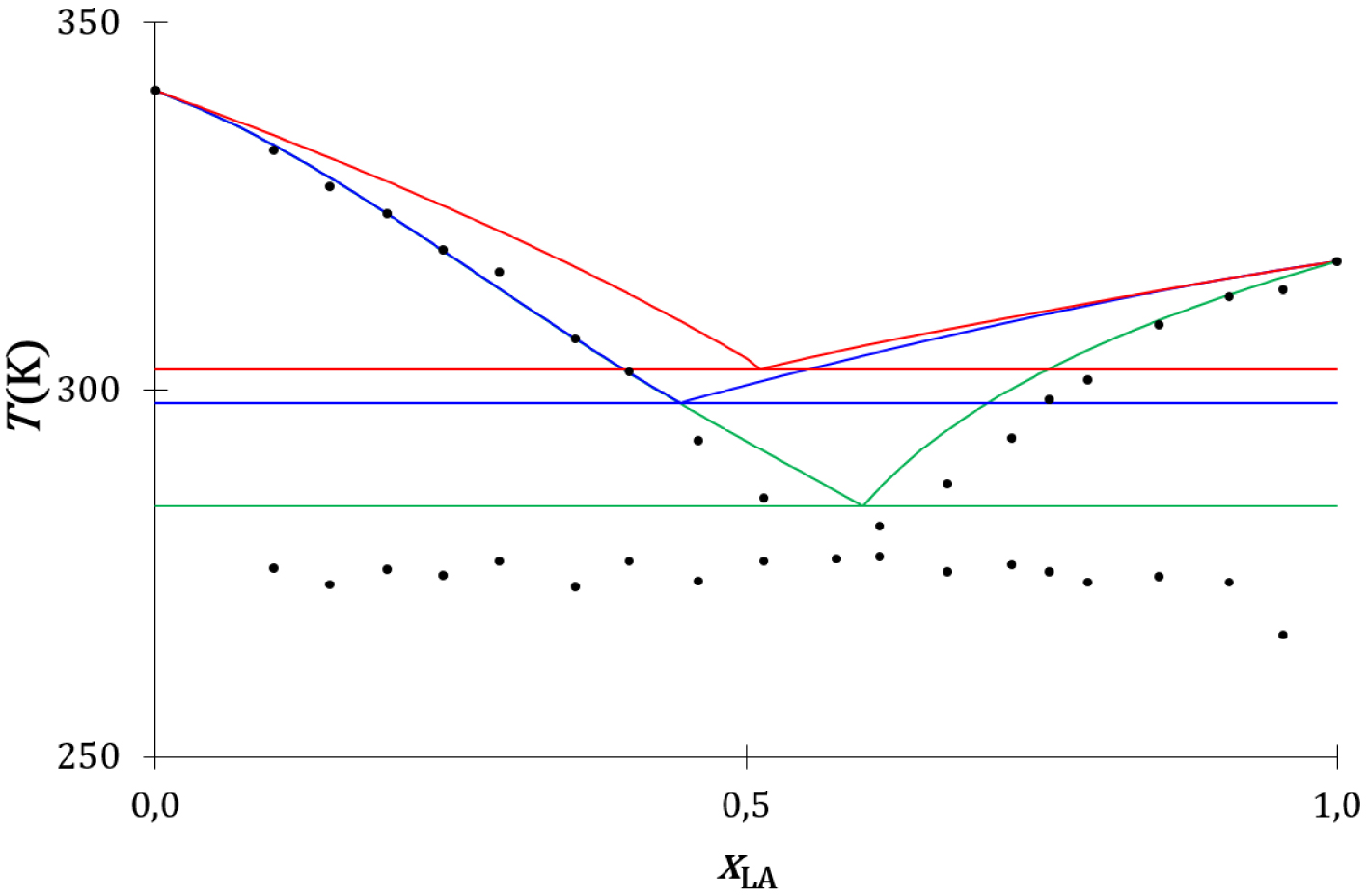
En revanche, la solidification de l’acide laurique ne peut pas se produire lors de la collision du complexe d’association avec la surface du cristal d’acide laurique, puisque le complexe d’association n’inclut pas le motif du cristal (Figure 30). Il convient donc de ne considérer que la fraction molaire d’acide laurique associé à lui-même dans l’équation du liquidus.
Le liquidus de l’acide laurique avec prise en considération de la formation du complexe acide laurique-lidocaïne est :
| (234) |
| (235) |
| (236) |
Le liquidus cinétique (traits verts) (Annexe 11) est proche du liquidus expérimental (disques noirs) (Figure 28). La description cinétique de l’équilibre de fusion de l’acide laurique dans son mélange avec la lidocaïne est concordante avec l’observation expérimentale.
Structure chimique des complexes d’association acide laurique-acide laurique (gauche) et acide laurique-lidocaïne (droite).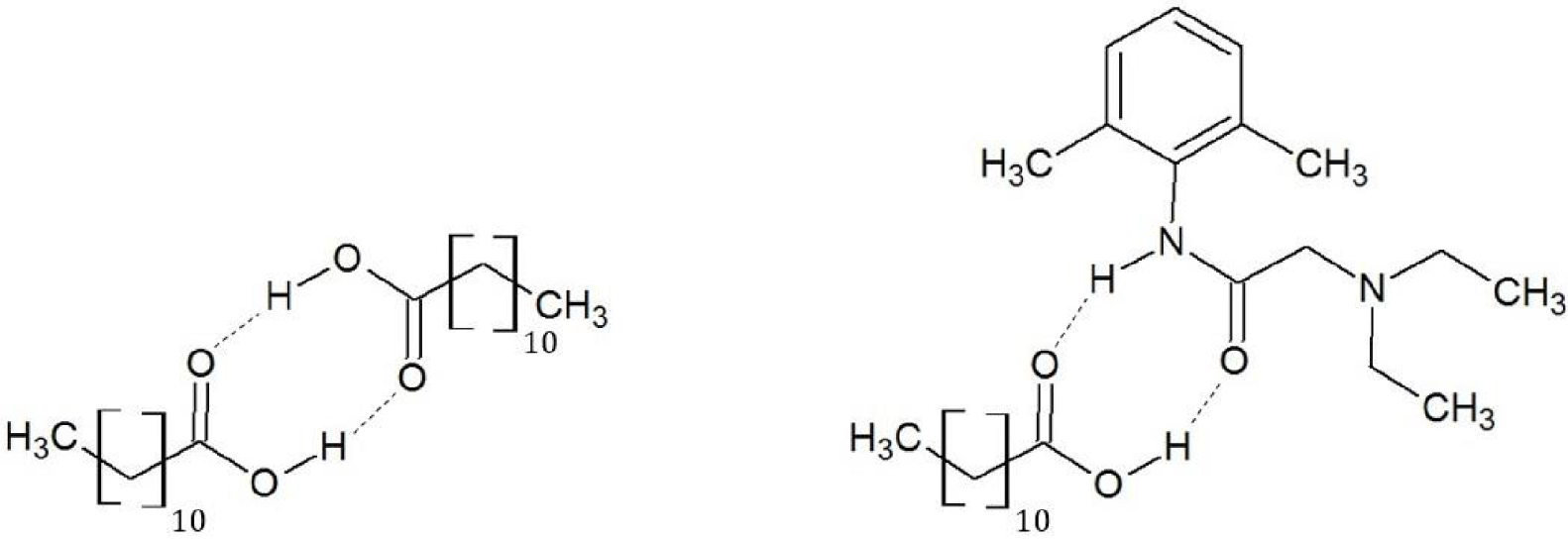
5.6. Entropie cinétique de mélanges binaires
La description cinétique des mélanges du chlorure de sodium avec la chlorure de rubidium ou l’eau d’une part, et des mélanges de l’acide laurique avec l’acide oléique ou la lidocaïne d’autre part, montre qu’il est possible d’établir une fonction analytique des courbes d’équilibre de fusion fondées sur le mécanisme collisionnel entre l’espèce chimique et la surface du cristal qui se produit en amont de la solidification. Ces fonctions cinétiques des courbes d’équilibre sont établies en substituant les fractions molaires des espèces par les fractions molaires des espèces qui entrent en collision avec la surface du cristal. Ainsi les fractions molaires du chlorure de sodium dans son mélange avec l’eau et de l’acide laurique dans son mélange avec la lidocaïne deviennent :
| (237) |
| (238) |
Tableau d’avancement de formation du complexe d’association acide laurique-lidocaïne
| Espèce | LC(l) | (LA − LC)(l) | |
|---|---|---|---|
| Etat initial | xLA | 1 − xLA | 0 |
| Etat final | 2⋅xLA − 1 | 0 | 1 − xLA |
Dans les deux cas, la fraction molaire de l’espèce chimique considérée diminue, ce qui induit une diminution de l’entropie de solidification. Ainsi, le chlorure de sodium dans son mélange avec l’eau est liquide dans un domaine de composition et de température, et solide dans ce même domaine dans son mélange avec le chlorure de rubidium. De manière analogue, l’acide laurique dans son mélange avec la lidocaïne est liquide dans un domaine de composition et de température, et solide dans ce même domaine dans son mélange avec l’acide oléique. Ces comportements sont illustrés sur les diagrammes cinétiques de fusion de la Figure 31.
