

 CC-BY 4.0
CC-BY 4.0
Broader context
Even if still a question of societal debate due to the vast international effort still ongoing; the fusion power is part of many country road maps toward decarbonized energies [Donné 2019]. Indeed, fusion energy can provide sustainable solutions to global energy needs for the future, requiring only abundant fuel and generating no long-lived radioactive waste. However, risks are inherent to any process and need to be addressed to engineer as safe as possible fusion technology.
Metallic particles of high specific surface areas will be present in the vacuum vessel of fusion reactor due to interaction between plasma and refractory plasma facing components. With the proved collector efficiency decrease of High Efficiency Particulate Air (HEPA) filters for particles in the 50–300 nm range, very small quantity of nanoparticles may be released. In addition, particles might be released in case of Lost Of Vacuum Accident (LOVA).
The aim of the study is to address the fate in biological media of W nanoparticles that might be released. The results proved that oxidative dissolution occurred in several aqueous/biological media (TRIS, LHC9 and Lung media). The mechanisms of dissolution involved W4+ and W6+ corroded layers prior to W6+ dissolution. Results of the paper enabled to better decipher the toxicity of W nanoparticles [George et al. 2019; Uboldi et al. 2019].
1. Introduction
The world has to face a major energetic challenge in the next years and various solutions to secure future energy supply that in the same time can minimize greenhouse gas emission, are developed. Among all potential solutions, fusion energy has the capability to provide a sustainable solution to global energy needs. In that context the ITER project (International Thermonuclear Experimental Reactor) has been initiated in 2006 and is the key facility of the European roadmap for instance [Donné 2019]. Upon many scientific and technological challenges of fusion, the selection of the refractory material needs to be improved. Indeed, in the past, Tokamaks (nuclear fusion reactor) were operating with carbon refractory materials as plasma facing components (PFCs) to sustain the huge energy outflow. However, due to large carbon erosion by deuterium plasma and to a large tritium trapping in carbon, huge quantity of tritium inventory was observed.
In order to reduce this phenomenon, tungsten (W) was foreseen as PFC to replace carbon because it exhibits low sputtering yield, a high melting point and a high thermal conductivity. In addition to cost reduction, W was recently selected for the construction of ITER [Pitts et al. 2013; Ueda et al. 2014]. Nevertheless, plasma-wall interaction process erodes this tungsten material and different processes lead to the creation of small particles ranging from tens of nanometres to tens of micrometres. As an example, W fractal particles of less than one micron are foreseen to be created during the ITER operation as it was observed in dust from plasma laboratory [Acsente et al. 2015]. More over with a very high Ductile to Brittle Transition Temperature (DBTT), tungsten is brittle in the operating temperature range, this brittleness inducing cracking and being at the origin of dust particles emission [Makhlaj et al. 2013].
Thus, in a fusion reactor such as ITER, metallic tungsten particles of variable sizes and high specific surface areas will be present in the vacuum vessel. In case of a breakdown of the first protection barrier (referred as LOVA (Lost Of Vacuum Accident)), they could be dispersed in the close environment. Under normal operating conditions, they can also be released in the open air in very small quantity when passing through High Efficiency Particulate Air (HEPA) filters used to purify the atmosphere. Indeed, considering the HEPA filters characteristics, it has been shown experimentally and theoretically confirmed that the collector efficiency (trapping efficiency) decreases in the 50–300 nm particle size range (e.g. [Huang et al. 2007; Steffens & Coury 2007]). The latter particles are called the most penetrating particle size (MPPS) [Bortolassi et al. 2017].
W and W compounds were considered for a long time as lowly toxic for humans [van der Voet et al. 2007] and of limited environmental liability [Koutsospyros et al. 2006; Strigul 2010; Strigul et al. 2005]. The low toxicity was associated to a supposed low solubility of metallic tungsten [Strigul 2010]. Such vision may not reflect the specific case of ITER-like particles. Indeed part of ITER released particles (especially released through HEPA) will range within the submicron size range and it has been shown that inhalation of poorly soluble nanoparticles may lead to toxic effects (e.g. [Poland et al. 2008]). In addition, the assertion concerning the low solubility of W particles found in the literature to explain the W low toxicity is questionable. Indeed recent results have shown that metallic tungsten particles can be dissolved in the environment and biological media and can cause adverse effects from soil acidification to direct and indirect toxic effects to living organisms as soil bacteria, fungi, plants [Koutsospyros et al. 2006; Strigul 2010; Strigul et al. 2005].
Although oxidative dissolution of tungsten was intensively studied for both acidic [Anik 2006; Anik & Osseo-Asare 2002; Johnson & Wu 1971; Lillard et al. 1998] and basic environments [Anik & Osseo-Asare 2002; Johnson & Wu 1971; Kelsey 1977], the rate and mechanisms of metallic tungsten oxidative transformation in biological solutions are not well documented. From the scarce literature, published data are related to corrosion of W electrode in neuroscience [Patrick et al. 2011] or coils in medical applications [Peuster et al. 2003].
The objective of the current paper is therefore to address the physical (particles agglomeration–aggregation) and chemical evolution of “ITER like” W particles in various aqueous and biological media prior to determine their toxic effects and their environmental transfer and transformation [George et al. 2019; Uboldi et al. 2019]. The aim of the paper is to focus on the 50–300 nm particle range, corresponding to the lowest trapping efficiency of HEPA i.e. particles with the highest probability to be released during ITER operations.
2. Materials and methods
2.1. W dust particles
2.1.1. Rationale for the W dust particles production and selection
One of the main issues of the current work is to address the fate of particles that might be released by ITER (“ITER like” particles). Up to now, no Tokamak is operating tungsten PFCs in the same plasma edge conditions that are expected during the ITER life. Then the production and selection of W metallic particles in terms of size, shape and structure remains extremely challenging. Several laboratories experiments are mimicking the ITER plasma-wall interactions and their consequences on the W dust generation [Acsente et al. 2015]. From them, relevant dust can be produced. In the present study, two laboratories techniques were selected to produce ITER like W dust particles.
Plasma-wall interactions in laboratory experiment were used as first technique [Acsente et al. 2015]. The production of W particles from the first technique (W_Plasma) enables to reproduce at best the ITER-like conditions. However, the complex technique failed in producing large amounts of powders to be tested (maximum 5 mg/h). The second selected technique is based on planetary ball milling (PBM) which is a common technique to produce small particles in relatively large quantities ( >g), controlling their chemical and physical composition (W_Milling) [Hendaoui et al. 2010]. However, the second technique leads to polydisperse particle sizes and requires a specific separation protocol to isolate particles within the 50–300 nm size range. The technique can also lead to surface amorphization of particles and affects particles reactivity.
2.1.2. W dust particle synthesis protocols
W_Plasma: the synthesis of tungsten nanoparticles powder using a gas aggregation cluster source based on a magnetron sputtering discharge was initially described [Acsente et al. 2015]. Briefly a magnetron sputtering gas aggregation cluster source (MS-GAS) combined to a vacuum deposition chamber was used to generate W_plasma particles. A tungsten target facing the cluster source at a distance of 90 mm served to simulate W PFC. W particles generated were collected on a Si(111) crystalline substrate in the vacuum deposition chamber.
W_Milling: high-energy planetary ball milling has been used to produce larger amounts of tungsten nanoparticles. The experimental protocol has been previously described [Dine et al. 2015; ?] and consists in using repeated high energy shocks to break down the initial W powders which are in the 3–13 μm range. In order to avoid shock-induced welding, the procedure has been carried out in ethanol. Milling efficiency is a function of the mill velocity, which has been set to 350 RPM (for the so-called solar plate, the jars rotating in the opposite direction at twice this velocity), of the milling balls density, where we chose to use tungsten carbide (also for its high hardness which reduces the risks of abrasion and therefore pollution of the powders produced), of the ball-to-powder mass ratio, which has been set to 40:1, and finally of the time, which has been chosen to 20 h.
2.1.3. Physical–chemical properties of the dry initial powders
W_Plasma: the particles were obtained during the same experiment as described by Bernard et al. [2019]. X-ray diffraction (XRD) indicated that W_Plasma metallic particles are crystalized under the beta phase with some traces of the alpha phase. From TEM, the size of individual particles obtained from a plasma varies from 50 to 150 nm [Bernard et al. 2019].
W_Milling: A complete description of the powders produced by high-energy planetary ball milling can be found in [Bernard et al. 2019]. XRD patterns indicated a crystallite particle size of 4–5 nm, although the particle size is larger, ranging in the aimed size range; particles are therefore polycrystalline. In the meantime, lattice micro-distortion ratio (micro-strains) is high, in the order of 0.3%. The crystal structure does not change from the initial powders, which are crystallized under the alpha phase structure (as opposed to the W_Plasma particles).
