

1 Introduction
The African Guineo-Congolian rain forest constitutes the second largest block of rain forest on earth and, although it is not as rich as the rain forests of South America or South-East Asia, hosts a remarkable biodiversity (ca. 6400 endemic plant species; Barthlott et al., 1996; Myers et al., 2000). The underlying cause of the species richness of rain forests has fascinated biologists for decades and remains a highly debated topic today. Recently, many new insights into the dynamics and behaviour of biological systems have been provided by molecular genetic markers. Indeed, molecular phylogenies have provided ever more detailed interpretations of the patterns of species diversification (e.g., Barraclough and Nee, 2001; Eckert and Hall, 2006; Manos et al., 1999). Additional insights come from the pattern and distribution of genetic diversity within species, e.g. phylogeography – the geographic distribution of lineages within species (or closely related species), that allows a reconstruction of the history of populations (Avise et al., 1987). Phylogeography can potentially detect past events of fragmentation, colonization, demographic expansion or bottlenecks (Hewitt, 1999; Knowles and Maddison, 2002; Petit et al., 2002; Taberlet et al., 1998). Such genetic approaches, when combined with information from other disciplines (e.g., palaeoclimatology, palaeobotany, palynology, archaeology, climate and vegetation modelling, biogeography), have proven their value to better understand the history of temperate biomes (e.g., Knowles and Alvarado-Serrano, 2010; Magri et al., 2006), and a few tropical ones (e.g., for the Atlantic forest of Brazil, Carnaval et al., 2009). However, until recently, few phylogeographic studies had been conducted on the African Guineo-Congolian rain forest biota, especially among plants. The accumulation of new genetic data on African rain forest trees over the past few years now enables a first synthesis for this biome.
This article reviews the phylogeographic patterns detected in Guineo-Congolian rain forest trees and discusses whether they support alternative hypotheses regarding the history of the African rain forest cover. Being designed for a broad interdisciplinary readership, this article starts with an overview of the African rain forest history, and of basic principles regarding the genetic tools used for phylogeography and of the phylogeographic patterns expected under different scenarios of historical population changes. Technical considerations, requiring a background in population genetics or phylogenetics, have been avoided and a few technical terms are defined in Box 1.
2 Biogeography and history of the Guineo-Congolian rain forest
The Guineo-Congolian rain forest was defined by White (1979, 1983) as a phytogeographic centre of species endemism (comprising ca. 8000 plant species of which ca. 80% are endemic), and was subdivided by the same author into three sub-centres (Fig. 1A): Upper Guinea (hereafter UG), Lower Guinea (hereafter LG), and Congolia (hereafter C). UG corresponds to the West-African rain forest extending from Sierra Leone to Ghana. LG corresponds to the western part of the Central African rain forest, from southern Nigeria to the south-western part of the Republic of Congo and of the Democratic Republic of Congo, and is crossed by the Cameroon Volcanic Line (hereafter CVL, Fig. 1A) in its western part. C corresponds to the eastern part of the Central African rain forest, including most of the rain forest from the Congo River basin. UG and LG are separated by the Dahomey Gap (hereafter DG, Fig. 1A), a ca. 200-km-wide savannah corridor in Benin and Togo, the product of a locally dry climate. LG and C are separated by the floodplain of the Ubangi River and part of the Congo River, an area dominated by swamp forests. This subdivision is justified by the observation that many plant and animal species are endemic to only one sub-centre (but see Linder et al., 2012, who do not distinguish LG from C). The origin of this biogeographic feature is an integral part of the history of the African rain forest, which is reviewed below (for additional details see e.g. Anhuf et al., 2006; Bonnefille, 2007; Jacobs, 2004; Maley, 1996; Morley, 2000).

Guineo-Congolian forest delimitation, subdivision and topography (A), and hypothetical range shifts in the Late Quaternary (B). CVL: Cameroonian Volcanic Line (Anhuf, 2000).