Collision du complexe d’association entre l’acide laurique et la lidocaïne avec la surface d’un cristal de lidocaïne (haut) et d’acide laurique (bas).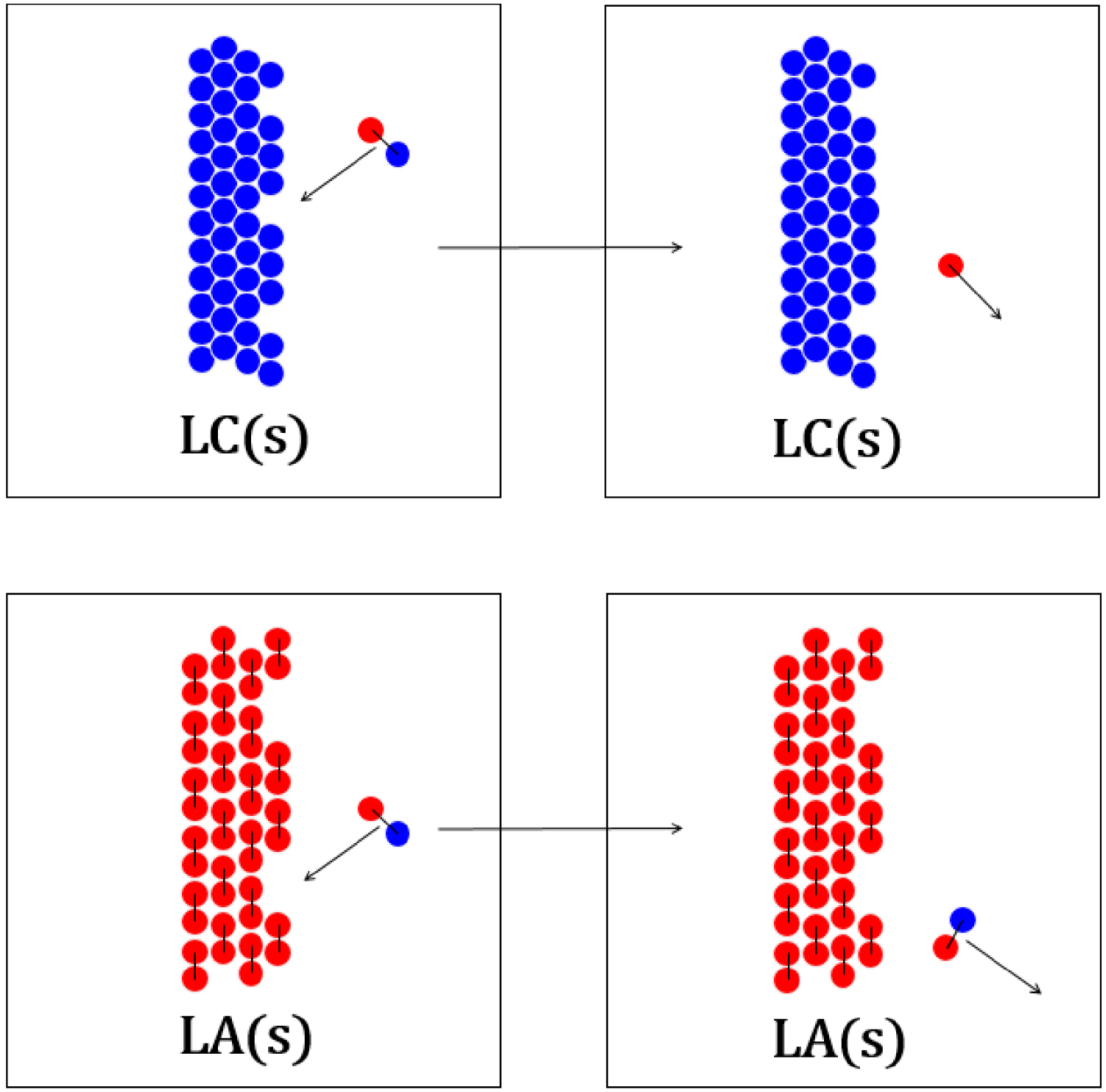
Diagrammes réactionnels cinétiques de l’équilibre de fusion du chlorure de sodium (gauche) et de l’acide laurique (droite) dans leurs mélanges.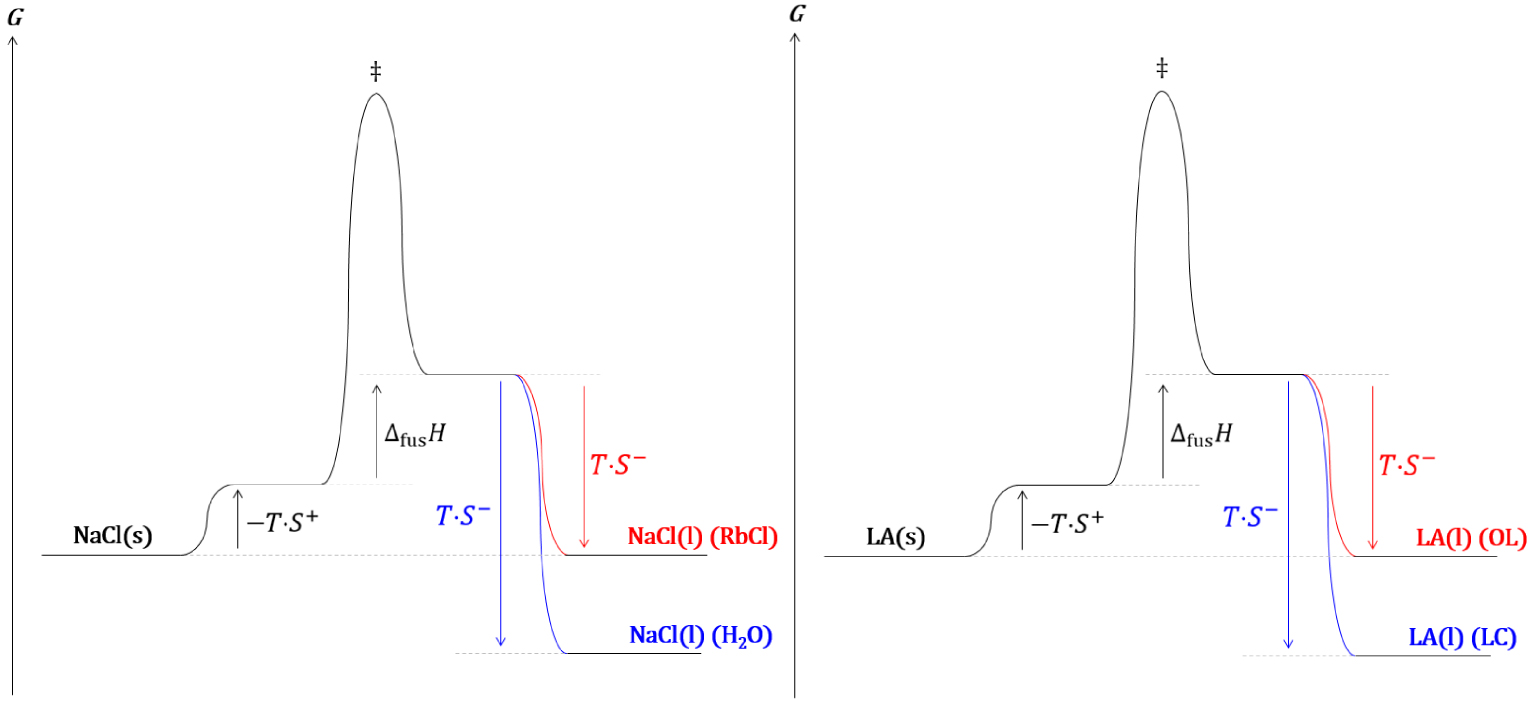
5.7. Mélange binaire de molécules chirales
Le mélange de certains couples d’énantiomères donne lieu à la formation d’un cocristal racémique plus stable que les cristaux de chaque énantiomère :
Topologie du diagramme de phases liquide–solide composition-température expérimental d’un système à invariant eutectique d’un couple d’énantiomères (A et A) avec son racémique (AA).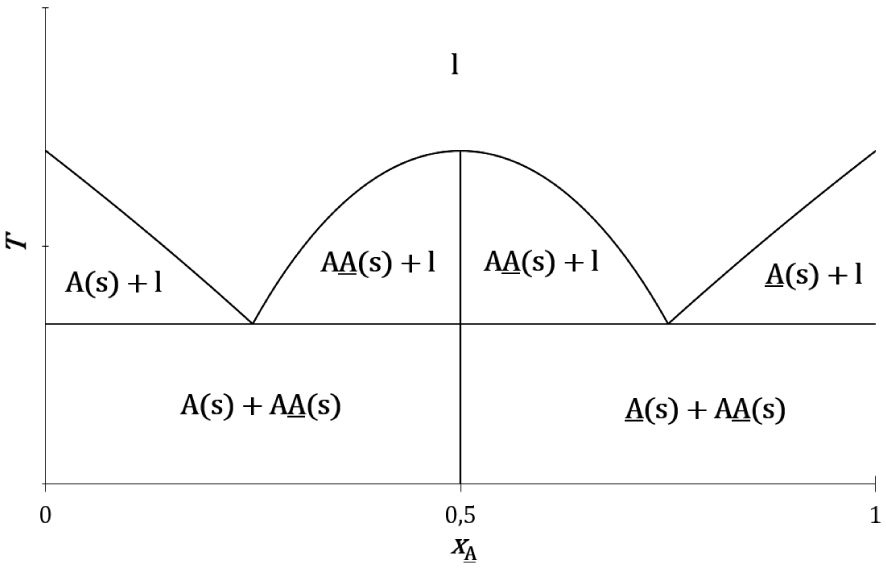
Trois équilibres de fusion sont impliqués :
Les équations cinétiques qui expriment ces trois équilibres de fusion sont :
| (239) |
| (240) |
| (241) |
Les deux premières égalités, qui concernent l’équilibre de fusion de A et A, mènent à :
| (242) |
| (243) |
Avec :
| (244) |
| (245) |
| (246) |
| (247) |
| (248) |
Les énantiomères A et A qui constituent le cocristal AA conservent leur structure moléculaire, il est donc possible d’exprimer l’équilibre de fusion du cocristal par deux équations cinétiques : une équation pour la fusion/solidification de A au sein du cocristal et une équation pour celle de A :
| (249) |
| (250) |
Donc :
| (251) |
| (252) |
Propriétés calorimétriques des formes cristallines des acides tartrique et mandélique
| Acide tartrique | Acide mandélique | |||
|---|---|---|---|---|
| 𝛥fus H° (kJ∕mol) | 𝛥fus H° (kJ∕mol) | |||
| Enantiomère (+)∕(−) | 443,4 | 36,3 | 404,8 | 25,5 |
| Racémate (+−) | 479,2 | 72,6 | 393,4 | 25,8 |
Le produit de ces équations de l’équilibre de fusion mène à :
| (253) |
| (254) |
| (255) |
Structure chimique de couples d’énantiomères de l’acide tartrique et de l’acide mandélique.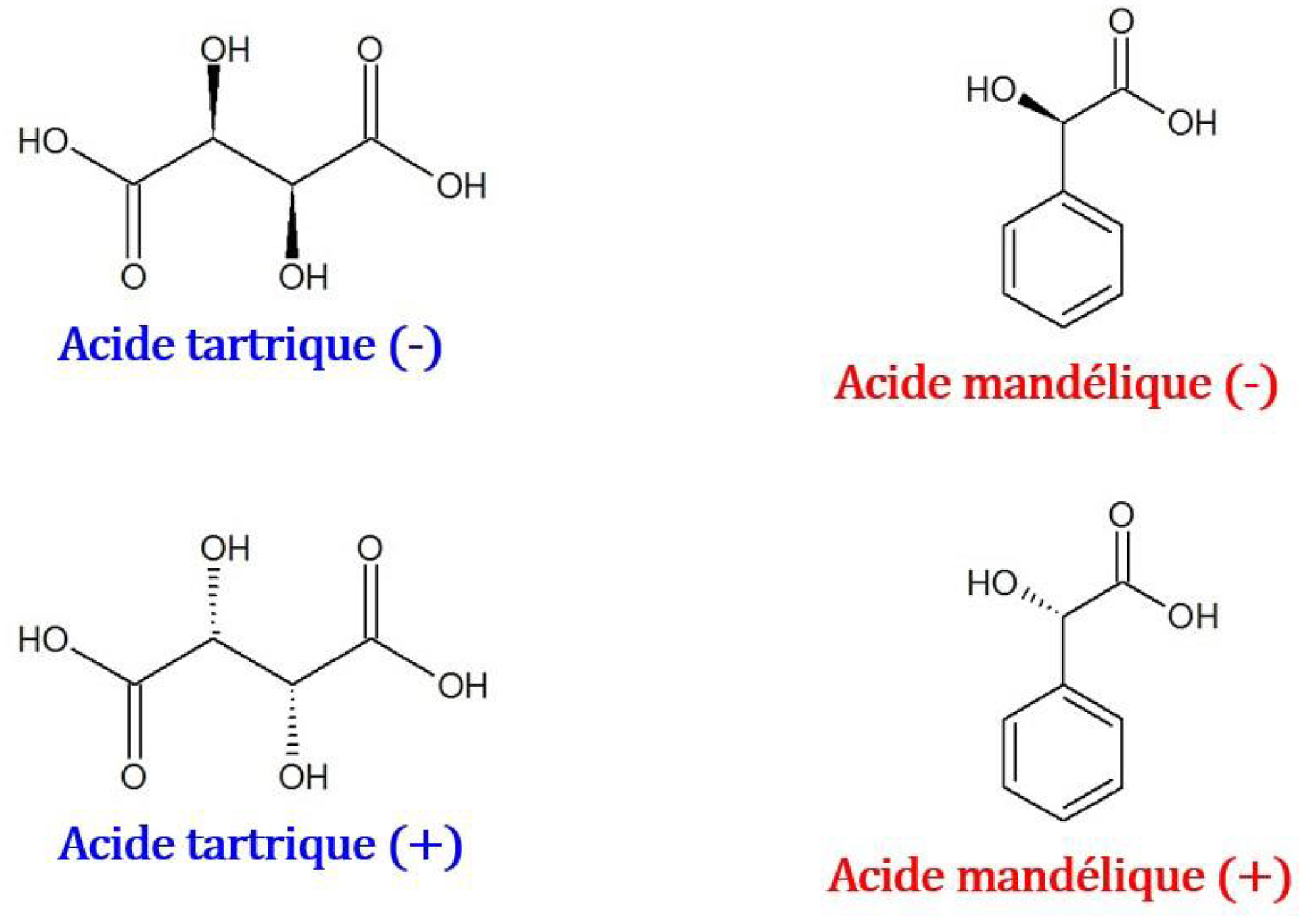
Les températures et enthalpies de fusion des énantiomères et racémates des acides tartrique [70] et mandélique [71] sont consignées dans le Tableau 12.