2.2. W particle size separation and dispersion protocol
In the case of W_Milling particles, it was necessary to develop a specific protocol to produce monodisperse suspensions with particle size ranging from 50 to 300 nm. The aim of the protocol was to remove both the largest and the smallest size fractions. The tris(hydroxymethyl) aminomethane (TRIS) dispersion medium was selected to avoid pH variations and store the produced stock suspensions.
The first step involved the weighing of the sample followed by the addition of an adequate amount of TRIS solution (5 mM, pH 8.5) to reach a concentration of 10 g W-particles/l. Then this suspension was sonicated using an ultrasound tip (Sonicator vibracell 72434, Amplitude 40%, 15 min). The ultrasound reactor was cooled using ice bath. The suspension was then filtered using a sterivex filter (0.45 μm) to remove the largest particles.
The next step comprised the centrifugation (3000g, 30 min) of filtrate to remove the smallest fraction of particles in the supernatant. The deposited solid at the bottom of the centrifuge tube was dispersed in a small volume of TRIS solution (11 ml) to produce the W_Milling stock suspension, that was sonicated again before their storage at −20 °C until use.
In the case of W_Plasma particles suspension, previous results [Acsente et al. 2015] suggested that the particle size perfectly fits the target 50–300 nm range. Therefore, it was not necessary to develop a specific separation protocol. The weighing step was followed by the addition of an adequate amount of TRIS solution (5 mM, pH 8.5) to reach a concentration of 10 g W np/L. Then this suspension was sonicated in the presence of an ice bath, using an ultrasound tip (Sonicateur vibracell 72434, continuous mode, Amplitude 40%, 15 min) and stored at −20 °C until use.
Protocol developed to generate W monodisperse particle suspensions with particles in the 50 to 300 nm range.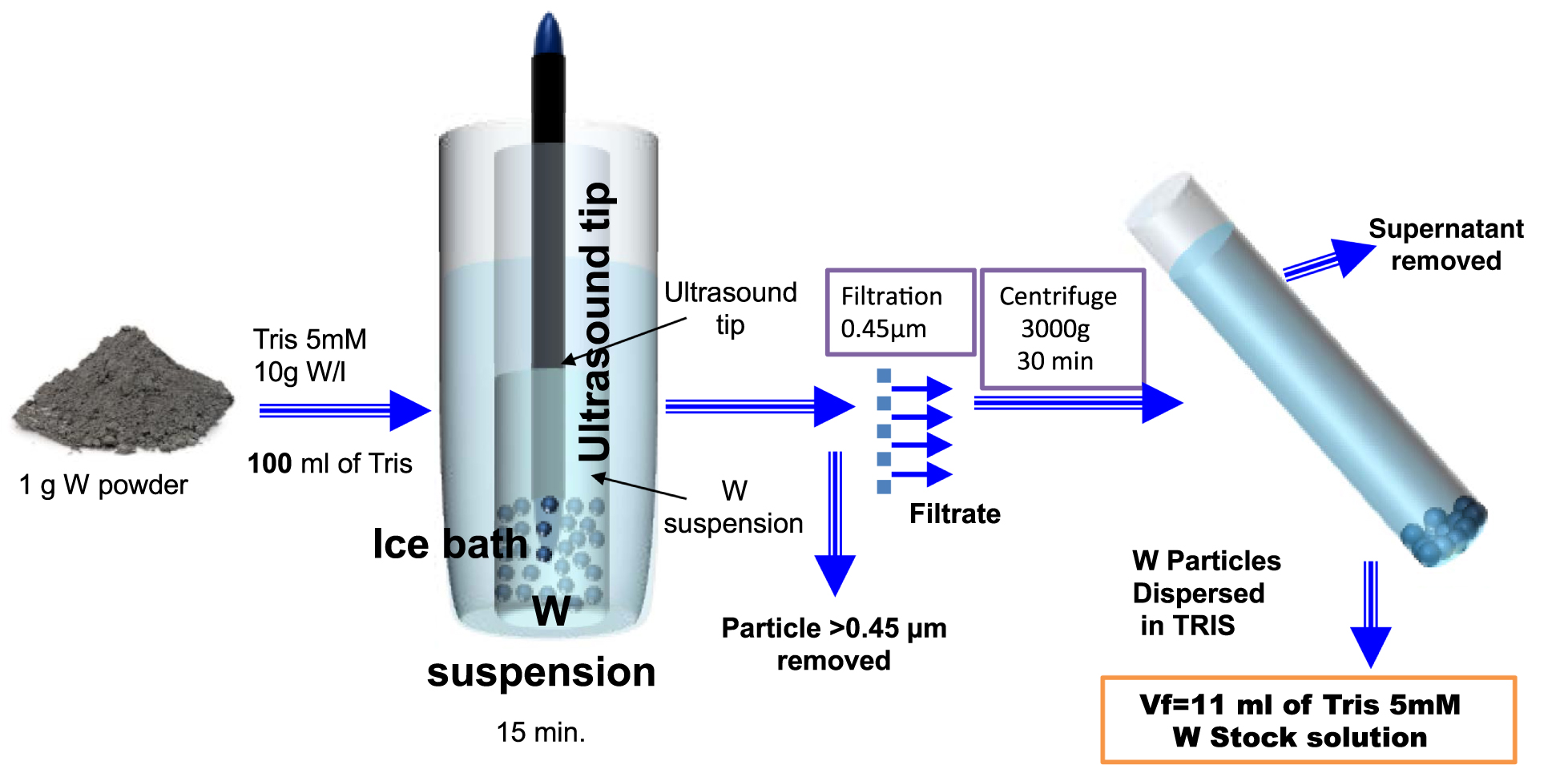
Several tests were carried out following the same protocol to verify its reproducibility in terms of chemical stability (dissolution process) and particle size distribution.
2.3. Experimental set-up
Several aqueous media were selected and tested in order to reproduce a large range of biological media used in toxicological tests performed on in vitro human cell models [George et al. 2019; Uboldi et al. 2019].
W powders were stored in TRIS stock suspensions and the oxidation kinetic as function of dilution was assessed. Two biological and a saline media were also tested accordingly to the lung models that were selected for the toxicological studies. The first in vitro model chosen to study toxicity of tungsten particles on human lung is the MucilAir® model (Epithelix Sarl). This model mimics the upper respiratory tract structure of the human lung and is grown on inserts with MucilAir serum-free culture medium (called “Pulmonary” in the paper) on the basal side. A saline solution (NaCl 0.9%, CaCl2 1.25 mM, Hepes 10 mM, pH7.4 called “Saline” in the paper), is used to wash the apical part of the tissue, to remove mucus. Both media are therefore in contact with W particles. The second toxicological model is a transformed human bronchial epithelial cell line, BEAS-2B, cultured in a dedicated serum-free medium: LHC9 (life technologies). The four aqueous/biological media (TRIS, Saline, Pulmonary and LHC9) were selected as they represent relevant biological media and were used in the Uboldi et al. [2019] and George et al. [2019] papers.
2.4. Protocol for the dissolution kinetic experiment
Two W concentrations were tested (10 and 100 mg/l) at t = 0, 2, 4, 24 and 48 h. Such concentrations are related to concentrations tested in toxicological studies [Uboldi et al. 2019; George et al. 2019]. The liquid media previously described were used. The main issue was to discriminate solid versus dissolved W. Different separation protocols were tested from ultracentrifugation to filtration using 20 nm seringe filters. The optimized protocol consisted in combining two filtration stages at 25 nm using an ester cellulose Ester cellulose Membrane MF-Millipore) for a first rough separation and 20 nm ceramic membrane (Whatman® Anotop®) to increase size selectivity will avoiding a fast fouling. Filtration was preferred since the operation procedure was faster than for ultracentrifuge avoiding any further transformation and oxidation of tungsten particles while samples were processed.
2.5. Characterisation methods
Particle size distribution
The size distribution was measured by Dynamic Light Scattering (DLS) using the Mastersizer S (Malvern, Worcestershire, UK). Concentrations of suspension were adjusted to avoid multiple scattering for the highest concentrations. Particle size distribution of large number of samples were analysed to test the sample preparation variability and compared based on Weibull function fits (mono-modal or poly-modal). The Zetasizer ES (Malvern, Worcestershire, UK) was used to determine the zeta potential of the particles by Electrophoretic Light Scattering.
Chemical composition
The chemical analysis of W in the solid and dissolved phases was performed within Inductively coupled plasma mass spectrometry (ICP-MS) (Perkin Elmer Nexion 300). The solid phases were digested via a total oxidation of W with H2O2 solutions at room temperature for 24 h. W content was determined by ICP-MS via quantitative analysis of 182W.
