

1 Variations du CO2 atmosphérique
1.1 Cycle global préindustriel du CO2
Ce cycle inclut l'atmosphère (730 PgC), les plantes (500 PgC) et sols (1500 PgC), l'océan (38 000 PgC), le C organique fossile et les roches carbonatées (Fig. 1). Les flux bruts de CO2 entre atmosphère et biotope terrestre s'élèveraient à 120 PgC an−1, tandis que ceux entre atmosphère et océan seraient de 90 PgC an−1. Chaque année, l'atmosphère échangerait donc avec le biotope terrestre et l'océan environ 30 % de son inventaire de CO2. Nous limitons notre attention à l'océan. Notons que les flux associés à d'autres réservoirs, tels que l'activité biosphérique et l'érosion chimique continentales, peuvent aussi avoir notablement contribué aux variations de CO2atm considérées ici.

Cycle préindustriel global du carbone: inventaires (PgC) et flux (PgC an−1). Les lignes en tiretés indiquent les flux de CaCO3(s) importants pour les variations de CO2atm sur de très longues périodes de temps (d'après [21]).
Global pre-industrial carbon cycle: inventories (PgC) and fluxes (PgC an−1). The dashed lines indicate the CaCO3(s) fluxes that are important for the CO2atm variations on very long timescales (from [21]).
Le CO2atm pénètre dans l'océan par dissolution. Le CO2 aqueux donne par dissociation HCO3− et CO32−. Le C inorganique dissous total CT=[CO2(aq)]+[HCO3−]+[CO32−] constitue plus de 95 % du C dans l'océan. Les variables mesurables du système chimique (en surface) sont la température (T), la salinité (S), CT, l'alcalinité totale AT, le pH et la pression partielle de CO2 (pCO2). Déterminer la spéciation du système requiert la connaissance de quatre variables : T, S et un couple parmi CT, AT, pH, pCO2. Par exemple, la linéarisation de pCO2 autour de petites variations de T, S, CT, AT donne :
La distribution de CT et AT en océan ouvert montre des fortes valeurs en profondeur et de faibles valeurs en surface. Ces gradients traduisent la présence de mécanismes ou « pompes », pouvant partiellement contrecarrer l'effet du mélange [47]. La pompe de solubilité résulte de la dissolution de CO2 dans les eaux superficielles froides des hautes latitudes et de son injection en profondeur par la circulation. La pompe organique tend à diminuer CT et à augmenter AT en surface par photosynthèse de matière organique ; elle tend à augmenter CT et diminuer AT en profondeur par sa reminéralisation. La pompe des carbonates tend à diminuer CT et AT en surface par production de CaCO3(s) et augmenter CT et AT en profondeur par dissolution de CaCO3(s). La calcite et l'aragonite, précipitées dans l'océan, sont thermodynamiquement stables dans la partie supérieure de la colonne d'eau et instables dans sa partie inférieure. Le C organique est respiré dans toute la colonne d'eau, tandis que la dissolution de CaCO3(s) est présumée apparaı̂tre dans sa partie inférieure seulement. À l'inverse du C organique, une partie importante du CaCO3(s) est préservée dans le sédiment. L'effet net des pompes biologiques sur pCO2 dépend notamment du rapport atomique C organique/CaCO3(s) du matériel exporté vers les eaux profondes. Notons que les cycles du C organique et des carbonates très simplement décrits ci-dessus n'incorporent pas plateaux et talus continentaux [52].
