

1 Introduction
Arsenic (As) is a toxic element that can lead to wide-scale poisoning in populations when present in waters (Anawar et al., 2003; Fendorf et al., 2010; Smedley and Kinniburgh, 2002). Since wetlands can be an important source of waters for both ground and river waters, they can transfer As to these hydrosystems for two reasons. Firstly, wetlands can be enriched in As-bearing Fe(III) oxides through erosion and deposition during high-flow periods of flooding (Kocar et al., 2008; Polizzotto et al., 2005). Secondly, the anaerobic conditions that periodically develop in these systems can lead to the reduction of the Fe(III) oxides and to As release within wetland waters, these latter being then able to pollute the water masses with which the wetlands are connected, namely the underlying groundwater and the adjacent river network (Anawar et al., 2003; Baeyens et al., 2007; Kalbitz and Wennrich, 1998).
In wetlands categorized as riparian zones (associated with streams), flows out of wetlands are preferentially performed by evapotranspiration, surface runoff, and sub-surface water outflow. In these waters undergoing oxidation–reduction events, precipitation or dissolution of carrier phases can play a major role on the mobility of dissolved trace elements. However, the biogeochemistry of the released As is still poorly constrained, particularly as regards to the role played by Fe(II) re-oxidation processes that may occur during the transport of the As from its wetland source to the hydrosystems. Indeed, the re-oxidation process could lead to the precipitation of secondary Fe(III) oxyhydroxide crystals that could immobilize the released As. This process of As scavenging by secondary Fe(III) oxyhydroxides could be a very efficient, naturally occurring attenuation process, particularly in oxygenated hydrosystems, such as streams and rivers. However, this attenuation role of secondary Fe(III) precipitation regards to As transport to streams and rivers could be challenged by the high dissolved organic matter (DOM) concentration of wetland waters that could stabilize the As in solution or lead to the formation of highly mobile Fe(III)-DOM colloids. In fact, several studies already showed that DOM could critically affect the aquatic geochemistry and the fate of As (Fendorf et al., 2010). The formation of As-DOM complexes has been already documented for various As species and DOM types, as well as for various physicochemical environmental media (Buschmann et al., 2006; Liu and Cai, 2010; Liu et al., 2011). The extent of As-DOM binding varies with DOM origin and metal loading (mostly Fe) of the DOM phase (Liu et al., 2011; Redman et al., 2002). Likewise, studies using ultrafiltration techniques have already demonstrated that most of the Fe occurring in wetlands or organic-rich solutions was found in the suspended colloidal fractions (Allard et al., 2004; Gaffney et al., 2008; Pédrot et al., 2008). These colloids may include large DOM species coagulated by chelated Fe and/or stabilized by ferric nano-oxyhydroxides (Pédrot et al., 2011; Pokrovsky et al., 2005). Several authors have recently shown that such Fe-DOM associations could be the carrier phases of As(V) in organic-rich and wetland solutions (Mikutta and Kretzschmar, 2011; Ritter et al., 2006). Arsenic may be either associated with Fe–OM colloids via Fe(III)-cationic bridging or via adsorption/incorporation in Fe(III) solid phases, or directly bound to OM molecules via functional groups, such as –SH (Gorny et al., 2015; Liu and Cai, 2010; Liu et al., 2011; Mikutta and Kretzschmar, 2011).
The occurrence of mixed Fe nano-oxyhydroxides-OM colloids has been assumed in several studies that focused on organic-rich waters (Pédrot et al., 2008; Pokrovsky et al., 2005; Rose et al., 1998). Recent studies dedicated to investigating the ability of DOM to control Fe speciation have demonstrated that Fe(II) oxidation in the presence of organic molecules can promote the formation of small amorphous Fe nano-oxyhydroxides colloids instead of Fe microparticles as DOM inhibits the development of large hydroxide crystals (Gaffney et al., 2008; Pédrot et al., 2011). The high sorption capacity of these neo-formed Fe nanoparticles makes them important potential transport vectors for contaminants in the environment (Warren and Haack, 2001). Their small size, due to their intimate association with DOM, confers them the capacity of also transporting the contaminants over long distances.
Despite the evidence of environmental occurrences of As–Fe–OM associations, no study has so far investigated in details the behaviour of As during the oxidation of Fe(II) and DOM-rich solutions, despite the potential environmental importance of those associations as potential carriers of As contamination in rivers fed by wetland waters. This study was dedicated to the reporting of a series of experiments, which were conducted to fill this gap. More specifically, oxidation of Fe(II) solutions were carried out with and without the addition of dissolved As and DOM (as humic acid) in order to specify the impact of DOM on the fate of As during Fe(II) oxidation. This was done by combining ultrafiltration experiments and NanoSIMS images. NanoSIMS – Nanoscale Secondary Ion Mass Spectrometers – are ion probes that can be used to obtain precise, spatially explicit, maps of elemental and isotopic distribution in complex matrices with a nanoscale spatial resolution (Herrmann et al., 2007). This technique offers many exciting opportunities for potential applications within the field of biogeochemistry (Herrmann et al., 2007; Moore et al., 2012), and this study may also be viewed as an experimental investigation of the ability of NanoSIMS instruments to map the distribution of As in both pure Fe and mixed Fe–OM colloids.
2 Materials and methods
Three types of Fe2+ oxidation experiments were carried out, namely (i) without humic acid (HA) and As, (ii) with As only and (iii) with HA and As. Humic acid suspension was obtained from standard humic acid (Leonardite, 1S104H, IHSS).