Solid state characterisation of the particles
Samples for X-ray Absorption Spectroscopy (XAS) analysis: Speciation and oxidation state of W were assessed by XAS. W suspensions were filtered on 20 nm Whatman® Anodisc inorganic filter membranes. The filtration leads to the formation of a deposition layer with homogeneous thickness. The fouled filter was frozen and frieze-dried under vacuum prior to XAS analysis. XAS spectra at the W LIII-edge were performed on the FAME BM30b beamline at the European Synchrotron Radiation Facility (ESRF), in Grenoble, France [Proux et al. 2005]. Beam damages can be critical for redox sensitive elements like W. A liquid helium cryostat was used to avoid photo-oxidation/reduction under the beam. Each spectrum was collected on a new zone of the sample to reduce beam damage. A Si(220) monochromator was used and the energy calibration was performed by setting the first maximum derivative of the XANES (X-ray Absorption Near Edge Structure) spectrum of a W metallic foil at 10,207 eV. Between 2 to 4 spectra were collected whether in transmission or in fluorescence mode for reference compounds and experimental samples respectively. A 30 elements array Ge solid-state detector (Canberra) and Si photodiodes were used for the fluorescence and transmission modes respectively.
The data reduction was performed with the ATHENA [Ravel & Newville 2005] and the CHEROKEE softwares [Michalowicz et al. 2013]. Metallic tungsten foil, WO2, WO3 minerals and amorphous H2WO4 were used as reference compounds. The oxidation states of W in samples were quantified using linear combination fit (LCF) of XANES and EXAFS (Extended X-ray Absorption Fine Structure) spectra of the experimental samples using the reference compounds as database. XANES LCF are detailed in Supplementary Information.
Particle size distribution of (A) W_Milling (red) and W_Plasma (blue) particles suspension at 2.5 g/l obtained from PBM and plasma-wall interactions respectively (variability obtained from the replicate of 5 stock suspensions prepared independently), (B) W_Milling particle suspensions before and after re-concentration (2.5 g/l TRIS stock solutions), (C) W_Milling particle suspensions before (TRIS stock solutions) and after storage at −20 °C (2.5 g/l), (D) W_Milling particles diluted ([W] = 100 mg/l) in various aqueous media, (E) W_Plasma particles diluted ([W] = 100 mg/l) in various aqueous media.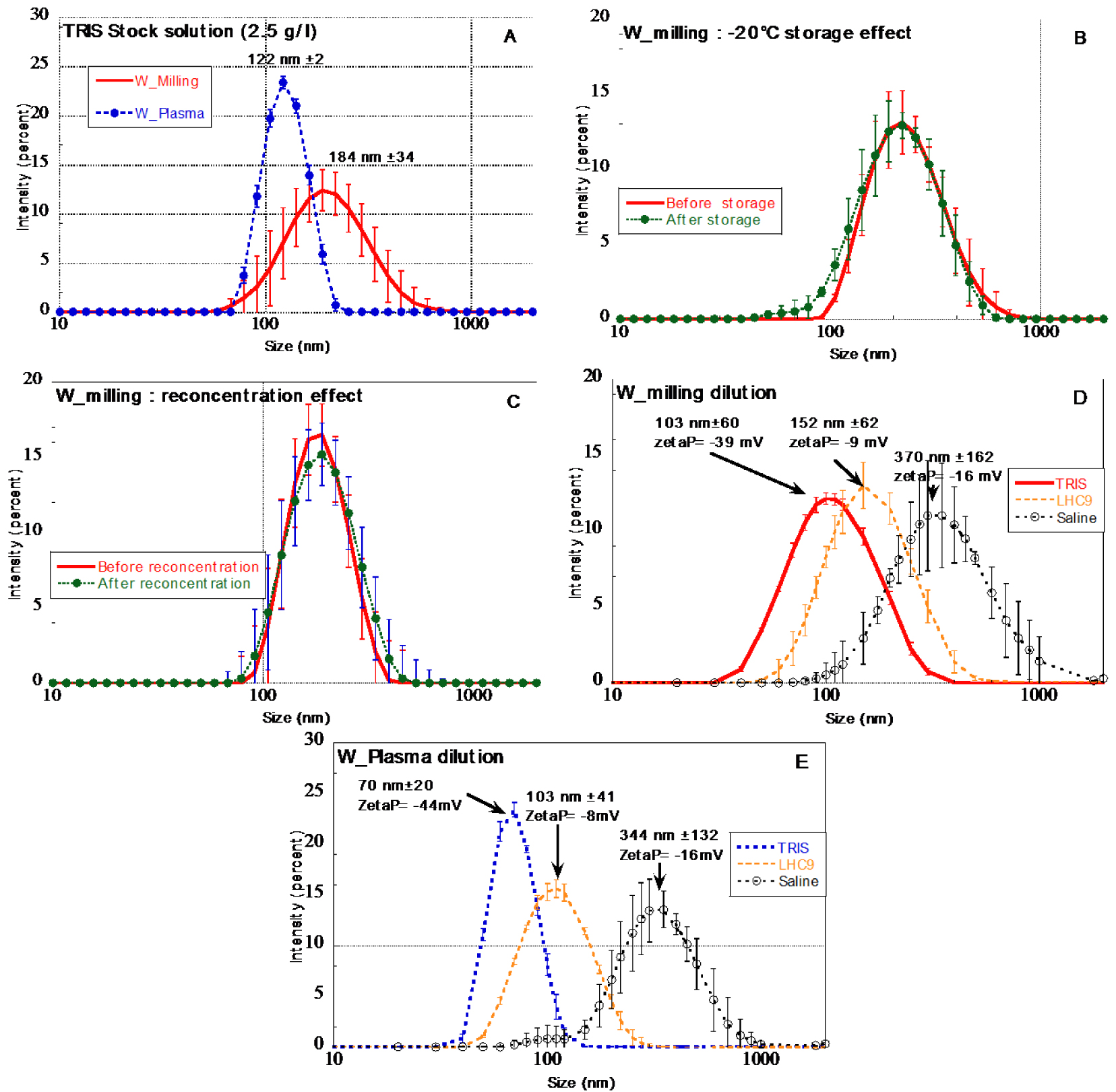
Samples for transmission electronic microscopy (TEM) analysis: 10 μl of W suspension was deposited on a carbon film after a glow discharge supported by a 300-mesh copper TEM grid and then dried. The corroded and oxidized shell thickness and structural morphology of W particles were then analysed using a transmission electron microscope (FEI Tecnai Osiris) working at 200 kV. Observations were carried out in STEM mode with HAADF (high angle annular dark field), DF2, DF4 (Dark Field) and BF (bright field) detectors and 4 EDX detectors (1 srad of solid angle). The point resolution in this mode is 0.8 nm. The microscope is also equipped with a Gatan Ultrascan 2k Camera and a Quantum GIF for EELS (Electron Energy Loss Spectroscopy) and EFTEM (Energy Filtered TEM) analysis.
3. Results
3.1. W suspension preparation protocol: size distribution and stability
Stock suspension: chemical and colloidal stability
The protocol developed to produce W stock suspensions from the planetary ball milling (PBM) powders (Figure 1) enabled to generate monodisperse W particle suspensions with particle size distribution fulfilling the initial objective. Indeed, a mono-modal size distribution was obtained with a mean size of 184 nm ± 34 nm. It is worth mentioning that the protocol was reproducible since the variability of the size distribution of five stock solutions prepared separately did not exhibit significant differences (red curve in Figure 2A). The storage at −20 °C did not affect the size distribution (Figure 2B).
The concentration of the obtained W suspensions varied between 1.4 to 2.1 g W/l. The variability was difficult to explain but it is supposedly based on the variability of the size distribution of the PBM initial powders. To avoid W concentration variability of the stock solutions it was decided to re-concentrate the more diluted suspensions to reach 2.5 g/l ± 0.1. Samples were centrifuged (3000g, 30 min). The supernatant was removed and the solid re-dispersed in lower volume of solution to reach 2.5 g/l. Figure 2C revealed that the re-concentration did not affect the size distribution of the W particles.
W_Plasma average particle size was slightly smaller than W_Milling particles, with an average of 122 nm ± 2 in accordance with TEM observations of [Bernard et al. 2019].
Dilution of W particles in various aqueous media
The dilution of W_Milling and W_Plasma stock solutions in various aqueous media prior to dissolution kinetic experiments affected the size distribution (Figure 2D and E). For both W particle types, the dilution (from 2.5 g/l to 100 mg/l) in TRIS medium lead to a slight decrease of the average size distribution at t = 0. On the opposite the dilution of the stock suspension in saline medium lead to a significant aggregation of particles with average sizes larger than 300 nm. The dilution in LHC9 medium does not significantly modify the size distribution.