1.2 Des reconstructions récentes
Indermühle et al. [19] rapportent une reconstruction de CO2atm basée sur une carotte de Taylor Dome (78° S, 159° E ; cercles rouges sur la Fig. 2). Les mesures montrent (i) une diminution d'environ 8 ppmv de 11 à 8 ka BP, et (ii) une augmentation d'environ 25 ppmv sur les 7 ka suivants. Fluckiger et al. [17] confirment essentiellement ces deux caractéristiques par des mesures dans une carotte de Dome C (75° S, 123° E). Monnin et al. [31] publient une reconstruction de CO2atm basée sur la carotte de Dome C (cercles bleus sur la Fig. 2). Les mesures montrent une augmentation d'une moyenne de 189 ppmv entre 18,1 et 17,0 ka BP à une moyenne de 265 ppmv entre 11,1–10,5 ka BP. Cette augmentation de 76±1 ppmv sur 6 ka n'est pas continue. Elle comprend quatre intervalles distincts, tous identifiables dans les mesures de CH4 pour la même carotte. Les mesures de CO2 dans la carotte de Vostok (78° S, 106° E) montrent qu'une augmentation d'amplitude comparable a accompagné les trois précédentes déglaciations [33,35]. Indermühle et al. [20] rapportent une reconstruction de CO2atm basée sur la carotte de Taylor Dome (cercles verts sur la Fig. 2). Les mesures montrent quatre oscillations, avec une amplitude pic-à-pic d'environ 20 ppmv entre 20 et 60 ka BP. Ces résultats ont été comparés avec des mesures de CO2 dans une carotte de la station Byrd (80° S, 120° W) [43].
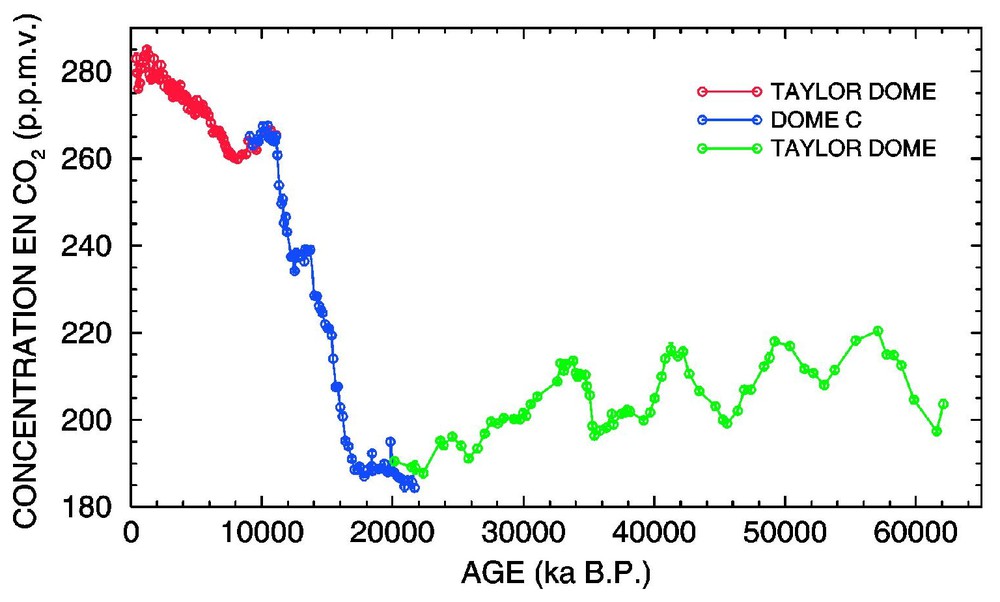
Reconstructions de CO2atm pour l'Holocène (rouge, [19]), la dernière déglaciation (bleu, [31]) et la dernière glaciation (vert, [20]). Les incertitudes sur les mesures sont comparables à la taille des symboles.
CO2atm reconstructions for the Holocene (red, [19]), the last deglaciation (blue, [31]), and the last glaciation (green, [20]). Measurement uncertainties are comparable to the symbols' size.