An acidified (HCl) 702 mg·L−1 Fe(II) stock solution was prepared with FeCl2. This solution was then added at 0.1 mL·min−1 with an automated burette (Titrino 794, Metrohm) in a thermostatic beaker set at 25 °C to three suspensions in equilibrium at pH 6.5, namely (i) a 0.01 M electrolyte (NaCl) solution for the Fe system (Exp. 1), (ii) a 0.01 M electrolyte (NaCl) solution containing As(III) for the Fe–As system (Exp. 2), and (iii) a 0.01 M electrolyte (NaCl) solution containing both As(III) and HA for the Fe–As–HA system (Exp. 3). These three suspensions, in contact with the atmosphere, were continuously stirred throughout the Fe(II) oxidation experiments. The buffering capacity of HA in equilibrium at pH 6.5 was tested in Exp. 3 to evaluate the hydroxyl consumption with no change in the pH. Simultaneously in these three suspensions, the pH was continuously maintained at 6.5 by progressively adding 0.1 M NaOH using a second automated burette programmed in a pH stat mode (Titrino 794, Metrohm). The accuracy of the pH measurement was ± 0.04 pH units. At the end of the Fe(II) addition (t = 250 min), the suspensions were left in contact with the atmosphere at pH 6.5 for another 750 min. Table 1 shows the Fetot, Astot, and DOCtot concentrations of suspensions at the end of Fe(II) addition for the three systems, as well as the volume of NaOH added during Fe oxidation–hydrolysis experiments.
Iron, As and DOC concentrations (mg·L−1) in the different investigated Fe systems at the end of the experiment.
| [Fe]final | [As]final | [DOC]final | Vol. of NaOH added (mL) | |
| Fe system (Exp. 1) | 111.4 ± 0.6 | / | / | 7.05 ± 0.03 |
| Fe–As system (Exp. 2) | 93.2 ± 1.1 | 13.1 ± 0.2 | / | 7.28 ± 0.13 |
| Fe–As–HA system (Exp. 3) | 94.7 ± 0.3 | 12.9 ± 0.5 | 63 ± 2.9 | 8.59 ± 0.35 |
Syntheses were performed in triplicate for Exp. 3 with HA and replicated five times for Exp. 1 and two without HA. The O2 concentration and Eh values were recorded during the running time of the synthesis experiment using an oxygen sensor electrode (Fisher Scientific Bioblock) and a combined Pt electrode (Fisher Scientific Bioblock), respectively.
The suspension was sampled at t = 1000 min for each experiment, filtered through a 0.2-μm pore size cellulose acetate filter capsule (Sartorius Minisart) to separate the particulate phase from the colloidal and dissolved phases, the filtrate being then ultrafiltrated to separate the colloidal and the dissolved fractions using decreasing pore size cuts. For this purpose, 15-mL centrifugal tubes (Sartorius Vivaspin 15R Hydrosart) equipped with permeable membranes of successively 30 kDa and 2 kDa were used. Each centrifugal filter device was washed and rinsed with HCl 0.1 N and ultra-pure water three times before use. Centrifugations were performed using a Jouan G4.12 centrifuge with swinging bucket at about 3000 g for 20 min for 30-kDa devices and 3750 g for 30 min for 2-kDa devices (Pédrot et al., 2010).
The dissolved organic carbon concentration was analysed on a Total Organic Carbon Analyser (Shimadzu TOC-5050A). The accuracy of the DOC measurement was estimated at ± 3% by using a standard solution of potassium hydrogen phthalate. The concentrations of Fetot and Astot were determined using a furnace atomic absorption spectrometer (SOLAAR M6, THERMO). The concentration of As species – As(III) and As(V) – were obtained using the hydride generation–atomic absorption spectrometry (HG–AAS) for As(III) determination, via an atomic adsorption spectrometer (SOLAAR M6, THERMO) equipped with a vapour injection system (Vapor System VP100 THERMO) to generate hydrides (Nielsen and Hansen, 1997). Always negligible, blank test performed to determine possible contamination were below 0.4 mg·L−1, < ql of 5 μg·L−1 and < ql of 1 μg·L−1 for DOC, Fe and As, respectively.
A sample of each experimental suspension was dried out at the critical point (Balzers Instruments, CPD010), and observed by Scanning Electron Microscopy (SEM). The samples were observed after having been coated, as required, with Au–Pd nanoparticles by cathodic deposition with a JEOL JFC 1100 sputter. The equipment used was a JEOL JSM-6301F Field Emission Gun Scanning Electron Microscope operated at 5, 7 or 9 kV. These analyses were performed at the CMEBA Analytical Facility at the University of Rennes 1. In Exp. 1, the Fe precipitates were washed 5 times in ultra-pure water, frozen, and then lyophilized at 0.120 mbar (Alpha 1-4, Christ, S4 Osterode, Germany) to allow X-Ray Diffraction (XRD) characterization. XRD scans were performed with a Philips X’Pert diffractometer, using a Cu source operating at 45 kV and 40 mA, and a Kevex Si (Li) solid-state detector. All samples were run in step-scan mode (0.04°/30 s) with the angle 2Θ ranging from 8.5° to 90°.