3.2. Dissolution kinetics
Dissolution kinetic was first addressed via the quantification of dissolved W from the initial 100 and 10 mg/l W suspensions. As mentioned, the dissolved W phase was separated from the solid phase using 20 nm filtration as smallest cut-off and quantified with ICP-MS.
W_Milling (A,B) and W_Plasma (C,D) particles dissolution as a function of time, initial W concentration and biological or aqueous media, (E,F) comparison between W_plasma and W_Milling particles dissolution for 2 media at 100 mg/l and 10 mg/l.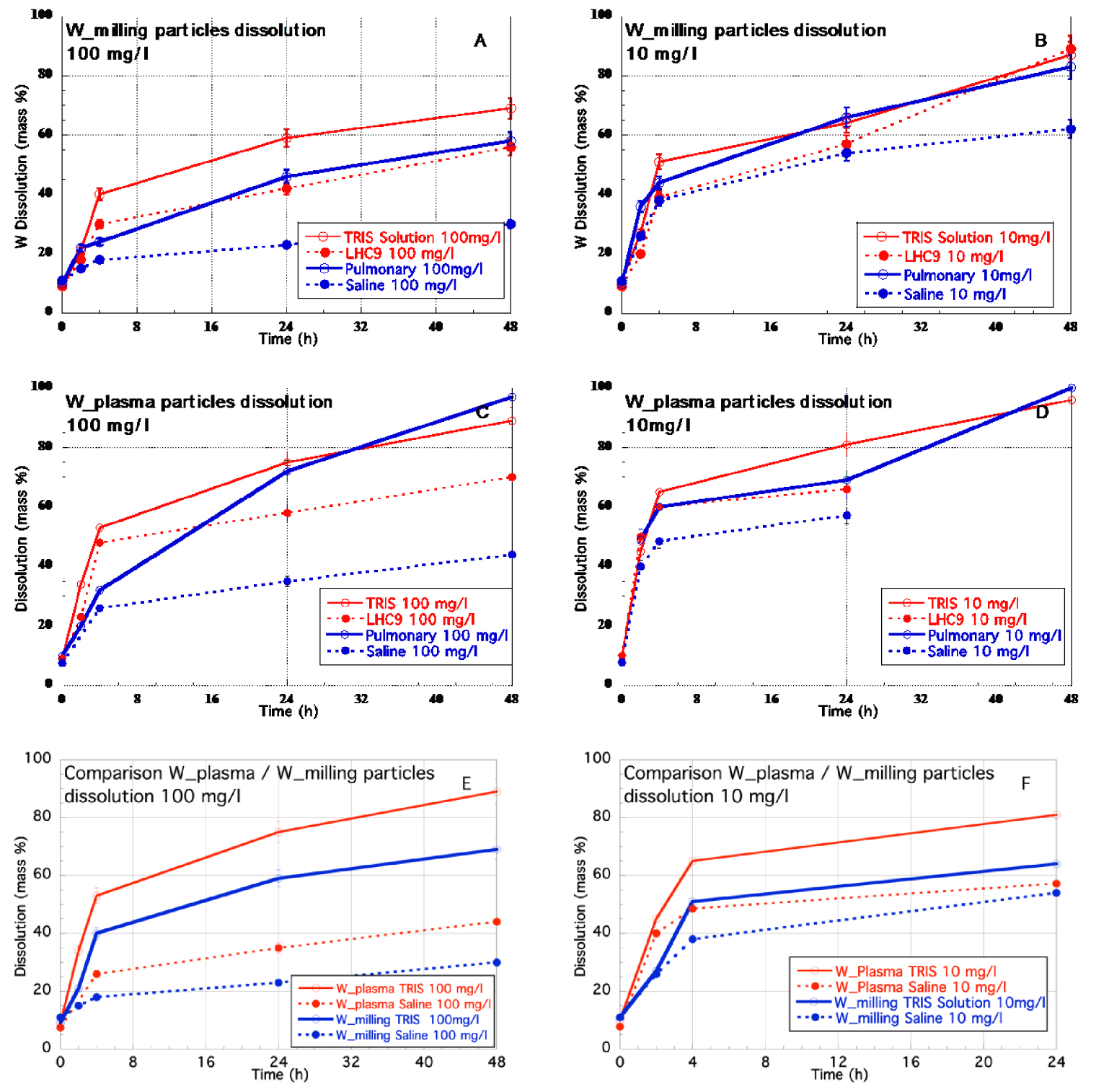
k2𝜒(k) EXAFS spectra of (A) reference compounds (B) particles from the kinetic experiment (plain line) and their corresponding fit (dotted lines).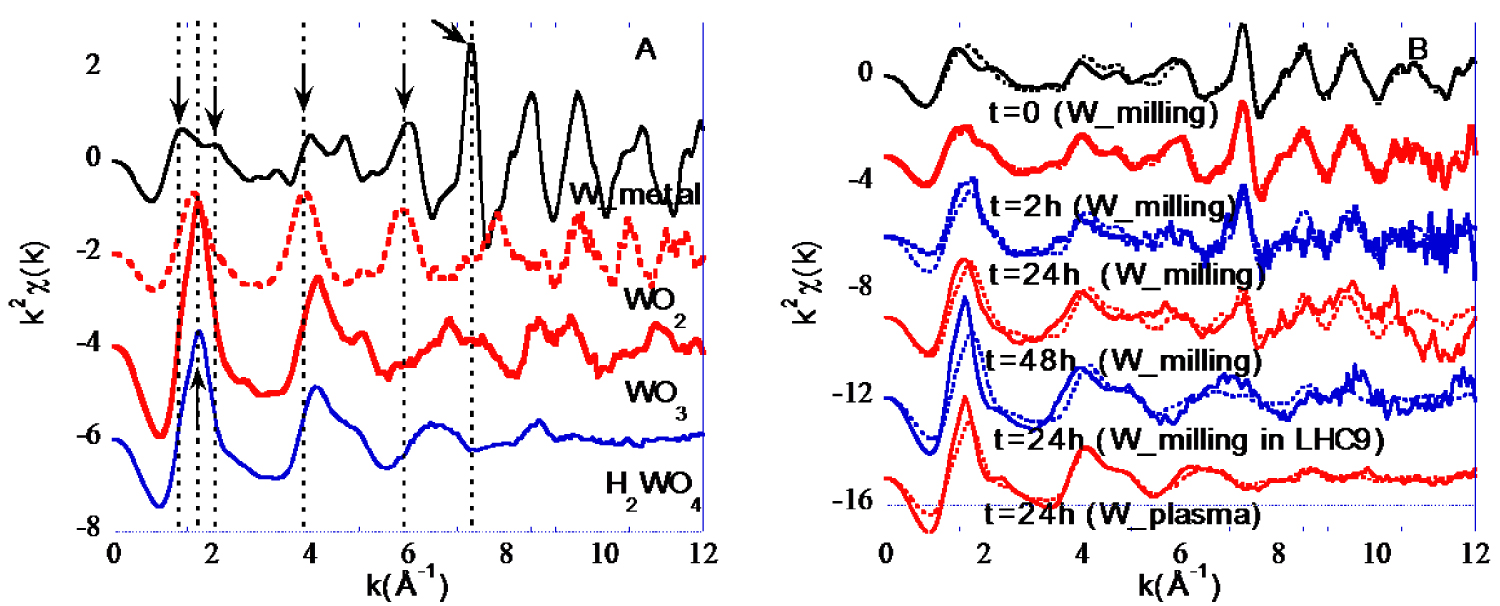
Whatever initial W concentrations and W particle types, TRIS, LHC9 and Pulmonary media lead to fast dissolution of W. More than 25% ± 1.2% of W is already dissolved after 4 h, and after 48 h the dissolution is higher than 55% ± 2.5% in mass and can lead to almost 100% ± 5 % (W_Plasma particle at 10 mg/l). On the opposite, Saline medium has the lowest effect, especially at 100 mg/l. Indeed only 30% ± 1.5% and 44% ± 2.2% of W are dissolved after 48 h for W_Milling and W_Plasma particles respectively. For a given initial concentration and a given medium, it appeared that the W_Plasma particles dissolved faster that the W_milling particles by at least 20% (Figure 3E,F).
The dissolution rate curves of W_Milling and W_Plasma particles whatever liquid media and initial W concentration (Figure 3) followed a “parabolic” law. Such rate-limiting dissolution curves are classically encountered for surface-controlled or diffusion-controlled dissolution mechanisms (e.g. [Holdren & Adams 1982]).