Les diagrammes de phases expérimentaux de ces deux systèmes concordent avec ceux déduits des analyses thermodynamique et cinétique (Figure 34, Annexes 12 et 13).
Diagrammes de phases liquide–solide composition-température des énantiomères de l’acide tartrique (gauche) et de l’acide mandélique (droite), expérimentaux (disques noirs) et déterminés par les modèles cinétique et thermodynamique : liquidus du cocristal (traits rouges) et liquidus de l’énantiomère (traits bleus).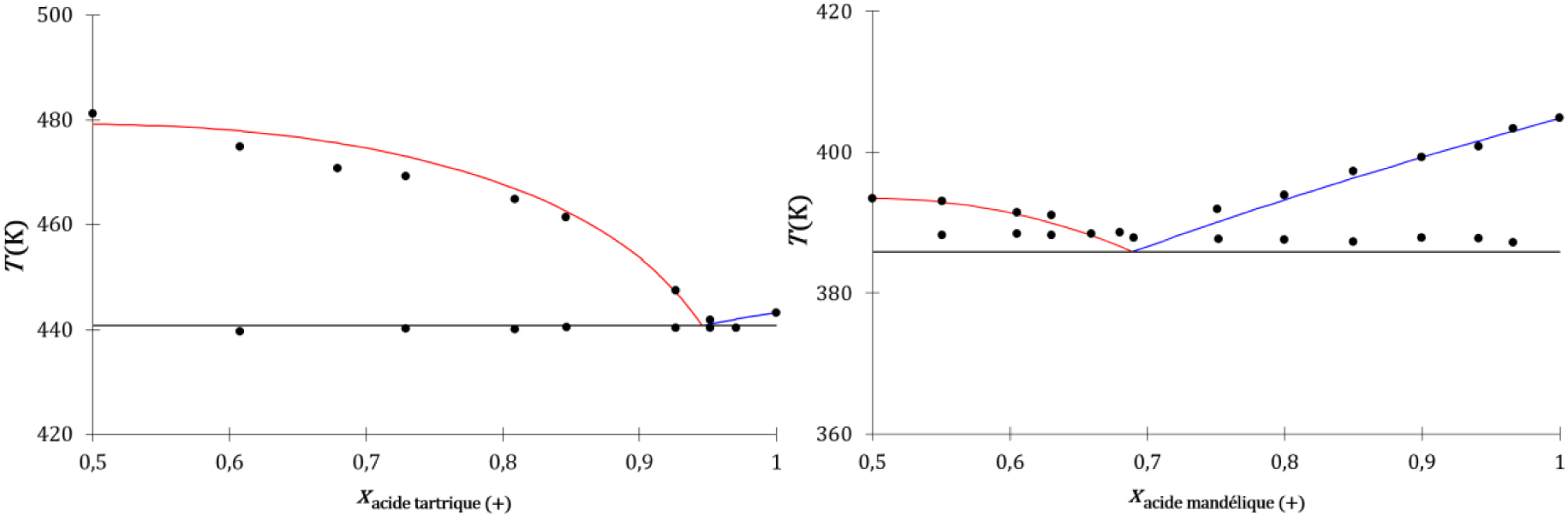
Les descriptions thermodynamique et cinétique de l’équilibre de fusion de systèmes à invariant eutectique à liquide idéal de mélanges d’énantiomères avec leur racémate sont équivalentes. Elles concordent entre elles et concordent avec le comportement expérimental de tels systèmes.
5.8. Cristaux atomiques et moléculaires
Les évolutions avec la masse molaire des températures et entropies de fusion standards des éléments de la classification périodique [72, 73, 74] qui cristallisent dans une structure compacte et des alcanes linéaires [75] sont présentées sur la Figure 35.
Températures et entropies de fusion standards des éléments de la classification périodique qui cristallisent dans des empilements compacts (hexagonal compact ou cubique à faces centrées) (bleu) et des alcanes linéaires de 2 à 22 atomes de carbones (rouge).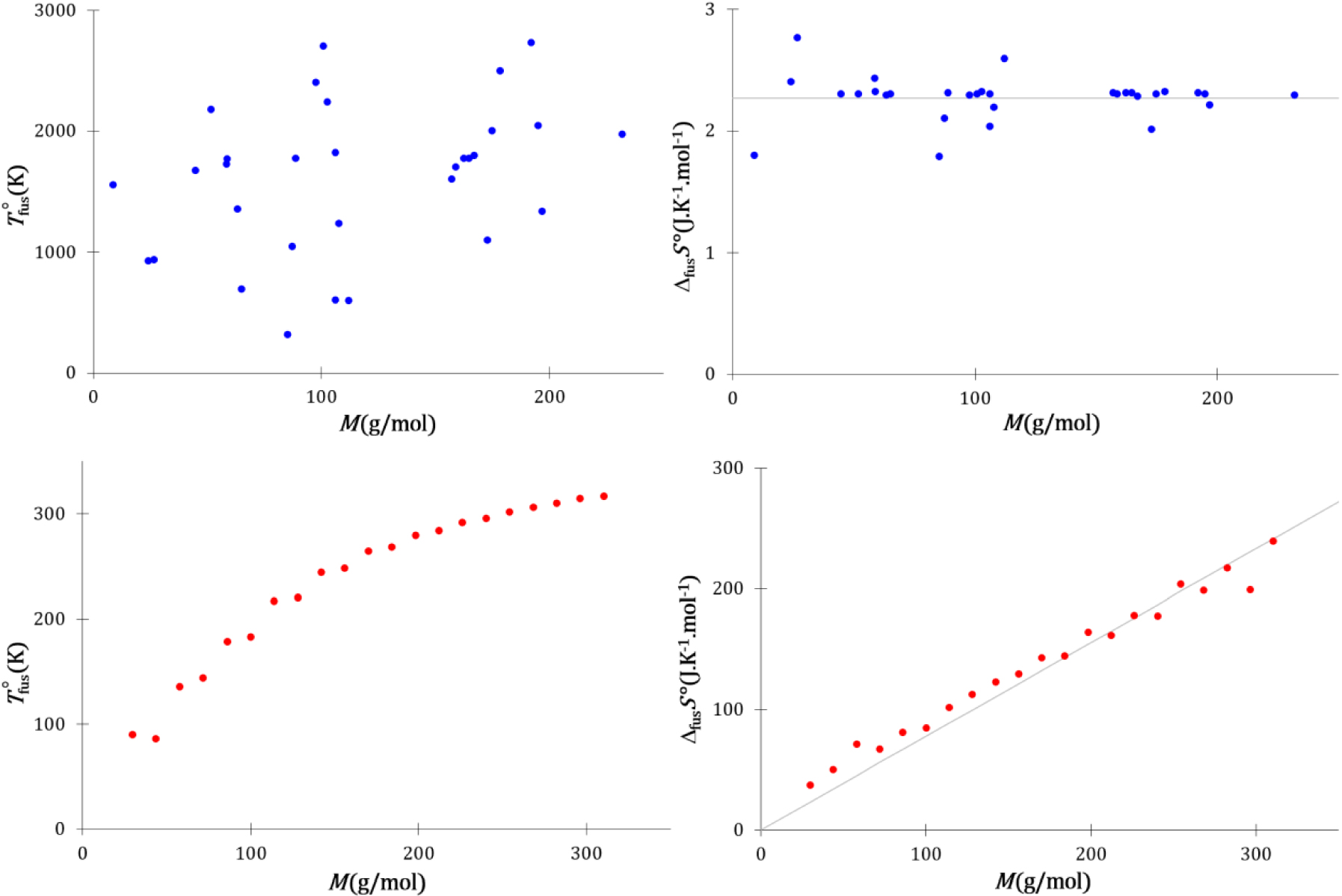
L’entropie de fusion des corps simples ne dépend pas de leur masse molaire, contrairement à celle des alcanes linéaires qui est proportionnelle à cette dernière. En thermodynamique statistique, l’entropie est liée au nombre d’états microscopiques :
| (256) |
| (257) |
Fusion et solidification d’un corps simple.
Pour les alcanes linéaires, la probabilité p− qu’une molécule qui entre en collision avec la surface du solide s’associe au solide est d’autant plus faible que la taille de l’alcane est élevée. En effet, il faut à la fois que la conformation, la position et l’orientation de la molécule au moment de la collision soient favorables à la cristallisation (Figure 37).
Températures de fusion et solidification de corps purs
| Corps pur | Acide laurique | Bi | Sn | Pb |
|---|---|---|---|---|
| 315,4 | 544,6 | 505,1 | 429,8 | |
| 304,6 | 516,4 | 480,6 | 429,1 | |
| Référence | Cette étude | Kano [76] | ||
Effet de la vitesse de refroidissement sur la température de solidification de l’eau liquide
| vrefroidissement (K∕min) | −0,3 | −0,5 | −1 |
| 265,3 | 264,1 | 262,2 | |
Températures de fusion et solidification standards de l’acide laurique
| Ecart type (K) | ||||
|---|---|---|---|---|
| Fusion | 315,3 | 314,6 | 316,1 | 0,5 |
| Solidification | 304,6 | 301,7 | 307,2 | 2,0 |
Temps de solidification d’échantillons du même lot de salicylate de phényle
| Echantillon | n°1 | n°2 | n°3 | n°4 | n°5 | n°6 | n°7 | n°8 | n°9 | n°10 |
| t (h) | 4,8 | 1,0 | 1,6 | 50,4 | 22,7 | 1,0 | 4,6 | 5,4 | 4,6 | 5,0 |
Par contre, la taille n’altère pas la probabilité p+ puisque la conformation, la position et l’orientation d’une molécule à la surface du cristal sont déjà définies. La probabilité de cristallisation d’une molécule lors d’une collision diminuera donc avec sa taille ce qui induit une augmentation de l’entropie cinétique de fusion avec la masse molaire. Les descriptions thermodynamique et cinétique de l’équilibre de fusion sont concordantes et en accord avec les observations expérimentales.
5.9. Hystérésis de la fusion et caractère métastable des nanocristaux
La température de solidification d’un corps pur est en général inférieure à sa température de fusion (Tableau 13).