1.3 Difficultés d'interprétation
Nous discutons de difficultés d'interprétation associées à la modélisation du cycle du carbone dans l'océan. Pour une connaissance de différentes interprétations, le lecteur consultera les références originales et des révisions [1,3,40]. Marchal et al. [27,28] et Schulz et al. [37] abordent les variations de CO2atm durant des changements climatiques rapides. Avant de discuter des difficultés de modélisation, notons le problème fondamental posé par l'interprétation des variations glaciaire/interglaciaire de CO2atm associé à l'existence de plusieurs mécanismes capables d'expliquer individuellement une grande partie de ces variations [25]. La même difficulté apparaı̂t probablement pour l'Holocène (0–11,5 ka BP) et la dernière glaciation. Saluons enfin les efforts récents pour mesurer à haute résolution le rapport 13C/12C du CO2 occlus dans les glaces polaires [16,26], qui devraient mieux contraindre les interprétations de CO2atm.
Modéliser le cycle du carbone dans l'océan passé comporte des difficultés majeures. Les champs superficiels de T, S, CT, et AT sont affectés par la circulation océanique générale (COG). Un premier type de difficulté est la modélisation de la COG passée et de ses variations. Notons les très faibles contraintes sur les conditions aux limites de surface (tensions de vent, flux de chaleur et d'eau douce). Une grande partie des éléments de la COG, divergences et convergences d'Ekman, intensité des courants de bord ouest, etc., est déterminée par le vent et son rotationnel. Il n'existe pas d'indicateur de paléo-vitesse de vent, qui puisse nous renseigner sur sa distribution à grande échelle. Malgré les efforts pour reconstruire les T et S superficielles, les incertitudes sur ces reconstructions sont trop larges pour permettre des simulations « robustes » de la COG [51]. Notons aussi la difficulté qu'il y a à représenter le mélange turbulent dans les modèles de COG. Le mélange diapycnal (au travers de surfaces d'égale densité) pourrait être le moteur de la circulation sous la base de la thermocline principale (à une profondeur d'environ 1 km) et donc dans la majorité du volume océanique [32]. Les modèles actuels paramétrisent le mélange de façon frustre, se basant sur des viscosités et diffusivités turbulentes. Le problème du mélange diapycnal est accentué dans les simulations paléoclimatiques, vu les variations probables, durant le récent passé géologique, de l'énergie disponible pour le réaliser [54]. Ces variations pourraient affecter l'efficacité des pompes décrites ci-dessus [1].
Un second type de difficulté concerne le transport de CO2 à l'interface air–mer et dans l'océan. Bien que la vitesse de transfert de CO2 à l'interface air–mer dépende d'une multitude de paramètres physiques, elle est usuellement exprimée comme une fonction de la vitesse du vent seulement. Cette fonction reste incertaine [49,50], et les vents pour les climats anciens sont inconnus. La modélisation des pompes biologiques se heurte à beaucoup de difficultés [14]. L'importance relative de différents facteurs dans la production de carbone organique exportée vers les profondeurs (teneur et nature d'éléments bio-limitants, lumière, mélange vertical, T, structure de l'écosystème pélagique, etc.) est mal comprise. Les facteurs contrôlant la production de CaCO3(s) sont très mal connus. Certains modèles supposent que cette production est proportionnelle à la production de carbone organique. Les estimations du rapport atomique Corganique/CaCO3(s) du matériel exporté vers les eaux profondes varient largement [36]. Reminéralisation de C organique et dissolution de CaCO3(s) dans la colonne d'eau sont très mal contraints. Certains modèles supposent que la reminéralisation épouse un profil vertical déterminé. Les profils rapportés dans la littérature diffèrent fortement dans la partie supérieure de la colonne d'eau [52]. La répartition du flux de dissolution de CaCO3(s) entre colonne d'eau et sédiments est mal connue. Certaines observations suggèrent une dissolution considérable dans les premiers 500 à 1000 m de la colonne d'eau [30]. Le rôle de la matière organique dissoute dans le cycle du carbone océanique est encore mal établi [18].
2 Variations du CH4 atmosphérique
2.1 Cycle global pré-industriel du CH4
À la différence du CO2, le CH4 ne met en jeu que deux réservoirs essentiels : l'atmosphère, contenant aujourd'hui 1745 ppbv, soit 4850 Tg de CH4 [21], et la biosphère continentale où ce dernier est continuellement produit, pour l'essentiel, par des bactéries méthanogènes.