3.3. Redox state of the tungsten particles in suspension: EXAFS results
While XANES is considered more sensitive to determine the redox state of elements than EXAFS, in the specific case of W it came out that EXAFS was more sensitive to detect all W redox forms as discussed in supplementary information.
EXAFS signals obtained for the references and the W suspensions are shown on Figure 4. Metallic W can be easily distinguished from any other W forms thanks to positive oscillations at 1.3, 2.08 and a very intense at 7.3 −1. The latter features are directly related to the atomic structure of metallic W and more specifically direct W–W bonds. The intense and sharp positive oscillation at 7.3 −1 does not exist for the oxide W forms. Beyond the 7.3 −1 feature specific to metallic W, W4+ and W6+ also exhibit some differences. Indeed, it is possible to discriminate W4+ oxide from W6+ oxide. WO2 spectrum exhibits a positive oscillation at 6 −1 while WO3 and H2WO4 EXAFS curves are close to zero. EXAFS spectra of the samples from the kinetic experiment (Figure 4B plain lines) reveal that the intense and sharp positive feature at 7.3 −1 specific to metallic W exists for all W_Milling samples but its intensity decrease from t = 0 to 48 h. The only W_plasma analysed sample (t = 24 h) does not exhibit such feature suggesting no or low amount of metallic W.
To quantify the proportion of the different W redox states within the solid samples, EXAFS spectra were fitted using LCF of spectra of reference compounds (Figure 4B dotted lines). Quantitative results are detailed in Figure 5.
Quantitative results of the LCF of EXAFS spectra of the W_milling particles samples using the reference compounds as database (the EXAFS spectrum of one sample from W_Plasma dissolution experiment was analysed).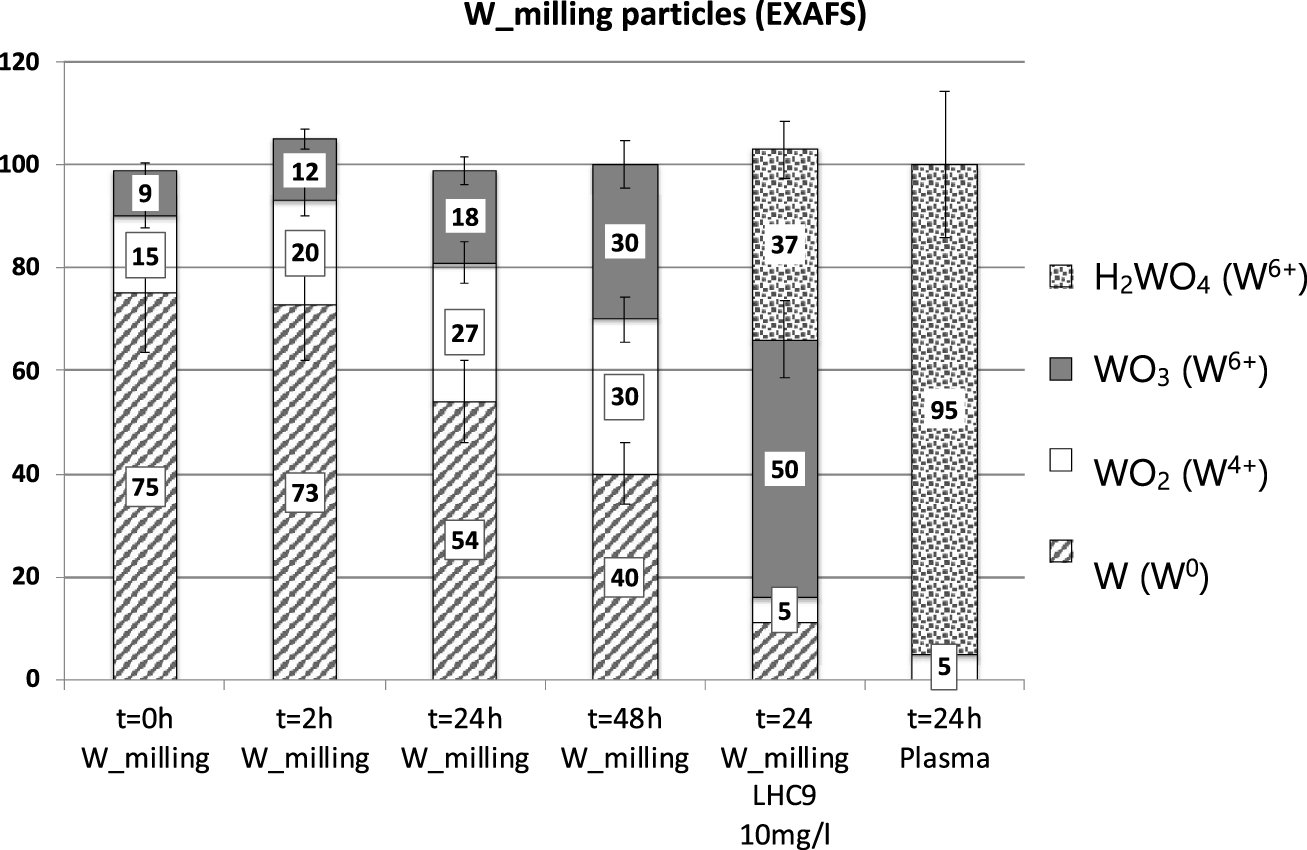
For all W_Milling samples diluted in TRIS, W0, W4+ and W6+ were detected and quantified. The proportion of metallic W in the solid phase remains constant during the first 2 h (68% ± 15) and decreases to 43 and 35% after 24 and 48 h respectively. The proportion of oxidized forms (W4+, W6+) increased from 32, to 33, 57 and 66% for t = 0, 2, 24 and 48 h respectively. It is worth noting that while the proportion of W4+ remained almost constant between 24 and 48h, the proportion of W6+ continued to increase. In addition to W6+ oxide, the W6+ oxy-hydroxide form appeared at 48 h. When dispersed in LHC9 media, only 10% of metallic W remained after 24 h and 84% of W was oxidized to W6+, with more than 50% under the oxy-hydroxide form. The W_Plasma particles at t = 24 h were composed by solely oxidized W with a probable core of WO2 and a large fraction of amorphous and/or hydrated W6+ oxy-hydroxide (95%).
3.4. Structure and size of the corroded layers of the W particles: TEM nano-chemical imaging
TEM was used to determine the location and size of the corroded/oxidized layers.
Transmission Electron microscope images for W_Milling (A–C) and W_Plasma (D–F) particles dispersed in TRIS media, t = 0 h. (A,D) HAADF (high angle annular dark field) images, (B,E) DF (Dark field) images. (C,F) EELS maps at the O (blue pixels) and W (green pixels) absorption edges. (G) Image processing: [DF HAADF] for W_plasma (E–D); (H) Oxygen (blue) and tungsten (dotted dark) profiles example from Figure 7C. (I) Oxygen (blue) and tungsten (dotted dark) profiles example from Figure 7F. (J) Profile example from Figure 7G.
When dispersed in TRIS and at t = 0 h, the formation of tungsten oxide was also proved by the EELS chemical maps. Oxygen surrounds tungsten particles (Figure 6(F)). The layer can clearly be seen on W_Plasma particles (Figure 6(F)) while on W_milling the O surrounding layer is less evident (Figure 6(C)). From the combined tungsten and oxygen EELS images it was possible to estimate the W-oxide layer thickness. The estimation was based on the position of the inflection point of the derivative curve of both oxygen and tungsten profiles (Figure 6(I)). In the case of W_Plasma the average thickness calculated from EELS images and 15 profiles was 4.15 ± 1.2 nm. In the case of the W_milling particles, the profiles were much noisier and difficult to interpret. Figure 6 indicated that the particle size and thickness variability is also contributing to EELS profiles and can be hardly deconvolved from the signal from the corroded layer. In many cases the EELS maps corresponded to several overlapping particles. A less accurate analysis was however attempted in the case of the W_milling particles. Oxygen and tungsten profiles were plotted, but no derivative curves were calculated, as too noisy. From the two elemental profiles, the shift in nm were extracted at various positions at the particle boundary (Figure 6H). The protocol repeated on 15 elemental profiles, leads to W-oxide layer thickness of 19 ± 6 nm. However, care must be taken concerning the estimation.
The combined analysis of both Dark Field and HAADF images can also provide information on the corroded layer thickness. Dark Field images are sensitive to the atomic order (amorphous vs. crystalized) and the composition of the particles while HAADF are mainly sensitive to the chemical composition and more precisely to the atomic number. Subtracting normalized HAADF to normalized DF images enabled to reveal the “transition” layer as shown in the case of W_Plasma particle (Figure 6G). The profile (Figure 6H) revealed the surface layer via two peaks. The peak thickness is correlated to both the curvature of the particle and the thickness of the W-oxide layer. Without deconvolution of the curvature effect, the estimation of the thickness surface layer is 6.3 ± 4.2 nm. The result needs to be compared with the estimation from EELS images (4.15 ± 2 nm). In the case of W_milling particle, the estimation was impossible, due to variability of particle thickness and size. It was impossible to unambiguously identify the corroded layers location with nanometric accuracy.