In Exps. 2 and 3 with As, the suspensions were analysed by NanoSIMS probe. These NanoSIMS analyses were performed on a NanoSIMS 50 standard ion microprobe (Cameca) at the University of Rennes-1. A Cs+ primary ion beam was used with 16 keV and focused to a spot of 100 nm in routine condition and scanned in a raster pattern on the surface sample. Secondary singly negatively charged secondary ions were sputtered away. They were extracted for mass analysis and collected in electron multipliers. Caesium images were recorded under a ∼ 15-pA primary beam (256 × 256 pixels, 55 × 55 μm, acquisition time = 11 min). A mass resolving power of ∼ 4000 (M/ΔM), enabling the removal of potential mass interferences, was used for all analyses. The chemical maps were determined by image processing using ImageJ software (1.38x version) and the NRIMS ImageJ analysis module. Line scans were generated with the function “Plot Profile” after having reduced the background noise of the isotopic distributions by applying a mean filter treatment, with a radius of 2 pixels for each pixel. Pearson's coefficient (PC) was calculated to determine the dependency of pixels in dual-channel images. PC is a coefficient where 1 and –1 represent a perfect colocalization and perfect exclusion, respectively, and where 0 represents a random colocalization. The colocalization analyses were performed under JACoP, a novel plug-in of Image J developed by Bolte and Cordelières (2006). A detailed description of this statistical treatment method can be found in Al-Sid-Cheikh et al. (2015).
3 Results and discussion
3.1 Impact of humic acid and As occurrence on the size and nature of the secondary Fe(III) precipitates
To investigate the impact of oxidation on the fate of dissolved Fe2+ and As(III) in a reduced solution, experimental studies on the Fe2+ oxidation–hydrolysis kinetics were performed with and without As(III) and humic acid (Leonardite Humic Acid). Concentrations of Fe(II), Fe(III), As(III) and As(V), Eh as well as the rate of hydroxyl consumption (V(t)) by pH and OH molecules were monitored. The final stoichiometry of the Fe2+ oxidation–hydrolysis reaction – calculated as in Fakih et al. (2008) and Pédrot et al. (2011) – suggested that Fe micrometric particles were formed without HA and amorphous Fe phases with HA (Table 2).
Normalized rates of hydroxyl consumption (V(t)) and proportion (%) of Fe, Astotal and DOC in the different cut-off fractions in the investigated Fe systems at the end of experiment. ql refers to the quantification limit (ql of 5 μg·L−1 for Fe, ql of 1 μg·L−1 for As and ql of 0.3 mg·L−1 for DOC).
| Proportion in fraction > 0.2 μm | Proportion in 0.2 μm > fraction > 30 kDa | Proportion in fraction 30 kDa > fraction > 2 kDa | Proportion in fraction < 2 kDa | ||||||||||
| V(t) | Fe | Astotal | DOC | Fe | Astotal | DOC | Fe | Astotal | DOC | Fe | Astotal | DOC | |
| Fe system | 2.02 ± 0.2 | 100% | < ql | < ql | < ql | < ql | < ql | < ql | < ql | < ql | < ql | < ql | < ql |
| Fe–As system | 2.03 ± 0.2 | 100% | 97.3 ±0.5% | < ql | < ql | < ql | < ql | < ql | < ql | < ql | < ql | 2.5 ± 0.9% | < ql |
| Fe–As–HS system | 2.53 ± 0.3 | 6 ± 0.5% | 1.1 ± 0.9% | 4.6 ± 1.1% | 94 ± 2.5% | 95 ± 1% | 85.8 ± 3.1% | < ql | < ql | 9.6 ± 1.9% | < ql | 4.1 ± 0.6% | < ql |
The different proportions of Fe, Astot and DOC are reported in the different cut-off fractions (fraction > 0.2 μm, 0.2 μm > fraction > 30 kDa, 30 kDa >fraction > 2 kDa fraction < 2 kDa) recovered by ultracentrifugation (Table 2). In pure Fe and in Fe–As experiments, the entire Fe pool was located in the fraction > 0.2 μm, which is consistent with the formation of Fe microparticles. Markedly different results were obtained for the Fe–As–HA system. In fact, most of the Fe (94%) occurred in the 0.2 μm > fraction > 30 kDa, with only a small part (6%) being found in the > 0.2 μm fraction.
Fig. 1 displays scanning electron microscope (SEM) pictures of the dried experimental suspensions. In the pure Fe system, a cover of thin sheet-like particles is observed (Fig. 1a). The characteristic size of the particles ranges from 400 to 800 nm. Because of the small thickness of the particles, needle-like structures can be locally observed, corresponding to the thin edges of particles. The particles are identified here by XRD as poorly crystalline lepidocrocite (β-FeOOH) (Fig. 2). Indeed, the XRD pattern displayed peaks at 1.18, 1.20, 1.26, 1.39, 1.53, 1.62, 1.73, 1.85, 1.93, 2.09, 2.37, 2.49, 3.30 and 6.90 Å, which are consistent with the d-spacings of lepidocrocite (Cornell and Schwertmann, 2003). Similar crystals have already been observed in natural environments or produced in the laboratory, when Fe2+ ions were oxidized under neutral or slightly acidic pH (Châtellier et al., 2001, 2004; Fakih et al., 2008; Fortin et al., 1993; Pédrot et al., 2011; Schwertmann and Taylor, 1979). When As is added, the shape of iron particles is less well-defined, fingerprinting the formation of a more amorphous, poorly crystallized ferric phase (Fig. 1b). The diversity and sequence of the short-range-ordered Fe(III) precipitates formed by Fe2+ oxidation at a near neutral pH depend on the water chemistry (Voegelin et al., 2010). Without interfering solutes, Fe(II) oxidation results in lepidocrocite formation. By contrast, the presence of oxyanions, such as silicates and phosphates favours the formation of short-range-ordered Fe(III) precipitates (Cornell and Schwertmann, 2003). In the present case, it is likely that the presence of As(III) and As(V) oxyanions also promoted disordered Fe(III) precipitates. Erbs et al. (2010) indeed showed that As coprecipitation in ferrihydrite leads to decreased crystallinity. Moreover, Voegelin et al. (2010) showed that, at a near neutral pH, when the molar As(V)/Fe ratio is < 0.5 as in this study (molar As(V)/Fe ratio ≈ 0.1), the high affinity of As(V) for Fe(III) precipitates leads to low dissolved As concentrations, which may prevent precipitate transformation into a more crystalline phase. Therefore, with As, the Fe(III) precipitates appeared to be not very crystallized and smaller than without As (Fig. 1b).