Dans l'atmosphère, 85–90 % du CH4 émis réagit avec le radical hydroxyle OH. Présent à des teneurs extrêmement faibles de l'ordre de 7×105 molécules cm−3 (soit ∼0,02 pptv), ce radical est continuellement produit par photolyse de l'ozone, générant de l'oxygène excité O(). Ce dernier réagit avec la vapeur d'eau pour conduire à deux radicaux OH. La constante cinétique de la réaction CH4+OH étant de 7,7×10−15 molécules cm3 s−1 à 15 °C, le temps de vie de CH4 relatif à cette réaction (temps nécessaire pour réduire son rapport de mélange d'un facteur e) est de 9,6 ans [21]. Cinq à 10 % du CH4 émis vers l'atmosphère atteint la stratosphère (altitude supérieure à 15 km environ) pour y être oxydé soit par OH, O(), ou encore le Cl radicalaire. Environ 5 % du CH4 atmosphérique retourne à la biosphère continentale par diffusion gazeuse dans les sols suivie d'une oxydation par des bactéries méthanotrophes. Ces puits conduisent à un temps de résidence atmosphérique moyen de CH4 de 8,4 ans [21].
Quels sont les compartiments de la biosphère continentale générant du CH4 hors contribution humaine (Fig. 3) ? Communément appelé « gaz des marais », le CH4 provient pour l'essentiel de la décomposition de la matière organique dans les zones humides, là où le sédiment ne contient pas d'O2. En réalité, une grande partie (jusqu'à 95 %) du CH4 produit dans le sédiment anaérobie est oxydée lors de son transport diffusif vers le sédiment aérobie [5]. Estimer la quantité de CH4 rejoignant l'atmosphère est un exercice périlleux (pour une synthèse récente, voir [11]). La difficulté tient à deux faits. D'une part, les zones humides constituent des écosystèmes diffus, éventuellement saisonniers, occupant les dépressions topographiques où l'eau peut stagner. Leur inventaire impose donc une approche à l'échelle du kilomètre voire moins et s'avère donc particulièrement délicat dans les zones continentales mal documentées (régions tropicales, par exemple). D'autre part, les facteurs environnementaux contrôlant les flux de CH4 d'une zone humide sont nombreux et mal contraints : température dans le sol, degré de saturation en eau, apport en nutriment, pH, échange avec l'atmosphère par diffusion ou bullage, épaisseur de la zone oxydante en surface du sédiment, etc. (pour une synthèse, voir [48]). Les estimations actuelles des flux globaux de CH4 depuis les zones humides demeurent donc entachées d'une large incertitude, variant de 115 à 237 Tg an−1 [21].