La température de solidification d’un corps pur dépend en général de la vitesse de refroidissement [77] (Tableau 14).
Les températures de fusion puis de solidification de l’acide laurique ont été mesurées à 10 reprises dans le cadre de cette étude afin d’illustrer le caractère partiellement aléatoire de la solidification. En effet, celle-ci se produit dans un intervalle de température plus large que la fusion (Tableau 15).
Fusion et solidification d’un alcane linéaire : la collision d’une molécule d’alcane avec la surface du cristal ne mène pas à la solidification si la conformation de l’alcane (I) ou la position de la collision (II) sont défavorables. La solidification ne peut se produire que si ces deux conditions sont favorables (III).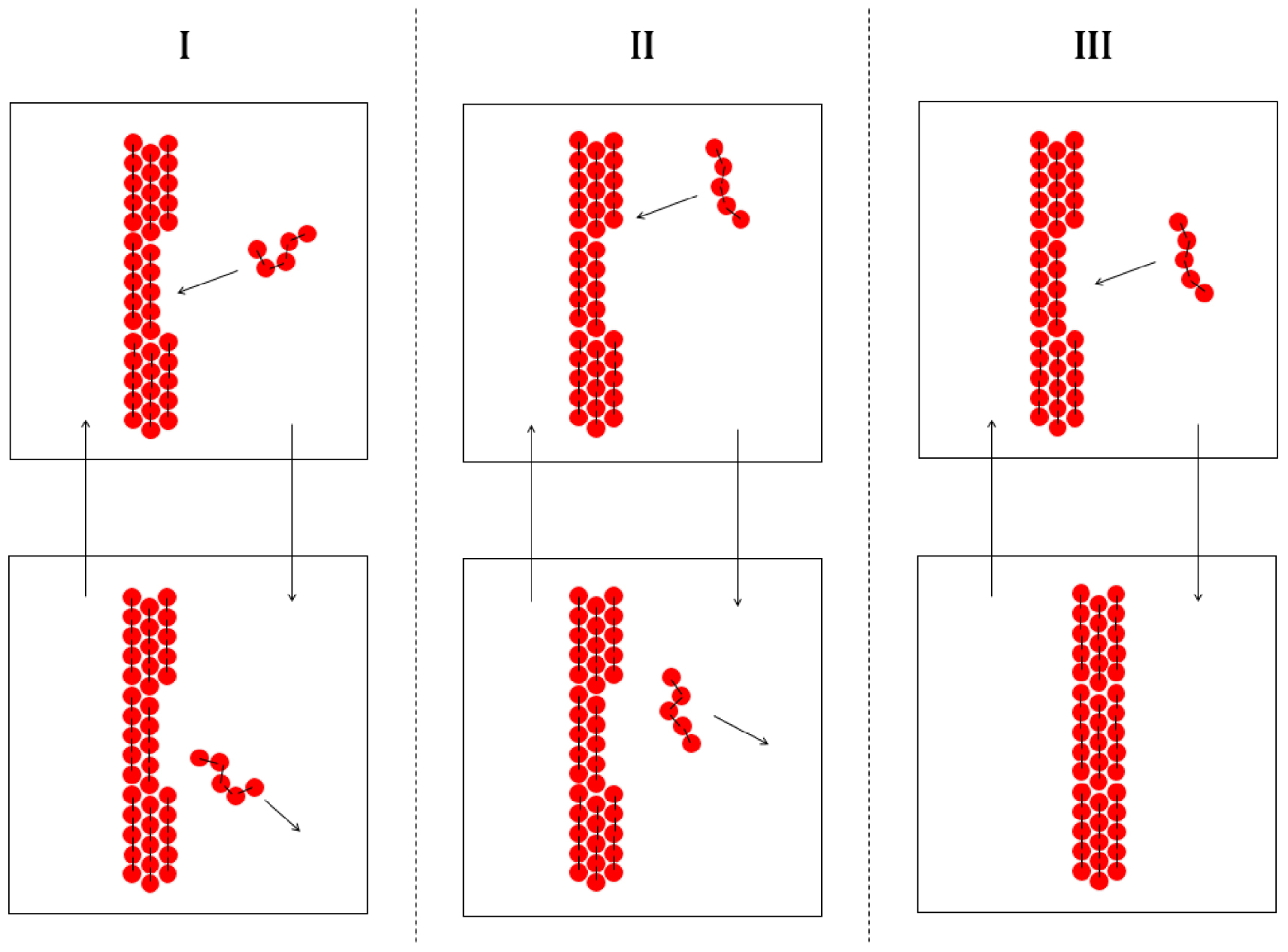
Une autre expérience qui illustre le caractère stochastique de la solidification de manière encore plus probante a également été menée dans le cadre de cette étude : 10 échantillons du même lot de salicylate de phényle ont été fondus. Les temps de solidification des échantillons présentent une dispersion importante (Tableau 16).
La solidification commence par la croissance d’un germe cristallin de dimensions nanométriques. La température de fusion d’un cristal nanométrique (nc) est inférieure à celle d’un cristal (c) (Tableau 17).
Températures de fusion de cristaux (c) et nanocristaux (nc) de corps purs
| Sn | Pb | Benzène | Naphtalène | |
|---|---|---|---|---|
| 506,1 | 600,6 | 280,8 | 356,1 | |
| Karmakar et al. [78] | Qi [79] | Zhang et al. [80] | Zhang et al. [80] | |
| 427,5 | 438,6 | 253,9 | 341,3 | |
| Qi [79] | Qi [79] | Zhang et al. [80] | Zhang et al. [80] | |
| Taille (nm) | 5,3 | 4,7 | 1,9 | 1,9 |
La température de fusion d’un nanocristal d’une espèce chimique diminue avec sa taille [80] (Tableau 18).
Température de fusion de cristaux de chlorobenzène de différentes tailles
| Taille | >1 μm | 13,0 nm | 7,8 nm | 4,3 nm | 1,9 nm |
| 229,1 | 225,7 | 222,2 | 216,1 | 201,2 | |
L’enthalpie de fusion d’un nanocristal diminue de manière encore plus marquée avec sa taille que sa température de fusion [80] (Tableau 19).
Enthalpie de fusion de cristaux de chlorobenzène de différentes tailles
| Taille | >1 μm | 21,9 nm | 10,9 nm | 5,6 nm |
| 9,8 | 6,3 | 5 | 3,6 | |
Soit la fusion d’un cristal (c) de grande taille (i.e. >1 μm) d’un corps pur A dans son liquide :
| (258) |
| (259) |
| (260) |
| (261) |
A la température de fusion, ces vitesses sont égales :
| (262) |
| (263) |
D’où :
| (264) |
Diagramme réactionnel thermodynamique (gauche) et cinétique (droite) de l’équilibre de fusion d’un cristal (c) d’un corps pur dans les conditions standards.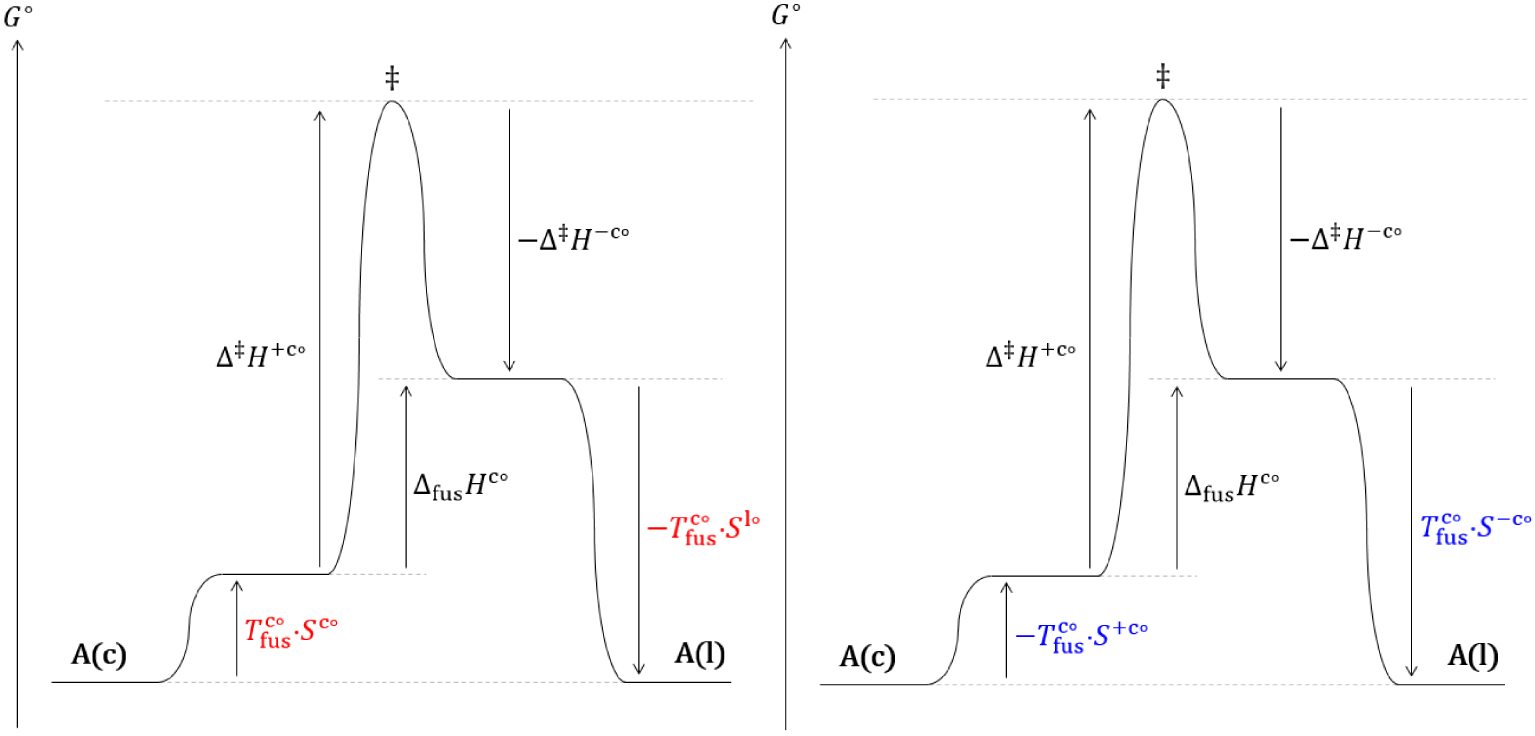
A une température inférieure à la température de fusion, la solidification est spontanée :
| (265) |
| (266) |
| (267) |
| (268) |
| (269) |
Diagrammes réactionnels thermodynamique (gauche) et cinétique (droite) hors équilibre de la solidification du cristal (c) d’un corps pur à une température inférieure à sa température de fusion.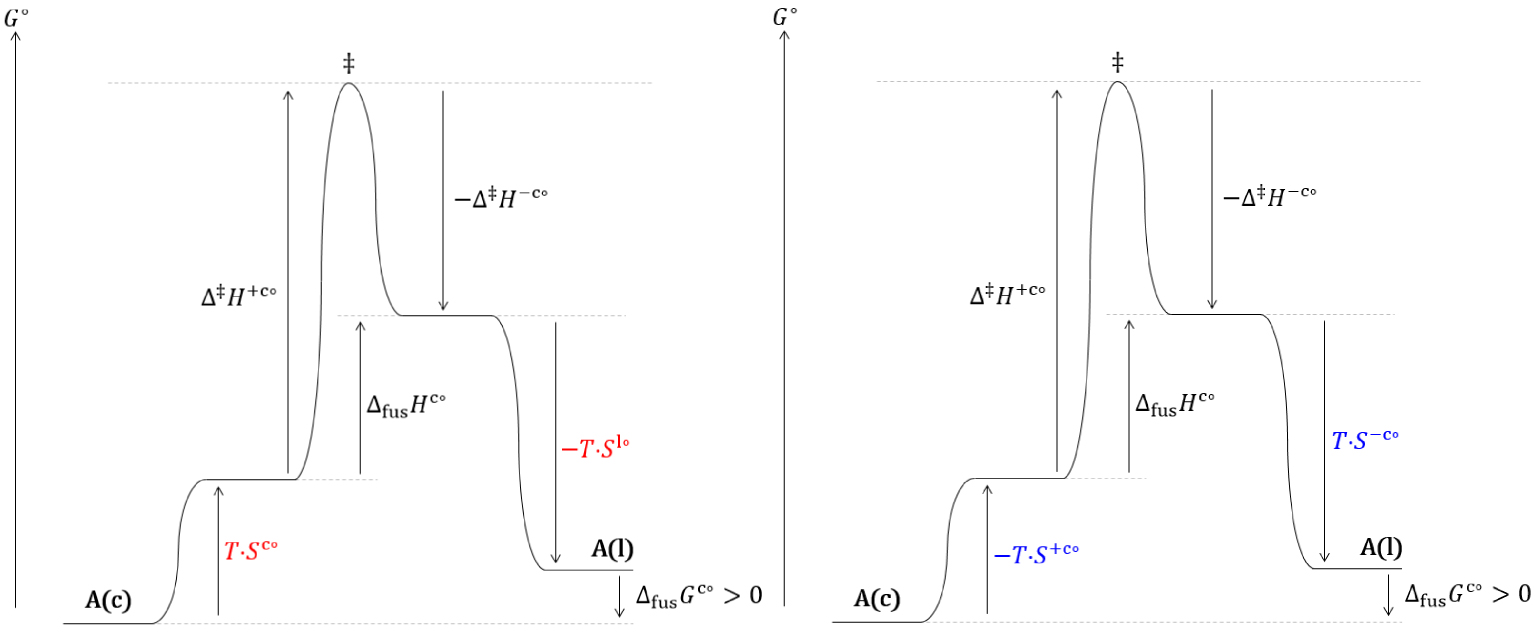
La cristallisation d’un corps pur débute par la formation d’un germe nanocristallin (nc) :
| (270) |
| (271) |
Diagrammes réactionnels thermodynamique (gauche) et cinétique (droite) hors équilibre de la fusion d’un nanocristal dans un domaine de température où le cristal est stable.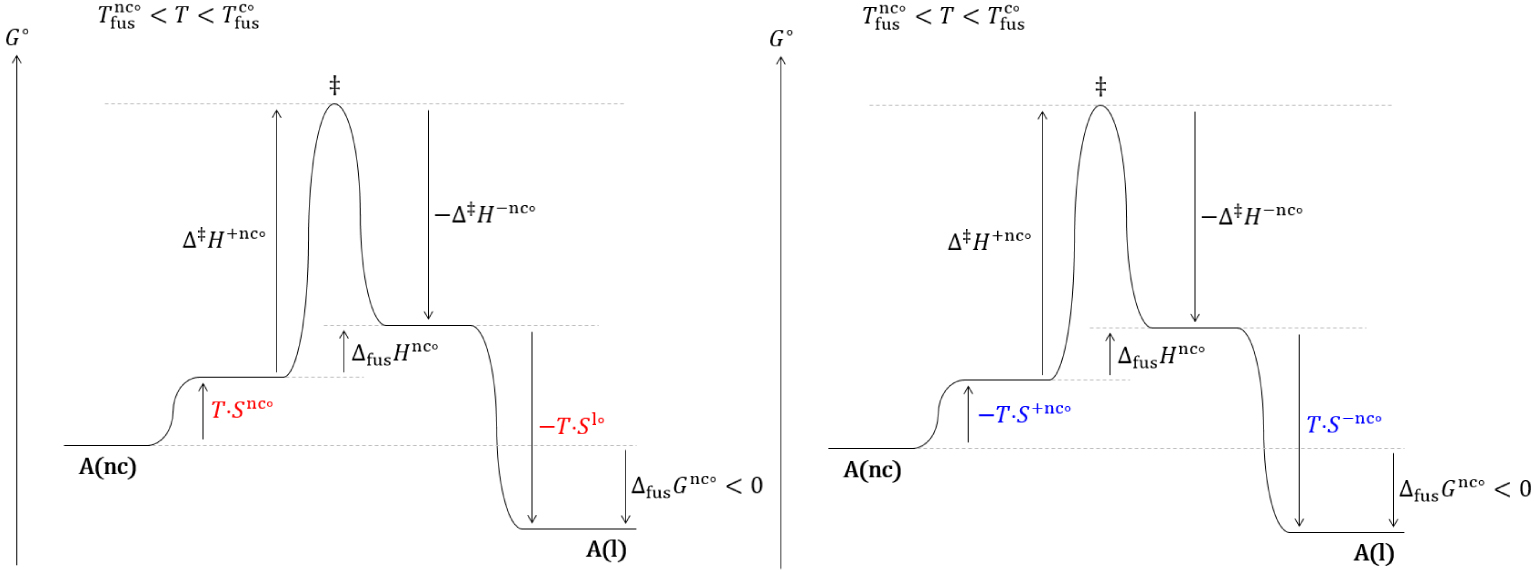