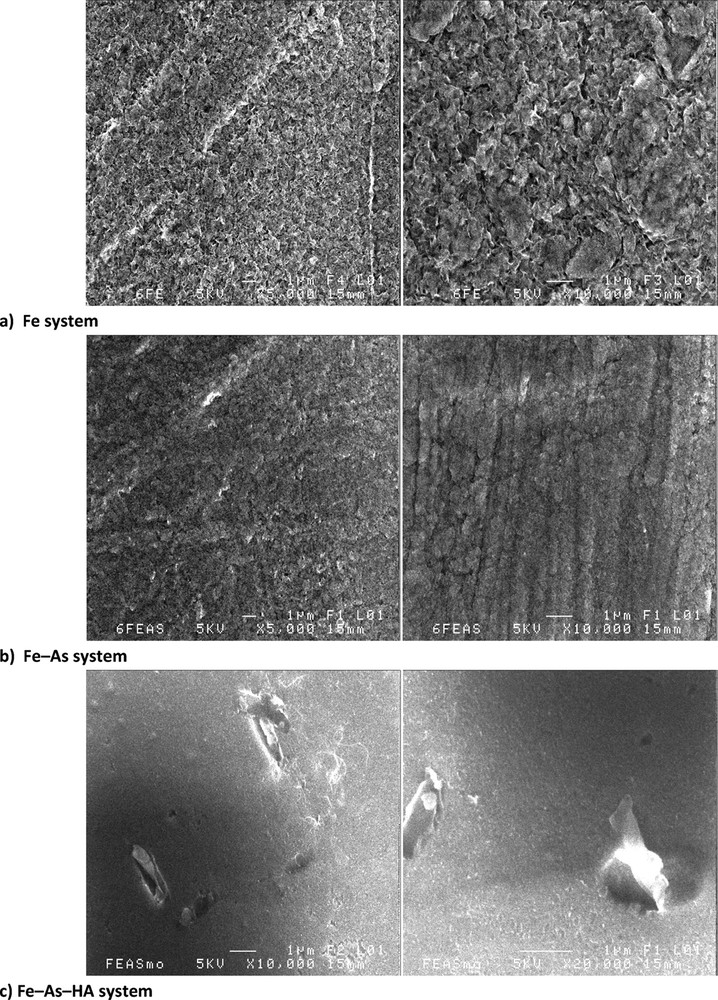
Scanning electron microscopy pictures of the different Fe systems. a: Fe system; b: Fe–As system; c: Fe–As–HA system.
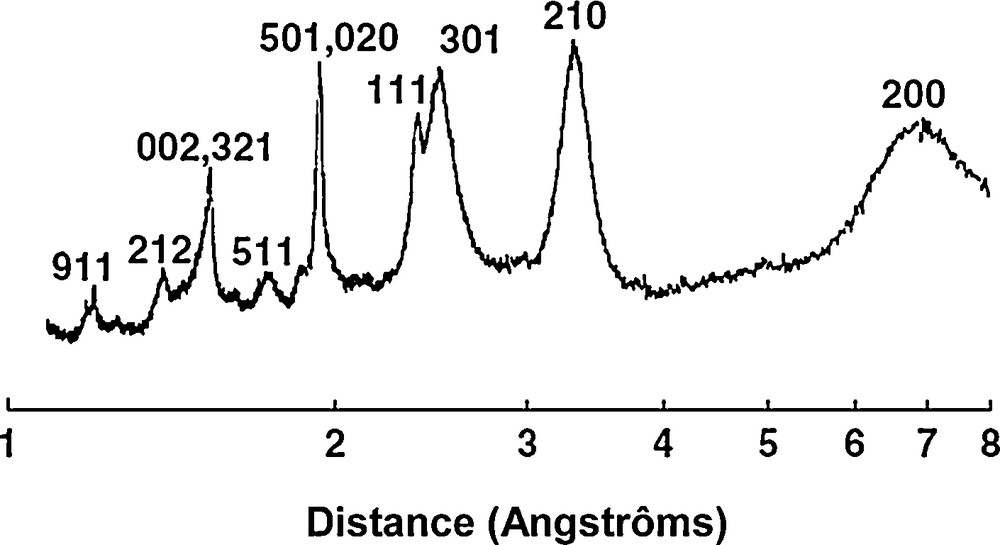
XRD patterns of Fe system. The hkl identification of the peaks is given as in Cornell and Schwertmann (2003).