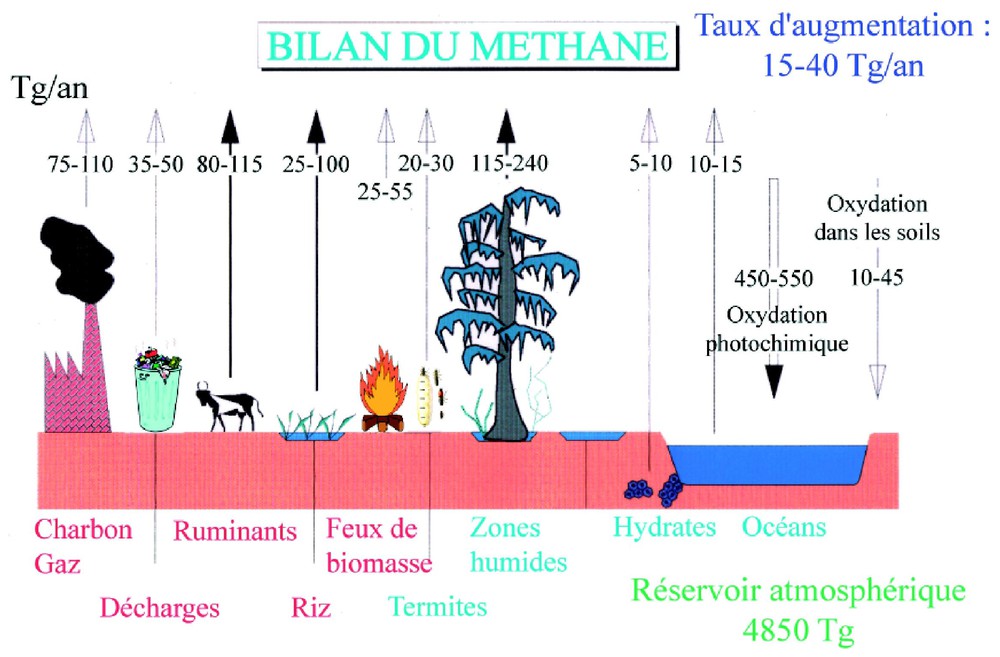
Sources et puits actuels de CH4 dans l'atmosphère, et leur importance relative (adapté de [21]).
Present-day sources and sinks of atmospheric CH4, and their relative importance (adapted from [21]).
Les autres sources naturelles de CH4 mettent en jeu une décomposition de matière organique en milieu anaérobie (fermentation dans la panse des ruminants, digestion des termites, réduction de l'acétate dans les couches anoxiques des océans), une décomposition thermique (feux de biomasse avec combustion incomplète), ou un dégazage de CH4 fossile (gaz naturel, charbon, volcans, hydrates en fond océanique ou dans le pergélisol). Ces sources représentent de 5 à 20 Tg a−1, selon les estimations les plus récentes [21].
Extrapoler les observations actuelles, déjà affectées de larges incertitudes, pour construire un bilan préindustriel du CH4 demande quelques précautions. (1) Le temps de vie de CH4 relatif aux radicaux OH dépend du rapport de mélange de ces derniers. Aucune mesure ne permettant d'estimer ce rapport dans le passé, on s'appuie sur des modèles photochimiques. Ceux-ci ([12,29,45] et références incluses) fournissent une gamme de variations depuis la période préindustrielle allant d'une perte de 4 à 60 %, à un gain de 6 à 15 %. Cette dispersion résulte des conditions initiales en terme de teneurs en oxydes d'azote et en hydrocarbures non méthaniques, elles-mêmes dépendant d'hypothèses non vérifiées sur leurs sources. (2) Certaines zones humides ont été asséchées au cours des derniers siècles [7]. (3) L'impact de l'activité humaine sur le bilan du CH4 était probablement non négligeable il y a 200 ans, d'une part du fait des activités agricoles (élevage de ruminants, riziculture), d'autre part du fait de l'utilisation généralisée du bois comme source de combustion [44]. Les quelques bilans ainsi réalisés et tenant compte peu ou prou des éléments énoncés ci-dessus font état d'une source globale préindustrielle située entre 180 et 250 Tg a−1, dans laquelle les zones humides contribuent à hauteur de 75 % [7,12,15,29,44].
2.2 Des reconstructions récentes
Durant les derniers 11,5 ka, les teneurs en CH4 montrent deux maxima voisins de 700 ppbv : durant les derniers 1000 ans et entre 10–11,5 ka BP. Ils encadrent un minimum de ∼570 ppbv centré sur 5,2 ka BP [2,7,17]. Superposée à cette tendance générale, une forte décroissance (∼100 ppbv) est observée il y a 8200 ans, concomitante à un refroidissement brutal observé dans plusieurs enregistrements climatiques [2,41].