3.5. W particles at t = 24 h
In the case of W_Milling particles after 24 h, from EELS images it was possible to estimate the thickness of the corroded layer. The estimation of the thickness of the corroded layer reached 34 nm ± 10 (n∗ = 10).
Transmission Electron microscopes images for W_Milling particles dispersed in TRIS at t = 24 h. (A) High angle annular dark field image HAADF. (B,C) Dark field images DF4. (D) EELS maps at the O (blue) and W (green) absorption edges.
4. Discussion
4.1. Chemical and physical stability of W particles in tested aqueous and biological media
The size of particles in the various aqueous and biological media can change. Even if at long term, dissolution will occur, short-term exposure level of living organism as well as environmental fate may be affected by the size and aggregation mechanisms.
The Pourbaix diagram of W at 10−4 mol/l ( = 18 mg/l) [Anik & Osseo-Asare 2002; Nave & Kornev 2017] confirmed that solid metallic tungsten is not thermodynamically stable at pH > 6 and that soluble specie should form. Even if our experiments focussed on short duration, the amount of dissolved W reached high level, up to 100% in the case W_Plasma particles in Pulmonary medium after 48 h. The kinetic of dissolution in Saline medium was slower, but data extrapolation suggests that full dissolution could occur, after longer time (≈54∕55 days). The current study found important amounts of dissolved tungsten when sub-micrometric tungsten powder was dispersed into aqueous solutions in accordance with some papers from the limited available results in the literature on tungsten solubility [Andersson & Bergström 2000; Dermatas et al. 2004; Peuster et al. 2003].
Environmental and health impact studies of tungsten particles must take into accounts the chemical stability of W and its kinetic of total or partial dissolution in order to clearly define the mechanism and mode of actions in the environment and in biological media (organisms, tissues). For instance, a fast oxidative dissolution of W to tungstate would affect the redox equilibrium and could create strong oxidative stress in biological tissue at very short term, while long-term effects would only be due to the presence of tungstate ions. A slower oxidation would certainly maintain oxidative stress longer in combination of constant presence of tungstate ions. But the concentration of released W and therefore the possible oxidative stress will vary as function of the kinetic. A very slow kinetic may lead to no specific effect.
4.2. Dissolution rate of metallic W nanoparticles changes as a function of biological media, initial concentration and particle size
Solid-to-liquid ratio (S/L)
For a given particle type and aqueous medium, the decrease of the solid-to-liquid ratio from 100 mg/l to 10 mg/l lead to the increase of the dissolution rate. The decrease of the S/L ratio leads to a relative increase of the proportion of reagent (oxidation and hydrolysis) per solid-surface unit and therefore accelerates the oxidative dissolution of W. It is also known that the rate of dissolution is related to the saturation of the solution with respect to the solubility product (R = f(1 −𝛺n), with R = dissolution rate, 𝛺 = saturation state, n = reaction “order”) (e.g. [Morse & Arvidson 2002]). The decrease of S/L will decrease the saturation and therefore it will increase the dissolution rate. However, the S/L ratio does not seem to affect to a same extend the various systems. Indeed, while for W_plasma in TRIS, the dissolved mass fraction increased by 8% after 24 and 48 h when the S/L ratio decreased from 100 to 10 mg/l, in Saline medium the dissolved mass fraction increased by more than 60%.
Dissolution rate as function of the initial crystal structure
The dissolution kinetic experiments revealed that the W_Plasma particles dissolved faster than the W_Milling particles whatever the biological media (Figure 3E,F). The difference can reach 20% between both particle types. One of the main differences between the two systems is related to the crystal structure. The W_Plasma particles are composed by beta phases while the W_milling particles are composed by alpha phases. The alpha phase has body-centered cubic (bcc) metal crystal structure while the structure of the β phase is called A15 cubic (space group–Pm(-3)n) [Kondo 1992; Shabalin 2014]. The crystal structure lead to an increase in the W–W distance from 2.74 Å for the alpha to 2.84 Å for the beta phase. In the literature the beta phase is less stable as the alpha when temperature increases [Shabalin 2014]. For instance, the beta phase is converted to the alpha phase at temperature higher than 600–650 °C. The structural differences seem to also affect the stability in water. It can therefore be hypothesized that the nature of the crystalline phase could affect the W stability in water. However, the specific surface area may also be involved, and it is not possible without complementary experiment to unambiguously determine the exact role of the nature of the crystalline phase in the dissolution rate.
Dissolution rate decreased with particle aggregation
The role of the aqueous and biological media on the dissolution kinetic of metallic tungsten is obvious from results shown in Figure 3. For most tested conditions, TRIS solution leads to the faster and larger extend of W dissolution while Saline media had the lowest effect. As shown by Figure 2C,D, the particle size distribution strongly varied between TRIS and Saline media. While TRIS keeps W particles with small average size and almost monodispersed particles, Saline medium produced aggregation and significant increase of the average size. Zeta potential of W particles in TRIS media reached low negative values ( − 39 and − 44 mV for W_Milling and W_Plasma particles respectively), which can explain a good particle dispersion. On the opposite, Saline medium strongly affects the surface properties of W particles with a significant decrease of the absolute value of the zeta potential. With an absolute value of the zeta potential in a 0–20 mV range, electrostatic repulsions are generally too low to avoid aggregation of particles [Bhattacharjee 2016]. The cases of nutritive media (LHC9 and Pulmonary) are more complex. Indeed, with zeta potentials of −9/−8 mV, particles should aggregate. However, the main difference between TRIS, Saline solutions and nutritive media is the presence of large size organic compounds such as proteins. It is highly probable that proteins adsorb at the W particle surface and therefore enable to maintain particle dispersion via steric interactions.
The effect of particles and/or aggregates size on the dissolution kinetic is related to the solid surface in contact with solvent (specific surface area) as stated by the Noyes–Withney equation for instance (e.g. [Hattori et al. 2013; Morse et al. 2007; Morse & Arvidson 2002]). The size decrease of particles inevitably leads to a specific surface area increase and therefore an increase of the dissolution rate.
4.3. Dissolution rate modelling
Figure 3 clearly indicates that the dissolution rate of W particles did not follow a linear function of time as it could have been inferred from literature and the dissolution proceeds via a “parabolic” rate law involving a limiting step. Indeed [Andersson & Bergström 2000] found that the WO3 surface layer on the oxidized WC particle dissolved readily at pH > 3 and that the concentration of dissolved W increased linearly with time. [Peuster et al. 2003] also found linear dissolution trend over time in the case of the dissolution of W coil used in medial application, in Ringer’s isotonic solution (pH = 5.8).
A basic concept in solid phase dissolution is that dissolution reaction follows different processes involving diffusive transport of reactants and products into the liquid phase and various surface reactions. As function of the limiting step, dissolution can be diffusion or surface controlled [Morse et al. 2007].
Various kinetic models exist to predict the reaction rate as function of limiting steps and then the dissolution curve profiles. Empirical or simplified models are based on hypothesis concerning the evolution of particle structure as function of time as shown in Figure 8.