With As and HA, the SEM observations did not point out well-defined Fe particles (Fig. 1c). However, ultrafiltration experiments showed that Fe and a high proportion of DOC were in the 0.2 μm > fraction > 30 kDa range (Table 2). The sorption capacities of dissolved organic matter–Fe range between 0.1 to 30 mmol Fe/mol C (Tipping et al., 2002) or are even lower (Hiemstra and Van Riemsdijk, 2006). Here, the Fe/C ratio reached 352 mmol Fe/mol C at the end of experiment in the fraction > 30 kDa (Table 2), suggesting that Fe were not only adsorbed, but that a significant Fe fraction also precipitated as amorphous nanosize Fe phase within the HA matrix. Dissolved organic matter inhibited the formation of large Fe oxyhydroxide aggregates, but stabilized Fe as ionic complexes or as colloidal form as previously suggested by Ritter et al. (2006) and Bauer and Blodau (2009). Therefore, the occurrence of HA in the present oxidation experiments favoured the formation of mixed Fe–OM colloids as observed in previously published experimental works (Pédrot et al., 2011).
3.2 Experimental and NanoSIMS constraints on the fate and distribution of As
NanoSIMS was used as a chemical mapping tool to gain further knowledge about the fate of As in the present experiments. Ion distribution images of 12C14N, 56Fe16O and 75As data were superimposed using an image processing software to identify the possible colocalization of As, Fe and HA. The mapping procedure, using secondary ion images of 56Fe16O and 75As, established the colocalization of As and Fe within the aggregated Fe oxyhydroxides formed in the Fe–As experiment (Fig. 3). Ultrafiltration data confirmed a nearly complete scavenging of the dissolved As by the neo-formed Fe oxyhydroxides microparticles, since the entire Fe pool located in the fraction > 0.2 μm contained also most of the As (97%) (Table 2). The chemical mapping showed a lack of hotspot or diffuse zone (halo) in As onto Fe oxyhydroxide particles, but rather a uniform diffusive distribution of As within Fe oxyhydroxide particles. The performing of line scans confirmed the concomitant increase/decrease of the intensities of 75As and 56Fe16O, providing evidence that the distribution of As was intimately linked with that of Fe (Fig. 3). These results suggested a homogeneous trapping of arsenic onto and within Fe particles implying 2D and 3D sorption processes. Thus, As sorption was performed by several processes, such as surface adsorption, coprecipitation and fixation (absorption), which involves the diffusion of an aqueous metal species into the solid phase.
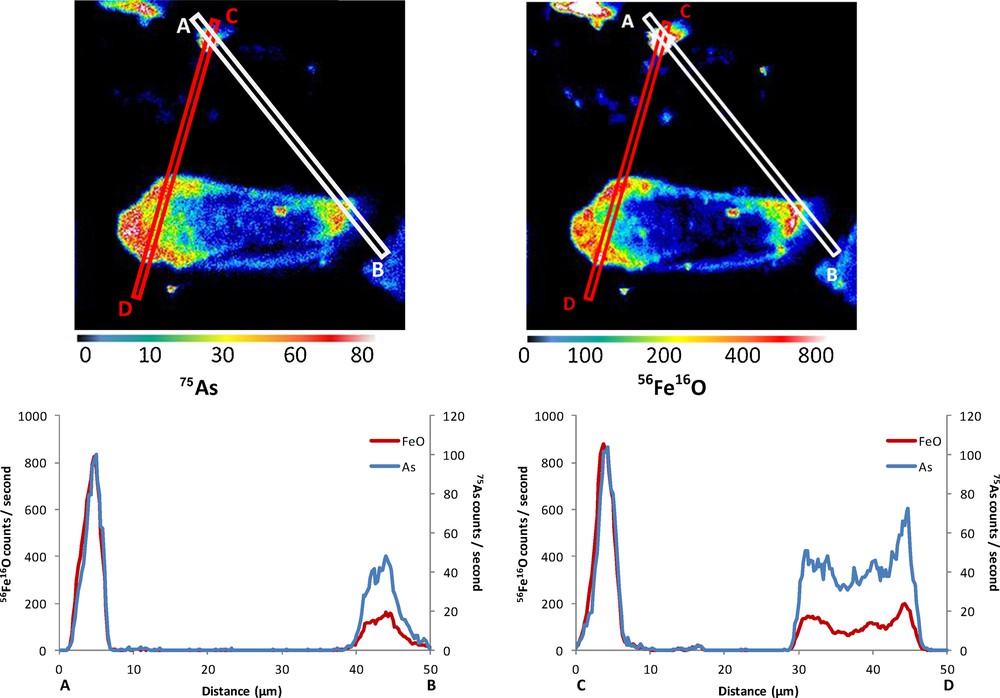
(Colour online.) 56Fe16O and 75As mapping in the Fe–As system. The colour scales of the isotopic distribution correspond to the counts per second measured by NanoSIMS. Line scans of 56Fe16O and 75As abundance data were performed from two A–B and C–D transects as displayed by the white and red rectangles for the A–B and C–D transects, respectively.