La comparaison des enregistrements groenlandais et antarctiques fait apparaı̂tre une différence interpolaire, résultant de la distribution inégale des sources en latitude et du temps de résidence relativement court du méthane dans l'atmosphère. Une différence moyenne de 44±7 ppbv est observée durant l'Holocène, avec des variations de l'ordre de 10 ppbv [8,9]. Son interprétation avec un modèle simple de l'atmosphère suggère une prédominance des sources tropicales ; les sources au nord de 30° N auraient largement contribué durant la période 11,5–9,5 ka BP [10].
Au cours de la dernière déglaciation, les teneurs en méthane doublent entre 18 et 11,5 ka BP, passant de ∼350 ppbv à ∼700 ppbv. La transition s'effectue en quatre étapes majeures : une augmentation lente de ∼100 ppbv jusqu'à 14 ka BP, suivie d'une augmentation brutale de ∼200 ppbv en un siècle, puis une chute tout aussi brutale et ample durant l'épisode froid du Dryas récent, et enfin l'augmentation rapide conduisant au début de l'Holocène, avec +220 ppbv en un siècle également [4,6,10,31]. Grâce à certains traceurs piégés dans la glace, on montre que les deux épisodes d'accroissement brutal prennent place simultanément aux réchauffements climatiques correspondants au Groenland, ou avec un très léger retard (30 ans) [39].
Cette correspondance entre CH4 et climat groenlandais à l'échelle de quelques décennies se retrouve tout au long de la dernière glaciation. Ainsi, chaque événement climatique rapide de type Dansgaard/Oeschger trouve son équivalent en CH4, avec des amplitudes de variations allant de 50 à 200 ppbv [4,7 (fig. 4), 13]. L'exception observée durant la période entre 60–80 ka BP (Fig. 4) a été récemment infirmée par des mesures très détaillées sur le forage North GRIP (J. Chappellaz, données non publiées). Les quelques mesures de gradient réalisées parallèlement sur certains événements rapides suggèrent une contribution dominante des sources au nord de 30° N [13].
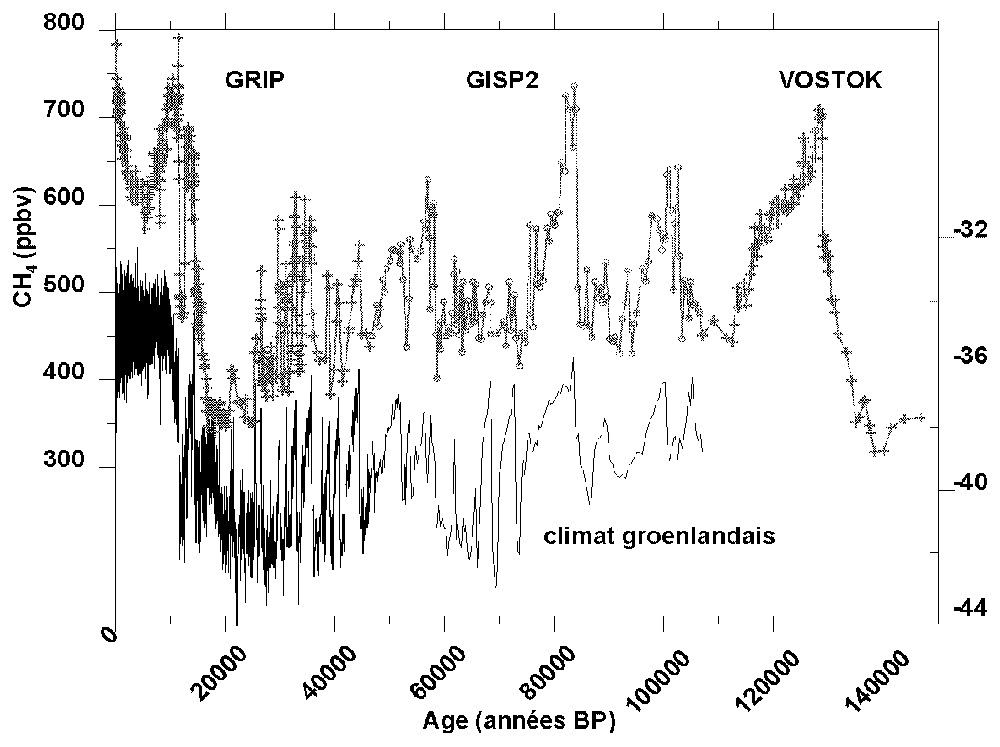
Évolution des teneurs atmosphériques en CH4 durant le dernier cycle climatique. L'enregistrement à la meilleure résolution temporelle pour chaque période est reporté (GRIP : [13]; GISP2 : [4] ; Vostok : [10]). La datation utilisée est celle du forage GISP2 jusqu'à 110 000 ans et celle de Vostok [35] au-delà.