Les descriptions thermodynamique et cinétique des fusions d’un cristal et d’un nanocristal mènent aux mêmes conclusions pour le signe de , la suite de l’analyse est donc valable pour les deux descriptions. La taille, et par conséquence la valeur de 𝛥fus G° d’un germe nanocristallin dans le domaine métastable est fluctuante (Figure 41-I). Ce nanocristal peut atteindre, par ce jeu de fluctuations, une taille critique à partir de laquelle la solidification est spontanée. La valeur de 𝛥fus G° de ce nanocristal augmente alors et ne fluctue plus de manière significative (Figure 41-II). Le nanocristal continue à croitre jusqu’à ce qu’il devienne un cristal, c’est-à-dire lorsque sa croissance n’affecte plus sa température de fusion, 𝛥fus G° atteint alors une valeur maximale. Comme la température du milieu est inférieure à la température de fusion d’un cristal de grande taille, le processus de solidification se poursuit jusqu’à la solidification totale de la phase liquide (Figure 41-III).
Les modèles thermodynamique et cinétique hors équilibre offrent une description cohérente de la solidification, de la naissance du premier germe cristallin nanométrique à la croissance spontanée d’un cristal de taille plus importante. La métastabilité des nanocristaux est à l’origine de l’hystérésis de la fusion-solidification et de la dispersion des températures de solidification : en effet, les fluctuations de la taille du nanocristal à l’origine de la cristallisation sont de nature stochastique.
Evolution conceptuelle de 𝛥fus G° du cristal d’un corps pur à une température inférieure à sa température de fusion : fluctuation de la taille d’un germe nanocristallin instable (I), croissance spontanée d’un germe cristallin stable de taille intermédiaire (II) et croissance d’un cristal de grande taille (III) jusqu’à la solidification totale (IV).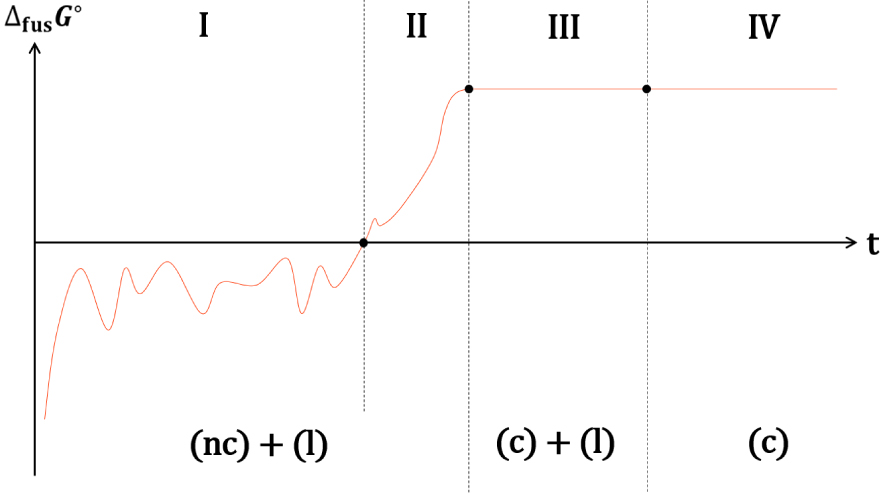
5.10. Formation de cristaux chiraux de sels ioniques
L’analyse présentée dans ce chapitre utilise les résultats obtenus dans le chapitre précédent sur la croissance cristalline d’un germe nanocristallin à un cristal de grande taille : les deux modèles, thermodynamique et cinétique, sont utilisables puisqu’ils mènent aux mêmes évolutions de la fonction 𝛥fus G. La cristallisation des chlorate (NaClO3) ou bromate (NaBrO3) de sodium en solution aqueuse présente un comportement singulier : alors que les cations en solution de ces sels sont achiraux, les cristaux présentent une chiralité [81]. Les formes chirales sont formées en proportions égales en absence d’agitation [82] :
Soit une solution sursaturée de chlorate ou bromate de sodium. La taille des premiers germes nanocristallins (A et A) fluctue (Figure 42-I) jusqu’à l’instant où un cristal d’une des formes chirales (A) atteint une taille critique à partir de laquelle sa croissance est spontanée (Figure 42-II). A partir de cet instant, la taille de ce cristal (A) croît de manière continue. La concentration de la solution sursaturée décroît de manière continue et homogène dans toute la solution en raison de l’agitation, ce qui défavorise la solidification des cristaux de l’autre forme chirale (A), qui vont progressivement se dissoudre jusqu’à disparaitre. La croissance du cristal (A) consomme les ions de la solution (Figure 42-III) jusqu’à établissement de l’équilibre (Figure 42-IV) : le solide d’une des deux formes chirales dans la solution aqueuse saturée. Un tel phénomène, pour lequel un changement mineur (formation d’un nanocristal de taille critique) entraîne la totalité du système à évoluer dans une des deux voies qui lui sont offertes (solidification de A ou A) est appelé en physique une bifurcation.