With regards to the fate and distribution of As in the mixed Fe–As–HA system, ultrafiltration data showed that As, at 80% under As(V) form, was predominantly concentrated in the 0.2 μm > fraction > 30 kDa, as was Fe and DOC. Only a small fraction of As was found in the < 2 kDa fractions (4.1%, Table 2). The Fe and DOC concentrations were not measurable in the fraction < 2 kDa (Table 2), suggesting that this small fraction of As occurred as true dissolved species. The high proportion of As observed in the 0.2 μm > fraction > 30 kDa established that As was maintained in this fraction as a colloidal form. NanoSIMS analyses showed that 75As occurred in a close spatial relationship with both 56Fe16O and 12C14N (Figs. 4 and 5). The line scans exhibited smaller 56Fe16O intensities as compared to the Fe–As system since the nanosize ferric phase is diffusely embedded within the HA matrix. In the experimental HA solutions obtained from standard HA, no Fe could be detected (ql of 5 μg·L−1 for the Fe signal) due to the low concentration of inherent Fe. Thus, all of the Fe evidenced by analysis and the NanoSIMS images do correspond to the Fe added during the Fe oxidation–hydrolysis experiment. Interestingly, we noticed that the spot zones showing the highest Fe intensities did not correspond to accumulation zones for As [regions of interest (so-called ROI) in Figs. 4 and 5]. Pearson's coefficients were calculated for the entire images and these ROI (Fig. 6), corresponding to spot zones. The lower colocalizations, between 56Fe16O−/75As were observed within these ROI. The same is true for the organic matter zones depleted in Fe, in which the As signal is low (line scans of Figs. 4 and 5). In fact, it seems that the As is concentrated only in diffuse zones, systematically displaying an association of organic matter and Fe. This may suggest preferential incorporation of As in the bulk of Fe–HA colloids during their formation rather than surface adsorption onto these colloids. Thus, the formation of mixed colloids, during the Fe(II) oxidation event in OM-rich waters, revealed a synergistic effect in trace metal mobility. Without HA, all As was trapped in the fraction > 0.2 μm and, by contrast with HA, organic sorption of As was low. However, when HA and Fe phases were associated, results showed that As was trapped in the bulk of Fe–HA colloids of size < 0.2 μm and, therefore, was more mobile.
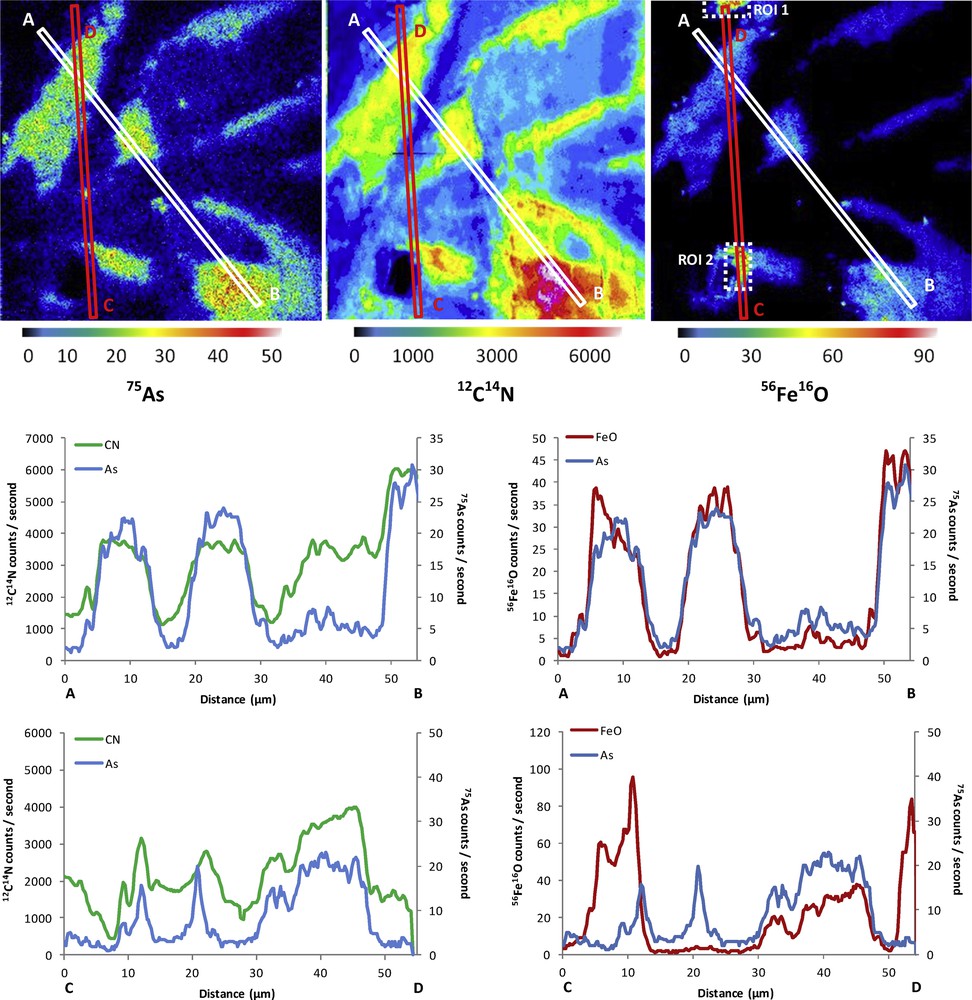
(Colour online.) 12C14N, 56Fe16O and 75As mapping in the Fe–As–HA system. The colour scales of the isotopic distribution correspond to the counts per second measured by NanoSIMS. Line scans of 12C14N, 56Fe16O and 75As abundance data were performed from two A–B and C–D transects as displayed by the white or red rectangles for the A–B and C–D transects, respectively. Regions of interest (ROI 1 and 2) are displayed by the white dotted rectangles within 56Fe16O mapping.

(Colour online.) 12C14N, 56Fe16O and 75As mapping in the Fe–As–HA system. The colour scales of the isotopic distribution correspond to the counts per second measured by NanoSIMS. Line scans of 12C14N, 56Fe16O and 75As abundance data were performed from two A–B and C–D transects as displayed by the red or white rectangles for the A–B and C–D transects, respectively. The region of interest (ROI 3) is displayed by the white dotted rectangle within the 56Fe16O mapping.