Evolution of the concentration of atmospheric CH4 during the last climatic cycle. The record with the highest temporal resolution for each period is shown (GRIP: [13]; GISP2: [4]; Vostok: [10]). The time scale is the one of the GISP2 ice core up to 110,000 years and the one of Vostok [35] beyond.
Enfin, à l'échelle des quatre derniers cycles climatiques, le méthane atmosphérique apparaı̂t largement empreint du métronome astronomique, avec de grandes variations glaciaire–interglaciaire dans une gamme 320–800 ppbv, ponctuées d'événements moins intenses durant les glaciations [35].
2.3 Difficultés d'interprétation
Les grandes tendances temporelles passées du CH4 reposent qualitativement sur trois mécanismes principaux. (1) Le premier d'entre eux est l'intensité du cycle hydrologique, conduisant à des étendues variables de zones humides, soit en zone boréale (localisation actuelle des grandes tourbières), soit en zone tropicale. Les variations de température du sol altérent au 2ème ordre l'intensité des flux d'émission et l'étendue des grands inlandsis modulant les surfaces continentales disponibles au nord de 50° N. (2) Les variations des teneurs en radicaux hydroxyles OH affectant le temps de vie du méthane constituent le deuxième de ces mécanismes, le dernier étant (3) la décomposition d'hydrates de CH4 sur les marges et talus continentaux et dans le pergélisol.
On conçoit aisément la difficulté qu'il y a à estimer l'évolution des zones humides aux grandes échelles de temps. Si quelques études régionales existent (par exemple, [34]) à partir de paléo-données, les estimations globales ne reposent que sur des outils de modélisation. Seules deux études portant sur le dernier maximum glaciaire et sur la période préindustrielle existent, l'une reposant sur des cartes de végétation pondérées par la topographie du sol [7], l'autre sur un modèle de végétation couplé à un modèle climatique [22]. La première aboutit à une réduction de leur étendue de 50 % au dernier maximum glaciaire, alors que la seconde propose une augmentation de 15 %. En termes de flux de CH4 depuis ces zones humides, elles concluent respectivement à une diminution de 35 et 24 %.
Les modèles photochimiques appliqués aux conditions glaciaires fournissent unanimement des teneurs en OH de 10 à 30 % supérieures à celles prévalant en conditions interglaciaires ([12,29,45] et références incluses). Surtout, ils nécessitent de modifier l'intensité des sources de méthane pour qu'en retour les teneurs en OH soient affectées et amplifient ce forçage initial. On a tenté d'utiliser les teneurs en formaldéhyde (HCHO) dans la glace antarctique pour valider ces modèles : elles conduisaient au contraire à une diminution des teneurs en OH durant le glaciaire d'un facteur 2 à 4 [42]. La complexité de la fonction de transfert entre le signal de HCHO dans la glace et celui prévalant dans l'atmosphère polaire rend hélas peu probant ce type d'étude.