Evolution conceptuelle de et de la concentration en A en solution : fluctuation de la taille de germes nanocristallins (A et A) (I) ; formation d’un cristal de taille critique d’une forme chirale (A) et solubilisation de la forme chirale énantiomère (A) (II) ; diminution de la concentration de la solution sursaturée (III) jusqu’à saturation (IV).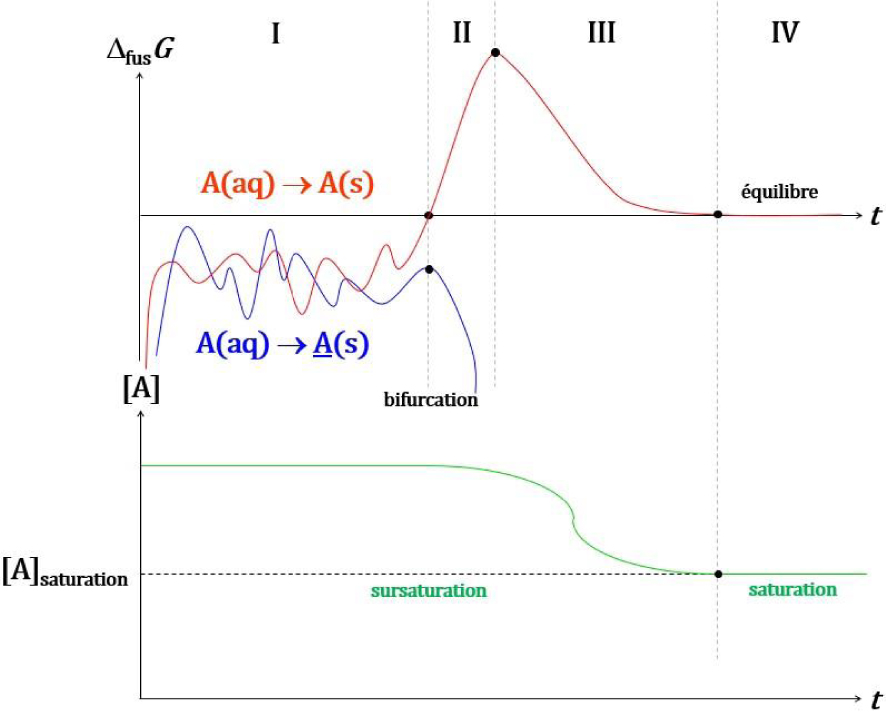
Si la solution n’est pas agitée, la modification de concentration induite par la croissance des germes cristallins ne se répercute pas sur celles des autres rapidement, ce qui laisse le temps aux germes cristallins des deux formes chirales d’atteindre la taille critique à partir de laquelle la cristallisation devient spontanée. Les modèles thermodynamique et cinétique hors équilibre permettent de comprendre pourquoi une seule forme chirale des cristaux de chlorate ou bromate de sodium se forme lorsque la solution est agitée et une seule lorsqu’elle ne l’est pas.
5.11. Synthèse
Les modèles thermodynamique et cinétique à l’équilibre et hors équilibre permettent de comprendre et quantifier la fusion de nombreux systèmes :
- i. mélanges binaires de sels ioniques et molécules organiques (NaCl–RbCl et acide laurique-acide oléique) ;
- ii. mélanges binaires de molécules chirales (acide tartrique et acide mandélique) ;
- iii. évolution de l’entropie de fusion de cristaux atomiques compacts et d’alcanes linéaires avec la masse molaire ;
- iv. lien entre le caractère métastable des nanocristaux et l’hystérésis de la fusion ;
- v. effet des conditions rhéologiques sur la formation de cristaux chiraux.
Pour d’autres systèmes, qui présentent un écart à l’idéalité selon le modèle thermodynamique, le modèle cinétique s’avère plus pertinent :
- i. mélange binaire du chlorure de sodium avec l’eau ;
- ii. mélange binaire de l’acide laurique avec l’acide oléique.
Alors que le modèle thermodynamique de tels systèmes ne peut s’affranchir de paramètres d’ajustement de nature empirique, le modèle cinétique permet de construire des fonctions analytiques des courbes de l’équilibre de fusion en se fondant sur la nature du processus collisionnel qui se produit en amont de la solidification. L’étude de ces deux systèmes, comparés à des systèmes qui partagent une des deux espèces chimiques du mélange, montre que l’entropie cinétique est totalement dépendante des espèces chimiques associées. Ainsi la solidification du chlorure de sodium dépend drastiquement de son partenaire, le chlorure de rubidium ou l’eau. De la même manière, la solidification de l’acide laurique en mélange avec l’acide oléique se distingue de celle en mélange avec la lidocaïne.
6. Théorie cinétique de l’équilibre
L’étude de systèmes réels des chapitres précédents, réactions chimiques et équilibres de fusion, corrobore le modèle cinétique de l’équilibre. Ce dernier chapitre présente le modèle cinétique de l’équilibre des transformations microscopiques dans un cadre théorique général.
6.1. Fonction cinétique d’évolution
Dans cette première partie, la fonction cinétique d’évolution d’une réaction chimique est établie. Il est important de préciser que cette fonction d’évolution est utilisable strictement dans le cas d’une seule transformation microscopique. Le cas de transformations microscopiques couplées sera traité dans le chapitre suivant. Soient deux molécules de gaz A et B qui peuvent réagir pour donner un ou plusieurs produit(s).
Système fermé constitué de deux molécules réactives A et B en mouvement dans une enceinte de volume V, de pression constante égale à 1 bar et dont les parois sont thermostatées à une température T constante.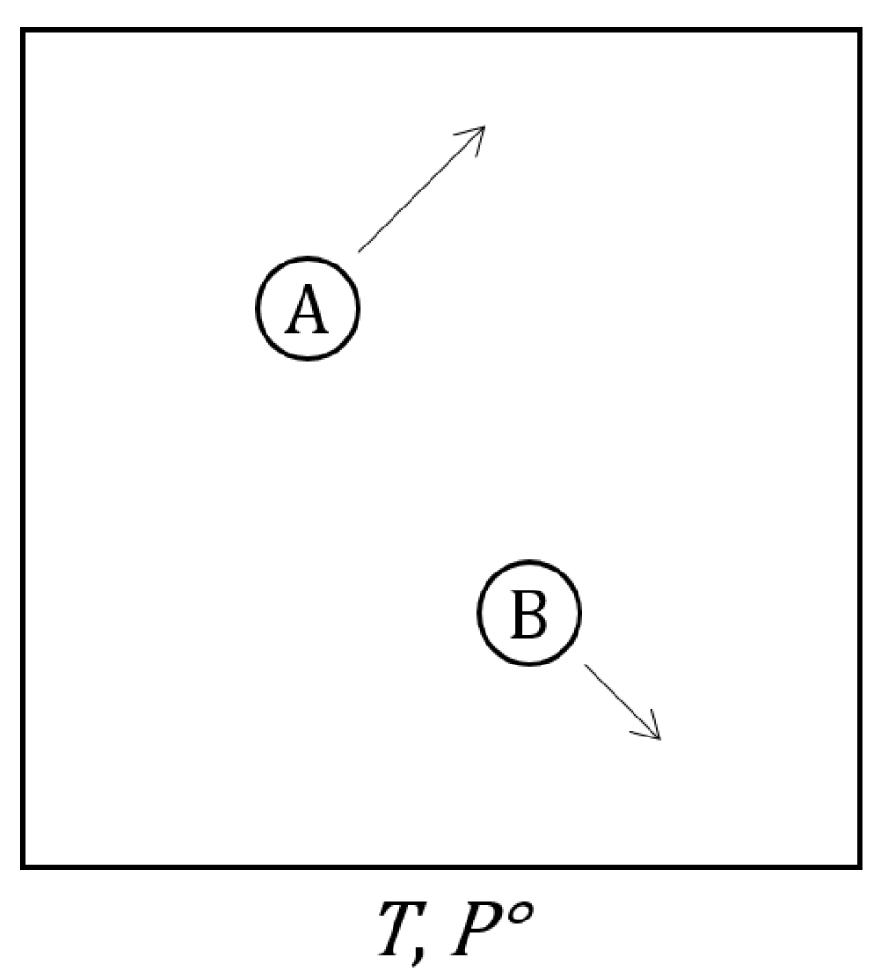
La pression de la boîte thermostatée est choisie suffisamment faible pour que la collision entre A et B ne se produise pas avant que l’énergie cinétique des molécules soit indépendante des conditions initiales. Autrement formulé, le nombre de collisions de A et B avec les parois de l’enceinte avant leur collision est grand devant 1. Dans ces conditions, la densité de probabilité de la norme de la vitesse des molécules A et B suit la loi de distribution de Maxwell–Boltzmann[83] :
| (272) |
| (273) |
| (274) |
| (275) |
| (276) |
| (277) |
| (278) |
Evolution de 𝜒° avec .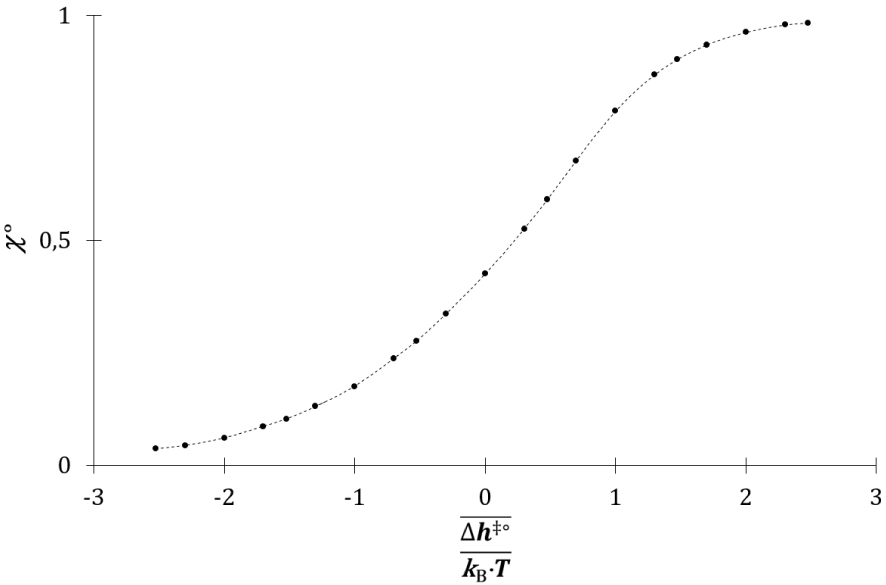
Il augmente avec le rapport , il tend vers 0 pour une valeur faible de ce rapport et vers 1 pour une valeur élevée de celui-ci. Le coefficient 𝜒° représente la fraction de l’énergie cinétique des molécules A et B engagées dans la collision. En effet, même lors d’une collision totalement inélastique, la variation d’énergie cinétique des molécules est inférieure à l’énergie cinétique totale. La vitesse v de la réaction chimique entre Ni molécules de A et Nj molécules de B, avec Ni et Nj grands devant 1, se déduit de la vitesse de réaction v(1) pour un couple d’espèces réactives : chacune des Ni molécules de A peut entrer en collision avec chacune des Nj molécules de B.