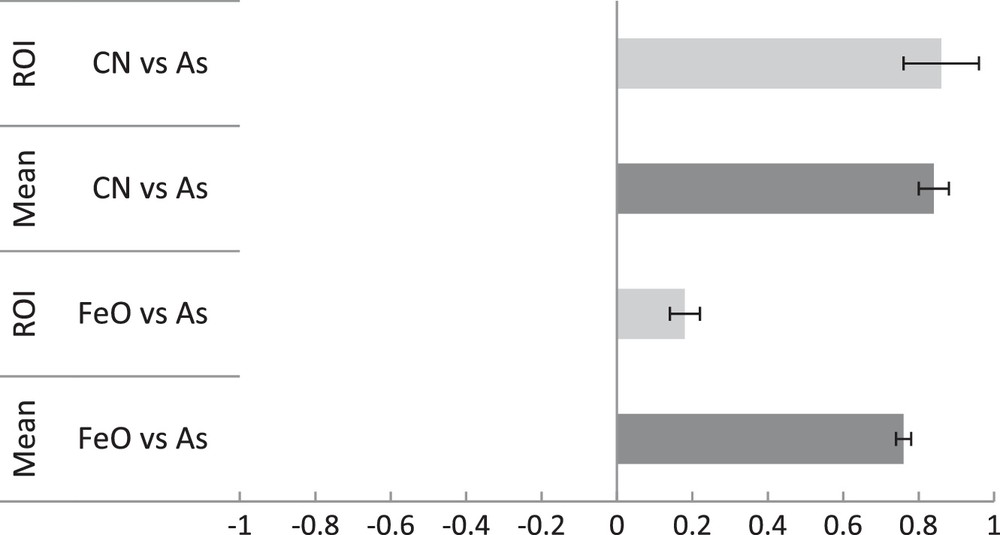
Colocalization of elements as evidenced through nanoSIMS mapping (Pearson's coefficient) within the whole image (mean) and within the Fe-rich regions of interest (ROI 1, 2 and 3) with low CN signal (ROI).
The coprecipitation process could allow explaining this arsenic distribution during the formation of mixed colloids. Indeed, by limiting the development and the crystallization of iron nanoparticles, HA conserves the ferric phase in an amorphous form, allowing this ferric phase to trap a high concentration of trace elements via coprecipitation during the oxidation–hydrolysis of Fe(II). Pokrovsky et al. (2005) suggested such process to occur in peat soil solution, implying an incorporation of trace elements in the bulk of Fe–Al–organic humus colloids via coprecipitation formed on redox fronts rather than through surface adsorption. A study of Sharma et al. (2010) suggested that As(V) is associated with combined Fe–OM phases, mainly via ferrihydrite-OM colloids, but also to a small extent in dissolved Fe–OM complexes via Fe-bridging. The transformation of the amorphous nanosize Fe phase to more crystalline phases potentially leads to a loss of sorption sites (Cornell and Schwertmann, 2003). This process can explain why the data do not show any arsenic association in zones characterized by high 56Fe16O intensities with low 12C14N intensities, which corresponds probably to an aging of amorphous nanosize Fe phase conducting to a more crystallized phase. Another possible hypothesis with regards to the inability of this latter to bind arsenic by surface sorption may originate in the fact that arsenic was previously trapped within mixed Fe–HA colloids. Thus, the arsenic incorporated into mixed colloids could not be considered to be readily exchangeable. Such a hypothesis should be however supplemented by further experiments in order to confirm that the formation of mixed Fe–HA colloids induces a synergistic effect with regards to the sorption capacity of dissolved trace elements.
Thus, both through the fractionation of Fe, As and DOC concentrations during ultrafiltration step and the NanoSIMS data, the results assessed (i) that the occurrence of HA and dissolved As during the Fe2+ oxidation–hydrolysis reaction promoted the formation of mixed As-doped Fe–OM colloids, and (ii) that As was especially incorporated in the bulk of Fe–HA colloids.
3.3 Environmental implications
Numerous studies have provided evidence that As solubilization in wetlands or aquifers is produced by the reduction of hosting mineral carrier phases, such as Fe oxyhydroxides (Smedley and Kinniburgh, 2002 and references therein). However, we should consider that re-oxidation could, in turn, strongly affect the behaviour of the released As, notably in such organic Fe(II)-rich waters where the presence of organic matter could lead to the formation of fine-scaled, highly mobile As-bearing colloids. Re-oxidation processes are expected to occur rather systematically at the interface between wetland and rivers. The question is then raised to know whether these zones could be zones of As immobilization due to the neo-formation of iron oxides or instead zones favouring the contamination of rivers by wetland-derived As. Considering the above experimental conditions, we demonstrated that Fe2+ oxidation–hydrolysis in organic matter-poor waters produces large Fe(III) oxyhydroxide crystals that are able to strongly bind dissolved As (for a molar As/Fe ratio ≈ 0.1). Thus, this study shows the major importance of freshly precipitated iron hydroxides for arsenic uptake, as observed in natural environments (Fritzsche et al., 2006). Moreover, SEM observations showed that the occurrence of As during Fe2+ oxidation–hydrolysis promoted the formation of smaller and less crystallized and potentially more reactive Fe(III) oxyhydroxides (due to the increasing surface area). Arsenic transfer has therefore to be strongly controlled by the Fe(III) oxyhydroxide dissolution/precipitation processes. These two processes could in theory immobilize the released As in wetlands. However, the situation could be much different in real wetland-river systems, due to the presence of high dissolved organic matter concentrations in wetland waters. In the presence of HA, the present results indeed showed that the oxidation–hydrolysis of Fe2+ allowed the trapping of a high amount of As within much smaller, mixed Fe–HA colloids (< 0.2 μm). This incorporation of As in the bulk of Fe–HA colloids rather than surface adsorption onto these colloids controls the transport of As. In order to remove As, the destructuration or solubilisation of the complex colloids in which they are incorporated may be required. Arsenic mobility was therefore controlled by the transfer process and stability of the mixed Fe–HA colloids. Thus, colloidal As–Fe–organic matter complexes formation in wetland soils could have a high environmental relevance concerning water quality with regards to the transfer of As from wetland soil horizons to rivers. The question of the stability and mobility of these ternary associations still needs to be addressed through further experimental contributions.