Compte tenu, d'une part, de la taille potentiellement gigantesque de ce réservoir naturel de méthane (plusieurs milliers de Gt) et, d'autre part, de la sensibilité du réservoir aux variations de température et de pression (niveau des mers), le scénario d'une contribution de dégazage d'hydrates de CH4 rencontre un succès croissant. Il s'est trouvé renforcé par l'observation sporadique d'anomalies isotopiques dans les tests calcaires d'organismes benthiques (pour une synthèse, voir [24]). Toutefois deux observations issues des carottes de glace s'y opposent :
- (1) le taux d'augmentation des teneurs en méthane au cours des quatre derniers cycles climatiques ne dépasse jamais 2 ppbv a−1 et les teneurs elles-mêmes apparaissent remarquablement stables une fois l'augmentation effectuée ;
- (2) l'étude détaillée de la relation de phase entre climat groenlandais et teneurs en CH4 n'indique nulle part une avance du méthane sur le climat, compatible avec un scénario de forçage par le gaz à effet de serre, suivi d'un effet boule de neige sur le système climatique [4].
3 Des recommandations
3.1 Pour le CO2atm
- (1) Toute interprétation de variation de CO2atm impliquant l'océan doit considérer quatre variables afin d'être cohérente.
- (2) Les conditions aux limites de surface de la COG passée doivent être bien mieux contraintes.
- (3) Il est nécessaire de garder à l'esprit le caractère arbitraire de la paramétrisation du mélange dans les modèles de COG. L'importance de la formulation ou tout au moins de l'intensité du mélange dans les modèles du cycle du C doit être examinée.
- (4) La description des pompes biologiques dans les modèles globaux est très naı̈ve. Des efforts ont été réalisés pour décrire de façon détaillée l'activité biologique en surface. Cependant, ces descriptions requièrent un grand nombre de paramétrisations et paramètres qui sont très mal contraints. Dès lors, il n'est pas évident qu'à une description plus détaillée de cette activité corresponde une simulation plus réaliste du cycle du carbone. Il est impératif, dans l'état actuel des connaissances, de maintenir une « hiérarchie » dans les modèles. Améliorer la connaissance des pompes biologiques nécessite des observations appuyées dans des milieux océaniques représentatifs.
- (5) Aucune donnée n'est parfaite et aucun modèle n'est parfait. Même les modèles de COG supposés les plus réalistes comportent des erreurs. Les procédures inverses permettent de traiter de façon explicite les erreurs inhérentes dans les observations et modèles [53]. Nous recommandons leur usage dans les études d'interprétation des variations de CO2atm (voir, par exemple, [25,46]).
3.2 Pour le CH4atm
- (1) Le développement de paramétrisations réalistes permettant de simuler la distribution des zones humides à partir des modèles climatiques constitue l'étape indispensable pour progresser dans l'interprétation des variations passées de CH4atm.
- (2) Le rôle de la chimie atmosphérique dans le bilan passé du CH4 demeure très mal contraint ; le facteur limitant concerne les hypothèses émises sur les bilans d'autres traces gazeuses réactives, en particulier les oxydes d'azote et les hydrocarbures non méthaniques. Le développement de paramétrisations adéquates pour ces composés sera nécessaire. Elle passe aussi par une meilleure compréhension des bilans actuels.
- (3) L'hypothèse des dégazages d'hydrates de CH4 nécessite d'être quantitativement abordée. Il faut développer des modèles thermodynamiques du sédiment superficiel sur les marges continentales et dans le pergélisol, mais aussi des modèles biophysiques tenant compte de la dynamique de recharge des réservoirs (soit par activité biologique, soit par diffusion depuis des réservoirs profonds) et du devenir du CH4 dans la colonne d'eau, une fois relargué.
- (4) Les contraintes d'observation sur le bilan du CH4 peuvent être accrues : évolution de la composition isotopique du CH4 (13C/12C : traceur possible des bandes latitudinales impliquées, D/H : contribution des hydrates de celle des autres sources), produits d'oxydation comme le monoxyde de C ou les acides organiques.
Remerciements
O.M. remercie le Penzance Endowed Fund of Assistant Scientists. J.C. remercie les programmes nationaux PNEDC et ECLIPSE (Insu–CNRS), ainsi que la Fondation de France et la Communauté européenne pour le soutien apporté aux études des archives glaciaires.