| (279) |
| (280) |
| (281) |
| (282) |
| (283) |
| (284) |
| (285) |
| (286) |
| (287) |
| (288) |
| (289) |
| (290) |
| (291) |
| (292) |
| (293) |
Evolution de 𝜒° avec 𝛥‡ H+°∕(R⋅T) et 𝛥r H°∕(R⋅T).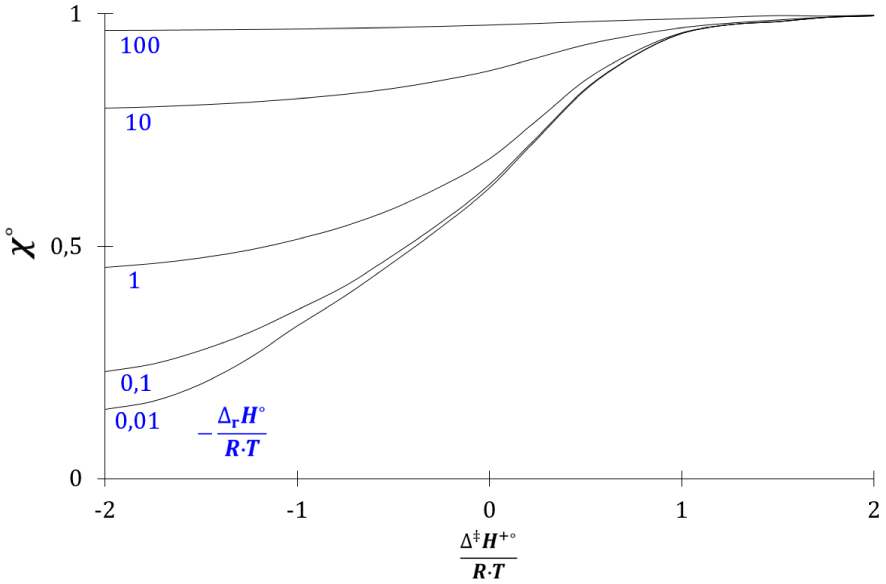
La grandeur 𝜒°, comprise entre 0 et 1, augmente avec les rapports 𝛥‡ H+°∕(R⋅T) et 𝛥r H°∕(R⋅T). L’enthalpie libre cinétique standard dépend donc de la grandeur 𝜒° :
| (294) |
| (295) |
| (296) |
| (297) |
| (298) |
| (299) |
| (300) |
| (301) |
| (302) |
| (303) |
De la même manière qu’aucun gradient de température macroscopique n’a jamais été observé dans un échantillon de matière à l’équilibre thermique, une réaction chimique parvenue à l’équilibre n’a jamais évolué spontanément et significativement vers la formation persistante d’un de ses réactifs. Comme l’entropie cinétique de réaction évolue vers la grandeur 𝛥r H°∕𝜒°⋅Ec, le caractère irréversible des transformations microscopiques peut se corréler à la diminution avec le temps de la fonction :
| (304) |
6.2. Couplage des équilibres thermique et cinétique
Soit un système constitué d’un nombre N grand devant 1 de particules sphériques, contenues dans une enceinte thermostatée, qui peuvent occuper deux états A et B d’énergies respectives et eB :
| (305) |
| (306) |
| (307) |
| (308) |
| (309) |
| (310) |
| (311) |
| (312) |
| (313) |
| (314) |
| (315) |
| (316) |
| (317) |
| (318) |
| (319) |
| (320) |
| (321) |
| (322) |
| (323) |
| (324) |
| (325) |
| (326) |
| (327) |
| (328) |
| (329) |
| (330) |
| (331) |
| (332) |
| (333) |
| (334) |
Les courbes de densités de probabilité d’énergie des états A et B sont présentées sur la Figure 46 pour quelques cas particuliers.
Courbes de quelques densités de probabilité d’énergie particulières des particules A et B.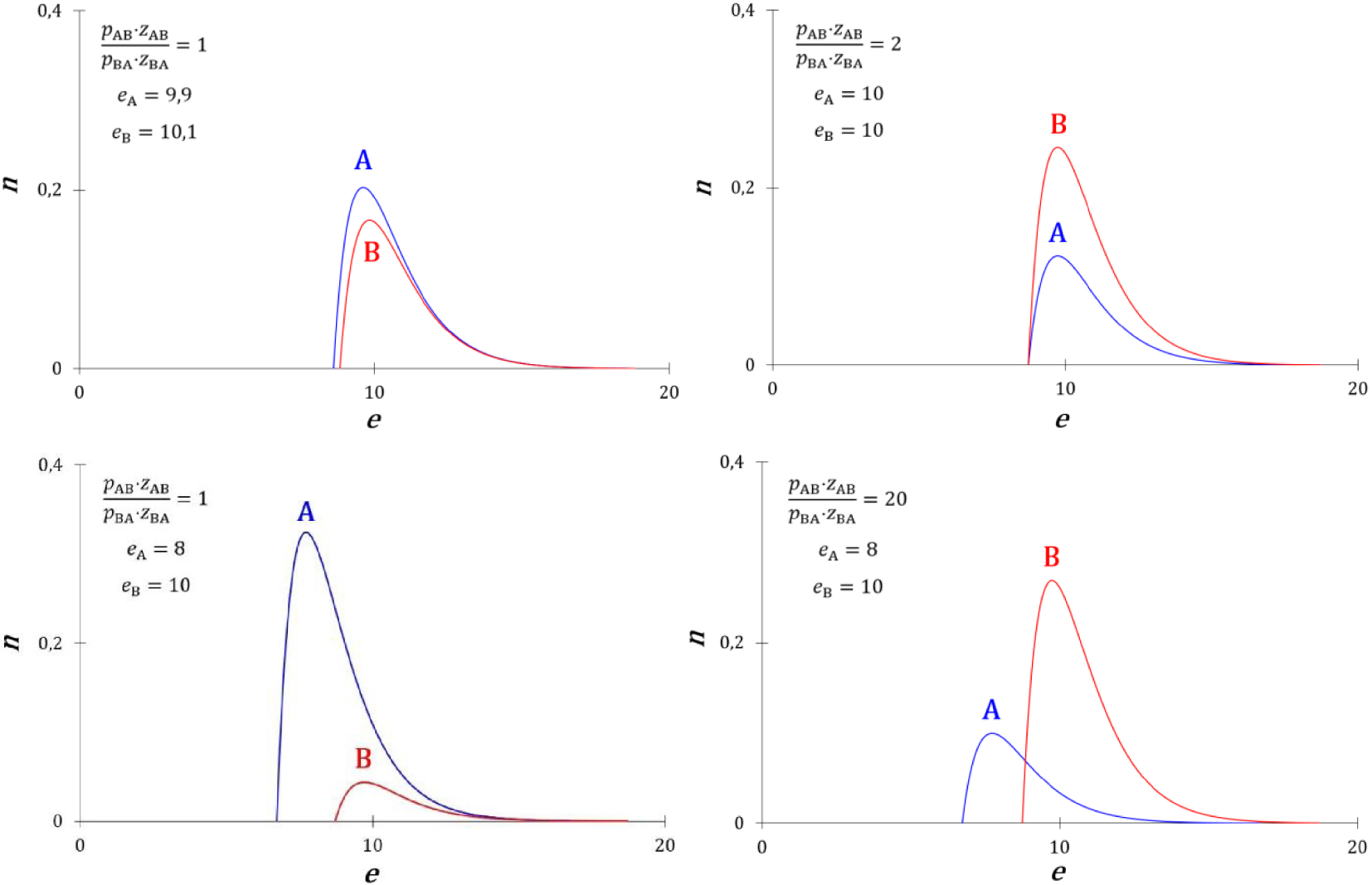
6.3. Equilibres couplés
Soit un système fermé délimité par une surface fermée, thermostaté à température T constante et de pression P ou volume V constant. Ce système est constitué de états impliqués dans q transformations élémentaires :
| (335) |
| (336) |
| (337) |
| (338) |
| (1) |
| (2) |
| (3) |
| (1) |
| (2) |
| (3) |
| (339) |
| (340) |
| (341) |
| (342) |
| (343) |
| (344) |
| (345) |
| (1) |
| (1) |
| (1) |
| (2) |
| (3) |
Le formalisme d’écriture des équilibres cinétiques présenté dans ce chapitre n’est en pratique pas intéressant, il est en effet inconfortable pour un utilisateur de devoir consulter un tableau de correspondance des états de chaque transformation du système : le dessein de ce chapitre était de montrer qu’il existe un système d’équations cinétiques adéquat pour étudier l’évolution de n’importe quel système de transformations.
6.4. Equilibres couplés cyclique et linéaire
Soit un système fermé délimité par une surface fermée, thermostaté à température T constante et de pression P ou volume V constant, constitué de N particules microscopiques qui peuvent occuper trois états. Les transitions des particules entre ces trois états sont permises (Figure 47) :
| (1) |
| (2) |
Système cyclique à trois états.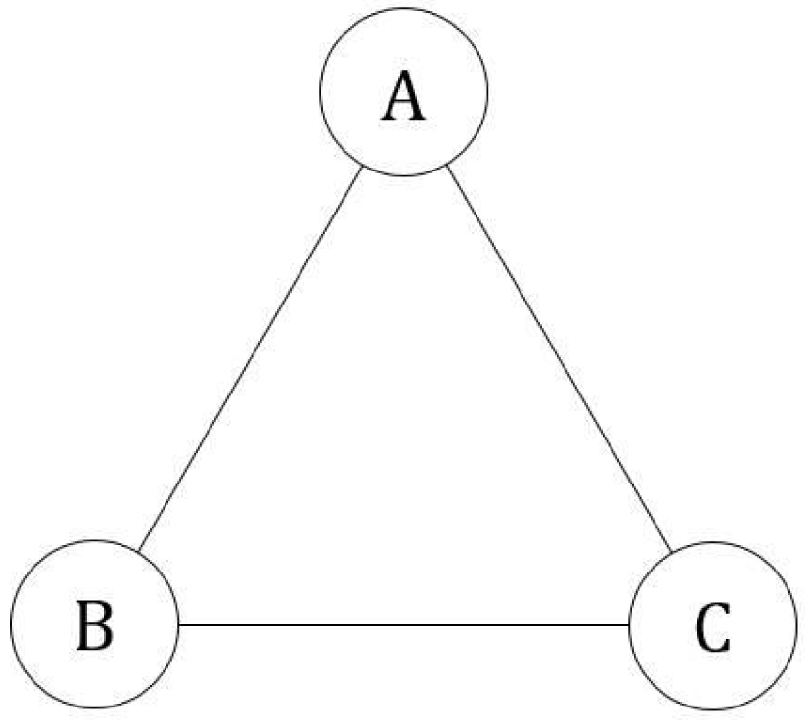
Le nombre de particules se conserve :
| (346) |
| (347) |
| (348) |
| (349) |
| (350) |
| (351) |
| (352) |
| (353) |
| (354) |
| (355) |
| (356) |
Système linéaire à trois états.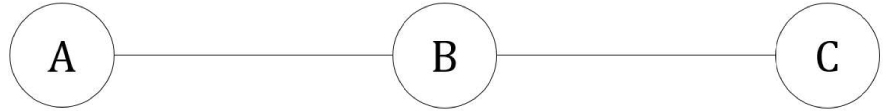
Ce système est le cas particulier du système cyclique à trois états lorsque et sont nuls :
| (357) |
| (358) |
| (359) |
| (360) |
| (361) |
| (362) |
6.5. Entropie cinétique et évolution
Soit une transformation renversable entre deux états microscopiques :
| (363) |
| (364) |
| (365) |
| (366) |
| (367) |
| (368) |
| (369) |
| (370) |
Schéma d’une transformation microscopique catalysée.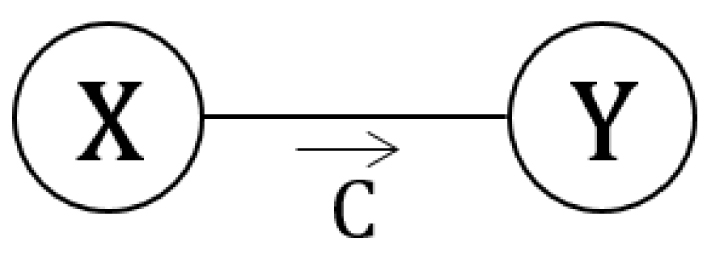
Soit un système constitué de particules engagées dans des transformations couplées dont certaines sont catalysées, les particules sont initialement dans l’état A (Figure 50).