Possible schematic shrinking modes of W particles dissolution.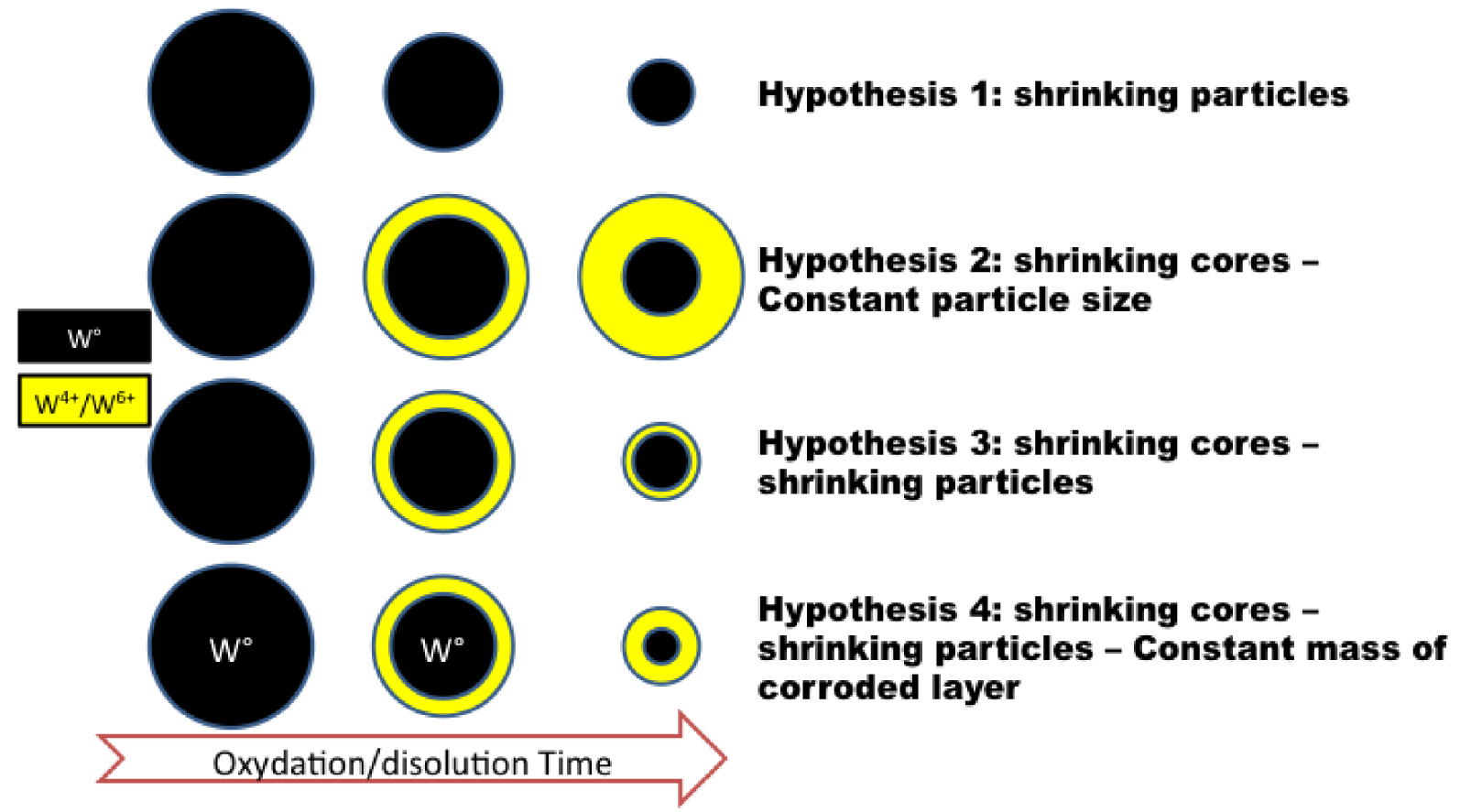
The rate of dissolution of solid in fluid can be controlled by different phenomena such as (i) surface chemical reaction (ii) diffusion through a “corroded/oxidized” surface layer or (iii) diffusion in liquid (fluid film). The equations relating the fractional conversion of solid (X) as function of reaction time (t) and kexp (experimental rate constant) are described below (e.g. [Costa & Sousa Lobo 2001; Demir et al. 2004; Reyes et al. 2017; Senanayake 2011]:
- Surface chemical reaction control:
(1) - Solid layer diffusion control:
(2) - Diffusion control:
(3)
Plotting the three F(X) equations as function of t (Figure 9A, B, C) may help to identify which mechanism controls dissolution rate of W in aqueous media as soon as one of the F(X) follows a linear trend with the slope representing kexp. In the case of W_milling at 100 mg/l, whatever the dispersion medium, experimental curves did not exhibit perfect linear trends in the case of (1) and (3) (Figure 9A,C). Figure 9B suggests that in the case of TRIS and Pulmonary medium (100 mg/l) the curves corresponding to the “solid layer diffusion control” (2) reached almost a linear trend. This is further highlighted in Figure 9D,E using linear fits. Linear fits of the experimental data plotted with y axis as F(X) equations for TRIS (Figure 9D) and Pulmonary media (Figure 9E) suggested that (2) lead to the best fit.
Then the assessment of various dissolution models suggested that the dissolution mechanisms may vary as function of the medium. In the case of TRIS and Pulmonary media it seemed that the dissolution is controlled by diffusion through a surface corroded/oxidized layer (“Dif Solid”). Even if the kinetic curve did not follow a perfect linear trend, this result is in line with the observation of the corroded/oxidized layer by TEM (Figures 6 and 7). For Saline and LHC9, none of the models were adapted.
Assessment of (1), (2) and (3), which correspond to the different controlling stages of the shrinking core model. In the plots, the slope represents kexp.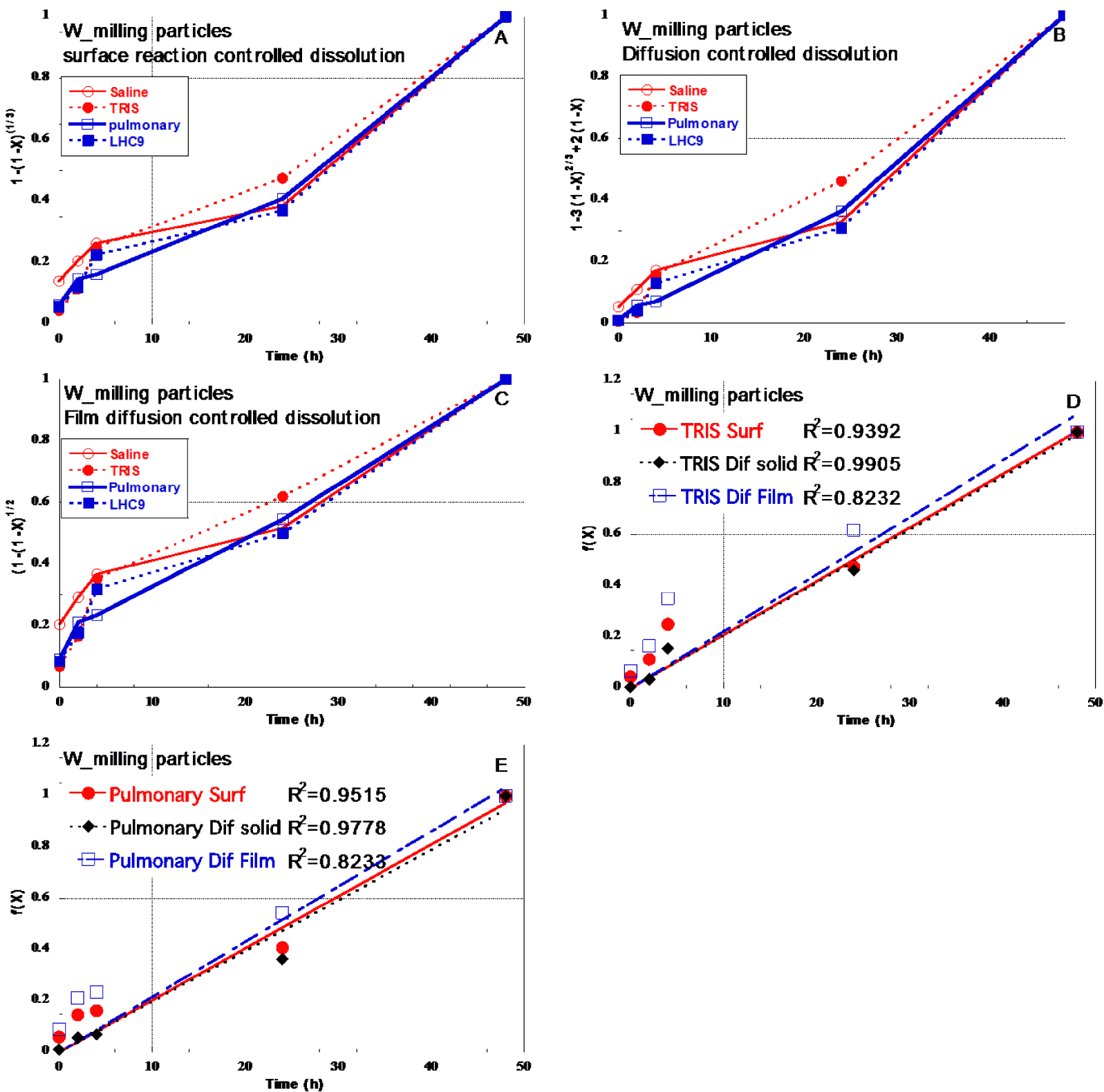
(A) Decrease of the solid mass of W0, W4+, W6+. (B) Calculated particles size and thickness of the corroded layer.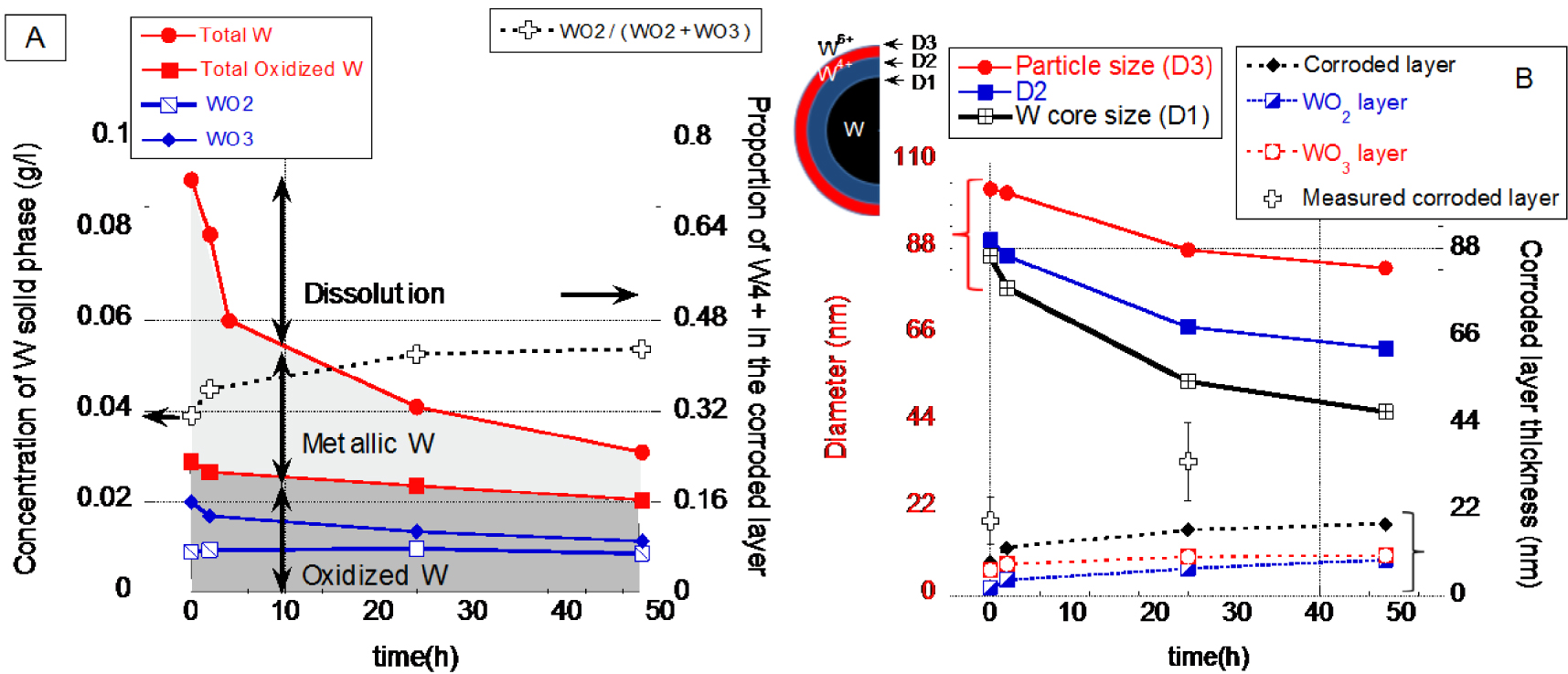
Even if the Solid layer diffusion control model lead to the best experimental curve fit, the linear fit did not perfectly adjust to experimental curve. Indeed, shrinking core models are developed on simplified systems (initial mono-modal size distribution of spherical particles, uniform particles shrinking that maintain spherical particles, homogeneous corroded/oxidized particle layer, etc) and did not perfectly describe the W particle behaviour. For instance, X-ray Absorption Spectroscopy (XAS) results clearly indicated that in the case of W_Milling particles in TRIS medium, the corroded/oxidized layer is not homogeneous and can be composed by both W+4 and W+6 oxides/oxy-hydroxides.
4.4. Dissolution mechanisms (the example of W_milling in TRIS)
The dissolution of metallic particles in aqueous system involves first the oxidation of the metallic surface and then the dissolution of the metal oxide form. Pioneer work concerning the oxidation of W [Johnson & Wu 1971] considered that oxidation of W involves the formation of a surface film of W2O5(s) further oxidized to WO3(s). Then as function of the pH and W concentration, WO3(s) can dissolve in water to form tungstate ions as follow:
However even if W dissolution requires oxidation, a solid W oxide layer is not always formed at the surface of the metallic particle. The dissolution of W micro-electrode in Phosphate Buffer Saline medium to simulate neural system, was shown to occur without the formation of stable oxides for instance [Patrick et al. 2011].
In our experiments, the solid W oxides were always detected for the initial products (t = 0 diluted W suspensions). TEM evidenced the presence of the W oxide layer at the surface of the metallic particles. The dissolution of tungsten particles (and any metallic particle) is therefore based on the oxidation of the W core to form a W-oxide layer, and the dissolution of the oxide surface later.