The transport of colloids through porous media strongly depends on the kinetics of the colloid deposition and release (Kretzschmar and Sticher, 1998). Advection is the main process that affects the transport of small particles (< 1 μm) in porous media. When the colloids are of small size, they are less filtered by the porous media. By contrast, when the particles display sizes larger than 1 μm, these particles can be intercepted or deposited. Thus, in media where porosity acts as an efficient filter, colloids cannot enter the whole structure and therefore are able to move faster than ion (phenomenon of size-exclusion chromatography, where particles are excluded from the smaller pore spaces and more tortuous flow paths; Ryan and Elimelech, 1996). As a consequence of their nanometric size, the mixed Fe–organic matter colloidal As carriers formed during the re-oxidation of wetland waters could favour the transport of As over long distances, thus contributing to the environmental dissemination of this toxic element. Indeed, several studies showed that the presence of interactions between DOM and nanoparticles reduced the deposition of nanoparticles and increased their transfer by a complex combination of interactions involved in their stabilization (Chowdhury et al., 2012; Johnson et al., 2009). This association has also a particular importance since these mixed Fe–OM colloids have been shown to represent the most bioavailable Fe source for Fe-reducing bacteria in anoxic area (Davranche et al., 2011; Pédrot et al., 2011). Quite clearly, the behaviour of these amorphous nanosize Fe phases, which strongly bind both As(III) and As(V) (Borch et al., 2010 and references therein), as well as other trace elements (Dahlqvist et al., 2007; Lippold et al., 2007; Pédrot et al., 2009; Pokrovsky et al., 2006, 2012; Vasyukova et al., 2012) must be taken more seriously into account than it is currently done, particularly as regards their impact on the streams and rivers that are hydrologically connected with wetlands. Thus, one of the key questions becomes: which conditions in the river systems will possibly breakup the As–Fe–organic matter associations, leading to re-solubilisation of the carried As? With regards to this question, chemical mapping at the nanoscale represents therefore an essential step towards a better characterization of the carrier phases with which the As released in wetlands could bind. The nature of these carriers could control its fate into the adjacent river systems with which the wetland waters could be connected. The present study and related dataset highlight the power of the NanoSIMS instruments to map associations of Fe, As and organic matter within newly formed, complex colloidal phases. As a consequence, such analysis could be performed in solid samples located in highly contaminated areas, such as, for example, acid mine drainages and/or areas submitted to redox oscillations as hyporheic zones. Such analyses should be however supplemented by smaller-scale investigation techniques, such as the STXM and/or EXAFS/XAS techniques, for a direct characterization of the molecular environment of As in these mixed components, including trace metals, oxyanions, mineral phases and organic matter.
4 Conclusions
The experiments carried out here allowed us to discuss the As fate in reduced wetland waters, once these waters become re-oxidized at their contact with river waters. Both the fractionation of Fe and As concentrations during the ultrafiltration step of the experimental solutions and the NanoSIMS analyses of the solid phases evidenced that most of the As occurring in the wetland-like experimental solutions was trapped within nanoscale Fe–HA mixed colloids (< 0.2 μm). The results evidenced that this behaviour was a direct consequence of the occurrence of large amounts of dissolved organic matter – which is a general characteristic of wetland waters – the experiments without HA showing that in organic matter-free conditions, As was bound to much larger, micro-scale Fe crystals (> 0.2 μm).
Thus, in wetland areas undergoing important redox alternations and associated dynamic biogeochemical processes, the fate of dissolved As is strongly constrained by the Fe oxidation event. Notably, the resulting formed mixed Fe–OM colloids, where complex colloids bind possibly a large amounts of trace elements, can strongly impact the possible transport of trace elements towards ecosystems with which wetlands are hydrologically connected, such as in river systems. Complex colloids could be indeed a potential vector of As due to (i) their nanoscale size, (ii) their strong capacity to trap high amount of As, and (iii) the occurrence of organic molecules coating their surface modifying their chemical fate. These findings highlight the power of NanoSIMS to precisely map the chemical distribution of metallic and metalloid contaminant, such as at the colloidal scale within precipitates occurring in such complex mixed-phase environments. The use of nanoSIMS on natural samples would determine the presence of metallic and metalloid contaminant and their carrier phases as well, whereas preserving the natural associations of mixed organic-mineral phases.
Acknowledgement
This work was supported by the ANR Program “ARSENORG”. We thank Joseph Le Lannic for performing SEM analysis at the CMEBA Analytical Facility at the University of Rennes-1 and Thomas Delhaye for performing NanoSIMS analysis in the “Microscopy–Rennes Imaging Center” (MRic) at the University of Rennes-1.