Système de transformations couplées dont certaines sont catalysées (flèches).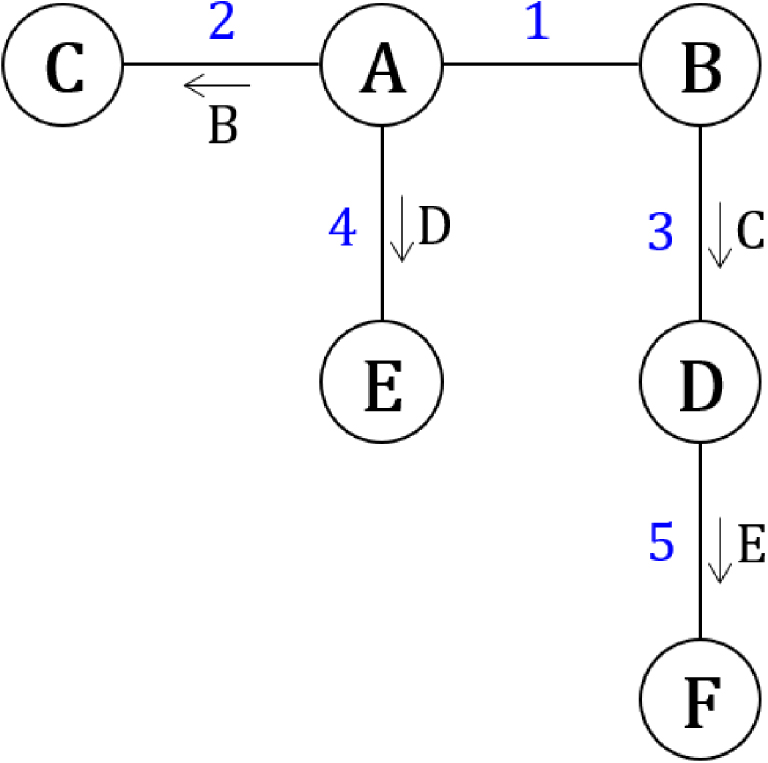
Si les constantes des équilibres avec catalyseur sont grandes devant celles des équilibres sans catalyseur, ce système évoluera de manière privilégiée en suivant l’ordre indiqué par les chiffres bleus. Le système ne pourra pas évoluer de A à C sans passer par B (étape 1), il ne pourra pas non plus évoluer de B à D sans passer par C (étape 2), et ainsi de suite. Dans le cas où un état est engagé dans deux transformations catalysées par des particules qui occupent un été particulier, le système évoluera de manière privilégiée vers la transformation dont l’espèce catalytique est apparue en premier (Figure 51), comme dans le cas de la cristallisation d’une seule des deux formes chirales de sels à partir du premier germe cristallin (Chapitre 5.9).
Evolution irréversible d’une transformation vers l’état occupé en premier.
Un tel phénomène constitue une bifurcation chimique.
6.6. Synthèse
La nature de l’entropie cinétique a été précisée dans ce dernier chapitre :
« Soit un système constitué de N particules engagées dans une transformation entre deux états, à pression (ou volume) et température constantes :
| (371) |
| (372) |
| (373) |
| (374) |
| (375) |
Une loi de Boltzmann thermique et cinétique a été établie :
« Soit un système constitué de N particules sphériques engagées dans une transformation entre deux états, à pression (ou volume) et température constantes :
| (376) |
| (377) |
Un système d’équations cinétiques permettant l’analyse de n’importe quel système de transformations couplées a été établi :
« Soit un système fermé délimité par une surface fermée, thermostaté à température T constante et de pression P ou volume V constant. Ce système est constitué depétats impliqués dansqtransformations élémentaires :
| (378) |
| (379) |
| (380) |
| (381) |
L’étude de systèmes de transformations couplées linéaire et cyclique montre que la connaissance de la constante d’équilibre d’une transformation isolée ne peut pas être utilisée pour déterminer la constante d’équilibre de cette transformation dans le cas général lorsque celle-ci est couplée à d’autres transformations. En revanche, la connaissance des constantes cinétiques est utilisable pour ce dessein. L’entropie d’un système de réactions couplées ne peut pas être isolée de l’énergie dans l’équation qui gouverne l’équilibre. L’existence d’une grandeur physique unique de nature entropique dont le sens de variation gouverne l’évolution d’un système est remise en cause par la description cinétique des équilibres.
Enfin, le modèle cinétique constitue un outil analytique permettant d’appréhender l’évolution de systèmes complexes incluant des bifurcations.
7. Conclusions et perspectives
7.1. L’erreur historique de Gibbs
Les différences entre le modèle cinétique de l’équilibre chimique présenté dans cette étude et le modèle thermodynamique sont colossales. En effet, les deux descriptions ne sont pas compatibles car les propriétés d’une des deux grandeurs fondamentales qui gouverne l’évolution, l’entropie, est fondamentalement différente. Le premier grand succès de Williard Gibbs, en 1873, fut l’étude de l’évolution de fluides avec une fonction mathématique de l’énergie et de l’entropie découverte quelques décades auparavant par Rudolph Clausius. Après le succès éclatant de ses travaux sur les fluides, en 1876, la tentation de Williard Gibbs fut d’utiliser l’entropie, de nature calorimétrique, pour prévoir l’évolution des réactions chimiques et transformation de phases, puisque ces transformations s’accompagnent dans le cas général d’échanges de chaleur. La question qui se pose est :
« Pourquoi l’entropie calorimétrique ne serait-elle pas appropriée pour décrire l’évolution de transformations microscopiques ? »
Autrement formulée :
« Pourquoi l’entropie cinétique serait plus appropriée que l’entropie calorimétrique pour décrire l’évolution de transformations microscopiques ? »
Un échange de chaleur se distingue fondamentalement d’une réaction chimique à l’échelle microscopique. En effet, lors de la collision de deux molécules ou atomes réactifs, l’échange d’énergie thermique (i.e. énergie cinétique) est un évènement certain alors que la réaction chimique est un évènement probable (Figure 52).
L’échange d’énergie des atomes ou molécules réactives A et B lors d’une collision est certain alors que la réaction est seulement probable.
Le modèle thermodynamique physique statistique ne prend pas plus en compte que la thermodynamique de Gibbs cette différence fondamentale : les fonctions d’état F et G sont établies en physique statistique pour le gaz parfait, puis extrapolées aux réactions chimiques et équilibres de phases. A contrario, le modèle cinétique de l’équilibre chimique est construit en introduisant un terme probabiliste dans l’équation élémentaire de l’équilibre : la réaction suite à une collision entre les atomes ou molécules A et B n’est pas certaine. L’équation de l’équilibre cinétique pour un nombre élevé de particules est fondée sur le comportement de ce système élémentaire et prend donc intrinsèquement en compte la différence fondamentale entre échange de chaleur et réaction chimique à l’échelle microscopique en termes de probabilités.
7.2. Les états inaccessibles
Au-delà des performances prédictives satisfaisantes de systèmes réels, la description cinétique des évolutions ouvre de nouvelles frontières. En effet, elle offre un degré de liberté de plus que l’approche thermodynamique chimique, celui de considérer que les produits d’une réaction chimique ne dépendent pas des stabilités intrinsèques des réactifs et produits, mais des vitesses à laquelle ils se forment et disparaissent, elles-mêmes dépendantes de l’environnement.
« Dis-moi à quelle vitesse tu te formes, je te dirais si tu es. »
« Dis-moi où tu te formes, je te dirais qui tu es. »
De ce fait, la description cinétique de l’équilibre chimique permet d’atteindre des états (molécule, polymorphe d’un cristal, conformation d’une molécule) qui sont inaccessibles, d’un point de vue conceptuel, à la thermodynamique, qui statue qu’une espèce chimique ou une phase est intrinsèquement stable ou instable.
7.3. Entropie cinétique et évolution
Un aspect intéressant qui émerge de la description cinétique des transformations microscopiques est la catalyse entropique, qui permet d’appréhender rationnellement le phénomène de bifurcation chimique d’un système de transformations microscopiques couplées : l’évolution d’un système vers une des voies qui s’offrent à lui est gouvernée par la nature de l’environnement chimique à l’instant où ces possibilités se présentent.
Remerciements
Les fondements utilisés dans le modèle cinétique de l’équilibre microscopique présenté dans cette étude sont issus de deux ouvrages de thermodynamique écrits en langue française : Eléments de thermodynamique statistiques [15] (1963), d’Adolphe Pacault et Thermodynamique, fondements et applications [4, 5, 83] (2001) de José-Philippe Pérez. La très grande clarté de ces deux ouvrages permet d’accéder sûrement à la connaissance des fondements de la thermodynamique à l’équilibre et de la physique statistique.
L’accès à des ouvrages récents et anciens et à des données expérimentales thermodynamiques a été possible grâce à l’accès ouvert de ressources numériques du World Wide Web, nous remercions le National Institute of Standards and Technology, la Dortmund Data Bank, la Bibliothèque Nationale de France pour l’accès Gallica aux fonds anciens et Ressource Canada.
Nous remercions les nombreuses personnes qui ont participé à l’éclosion de ce modèle. Nos collègues chercheurs pour leur écoute et leurs critiques constructives. Sophie Griveau, Virginie Lair et Gérard Côte de Chimie ParisTech, Jean-Christophe Lacroix, François Maurel et Michel Petitjean de l’Université Paris Diderot et Bernard Roques de l’Université Paris Descartes. Nos collègues du service de Technologies de l’Information et de la Communication pour l’Enseignement de la Faculté de Pharmacie de Paris qui ont réalisé le film de la première conférence de la description cinétique des équilibres le 12 janvier 2017[84], Stéphane Carraz, Alexandre Lacroix et Damien Renouf. Nos collègues des services de communication pour en avoir assuré sa diffusion, Samantha Conti de la Faculté de Pharmacie de Paris, Gwenaëlle Hennequin de Chimie ParisTech et Myriam Denis Ledru de l’Université de Versailles Saint-Quentin-en-Yvelines. Les documentalistes associés à nos établissements pour leur pugnacité et leur infatigabilité à localiser éditions rares et ouvrages anciens, Didier Partouche, Alain Delaforge et Eric Varon de la Bibliothèque Universitaire de Santé de Faculté de Pharmacie de Paris, Pascale Boi-Cuzin, Amélie Dessens et Sandrine Mouret de Mines ParisTech et Nathalie Queyroux de la Bibliothèque de Sciences Expérimentales de l’Ecole Normale Supérieure de Paris. Dominique Vichard, de l’Université de Versailles Saint-Quentin-en-Yvelines pour son soutien et Isabelle Gonne pour la relecture consciencieuse de ce manuscrit.
Annexes
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.40.
1L’énergie interne est notée E dans la formulation historique du potentiel chimique.
2La notation permet de distinguer la température du terme intégré de celle de la borne supérieure de l’intégrale.
3La nature de la phase ne sera pas précisée dans ce chapitre, afin de ne pas alourdir le formalisme des équations, puisqu’il n’y a qu’une seule phase.
4Les équilibres de fusion entre un polymorphe métastable et un liquide peuvent être décrits par l’approche cinétique présentée dans ce chapitre, en revanche la description cinétique de la transition solide–solide entre un polymorphe métastable et un polymorphe stable n’est pas abordée.
5La constante de proportionnalité du terme 𝜒 de cette équation, comparativement à celui de l’équilibre chimique précédent, a été modifiée de manière à exprimer l’équation d’évolution cinétique en fonction d’une fraction de l’énergie cinétique.