From all results it was possible to propose a simplified mechanism of W_Milling particle dissolution in TRIS medium. By combining the dissolution results (Figure 3) and the W speciation in the solid phase (XAS), the concentration of the metallic and oxidized particulate forms of tungsten was calculated (Figure 10). The mass of solid metallic W decreased over time, which corresponds to a shrinking of the particles core. The presence of oxidized W in the solid phase clearly indicated that the shrinking particles model (hypothesis 1 Figure 8) can be discarded. However even if the concentration of solid metallic W decreased, the concentration of oxidized W remained almost constant as function of time with only a slight decrease from 29 mg/l ± 5 at t = 0 h to 26.7 ± 5, 23.7 ± 5 and 20.5 ± 5 after t = 2, 24 and 48 h respectively.
Hypothesis 2 (Figure 8) with a constant particle size, can also be discarded, since it would imply an increase of the mass of oxidized W with time. Hypothesis 3 will neither describe the mechanism of dissolution since a constant thickness of the oxidized layer associated with a decrease of the core size, would lead to a decrease of the mass of oxidized W in the solid phase. In our case, a (almost) constant mass of oxidized W suggests that the size of the entire particle decrease, but the layer’s thickness of the corroded layer increased (hypothesis Figure 8). This can be illustrated with a simple model of particle dissolution. By considering spherical particles with the same size, W, WO2 WO3 and H2WO4 density and solid mass as function of time (XAS results), uniform dissolution, it was possible to estimate the particle size decrease as well as the thickness of the corroded/oxidized layers. With such simplified system and hypothesis, it came out that the size of the metallic core decreased faster than the size of the entire particle. This means that the thickness of the corroded layer increased. With our simplified model, we roughly estimated that the thickness of the corroded layer increased from 8.6, 12.0, 16.7 and 18.2 nm at t = 0, 2, 24 and 48h respectively. While at the same time the diameter of the core of the particles decreased from 86.0, 78.0, 54.2 and 46.6 nm respectively. The rough estimation of the corroded layer did not differ too much from TEM results i.e. 19 ± 6 nm and 34 nm ± 10 at t = 0 and 24 h respectively.
The dissolution mechanism of the W_milling particles in TRIS is following a more complex pathway compared to the four shrinking models described in Figure 8. Three interfaces can be defined between (1) W and WO2, (2) between WO2 and WO3 and finally (3) between WO3 particle surface layer and aqueous media from t = 0 h up to 48 h. A forth interface may also exist with the highly probable hydration of WO3. Therefore, even if WO2 solid phase is not thermodynamically stable under our tested conditions, it can be considered as an important intermediary product during the tungsten oxidation and dissolution.
5. Conclusion
The chemical and colloidal stability of W_Milling and W_Plasma particles in various aqueous and biological media was assessed for short-term dilution experiments, in accordance with the duration of exposures that were performed in particle toxicity tests on cellular models as detailed in [Uboldi et al. 2019; George et al. 2019]. The dilution of particles in TRIS, LHC9 and pulmonary media did not strongly affect the average size of the particle while the dilution in Saline medium lead to substantial aggregation. With increasing time, W particle dissolved in all tested media and a total dissolution can be emphasized from dissolution curve extrapolation over time. From the different dissolution rates as a function of the tested media, it seems that the oxidative dissolutions are rate limited by diffusion in the solid oxidized layer surrounded the metallic core of particles. The mechanisms of oxidative dissolution involve the formation of a heterogeneous oxidized layer composed by both WO2 and WO3∕H2WO4.
Knowledge provided by these dissolution experiments will help to determine the environmental mobility and persistence as well as the bio-durability of these tungsten sub-micronic materials. As dissolution has potential to influence the toxicity of particles, it is an important parameter in the risk assessment of particles as discussed in parent papers [George et al. 2019; Uboldi et al. 2019].