Délimitation, subdivision et topographie de la forêt Guinéo-Congolaise (A) et changements de distribution hypothétiques au cours du Quaternaire récent (B). CVL: Ligne volcanique camerounaise (Anhuf, 2000).
Climate fluctuations throughout the Tertiary and Quaternary, as well as tectonic plate movements, have largely determined the dynamics of the distribution of biomes on earth. In Africa, biome reconstructions indicate that, while the rain forest covered from 22 to 15 million km2 between 55 Ma (Eocene) and 11 Ma (Miocene) ago, this area dropped to ca. 10 million km2 3 Ma ago (Pliocene) and to 3.4 million km2 today (Kissling et al., 2012). This general trend of a reduction in the area of the African rain forests throughout the Cainozoic might explain their current lower species richness compared to Neotropical or South-East Asian rain forests. This longer-term range contraction was also accompanied by shorter cycles of range reduction and expansion, for example between the Late Miocene and Early Pliocene, and especially with the onset of the glacial–interglacial cycles of the Quaternary. Indeed, during the cooler and drier glacial periods, the African rain forest became fragmented, remaining in “refuge” areas while savannah and/or mountain forests expanded. According to palaeoenvironmental reconstructions (e.g. Fig. 1B for the Last Glacial Maximum, hereafter LGM), the reduction in the rain forest cover during the glacial maxima would have been much more important in Africa than in Amazonia (Anhuf et al., 2006). The rain forests re-expanded during the interglacial periods, eventually forming a nearly continuous forest block. For example, during the Holocene African humid period (ca. 9000–5500 years BP; Fig. 1B), the DG was forested (Hély et al., 2009; Salzmann and Hoelzmann, 2005). At the Late Holocene, ca. 3000 years BP, a general degradation of the rain forest is documented in many pollen records from lake or marsh sediments, coinciding with both the onset of a drier climate and the expansion of Bantu farmers into the rain forest (Bayon et al., 2012; Maley et al., 2012; Neumann et al., 2012; Ngomanda et al., 2009; Schwartz, 1992).
The occurrence of three sub-centres of endemism within the Guineo-Congolian rain forest might suggest that these areas were most often isolated from each other during recent geological history (Pleistocene, and maybe Late Pliocene); otherwise species would have had an opportunity to spread over the whole rain forest area. Using this logic, smaller areas hosting a particularly high number of endemic species have often been interpreted as ancient forest refugia that existed during the glacial maxima (e.g., Robbrecht, 1996; Sosef, 1994). Compiling these patterns of endemism and palaeoenvironment (e.g., Maley and Brenac, 1998), Maley (1987, 1996) proposed a map of the main LGM rain forest refugia (Fig. 1B). Most of these refugia are located in hilly areas, which would have precipitated the humidity brought by low stratiform clouds, favouring the maintenance of a hygrophilous forest in the valleys (Elenga et al., 2004). Forest refugia could have acted as museums of biodiversity (low species extinction rate – museum hypothesis) and/or as drivers of allopatric speciation (high speciation rate – species pump hypothesis) (Fjeldså and Lovett, 1997; Sosef, 1994, 1996; Stebbins, 1974; for a review see Plana, 2004). Dated phylogenies indicate that the diversification of many rain forest animal genera would have occurred prior to the Pleistocene, which supports the museum hypothesis (Moritz et al., 2000). However, some rain forest herbs seem to have diversified at least partly during the Pleistocene (Plana et al., 2004), which supports the species pump hypothesis. Under Maley's refuge hypothesis for the LGM, geographic gradients of diversity and endemism within each sub-centre of endemism are also explained by the history of the rain forest in response to climate change.
A difficulty with Maley's hypothesis is that it assumes that forest refugia can be identified by the high levels of endemic forest species, while such distribution patterns can also result from ecological species sorting for particular isolated habitat types, as expected for example for hilly landscapes. As most proposed forest refugia occur in hilly areas, the origin of their high species endemism is controversial. In fact, although there is no doubt that vegetation composition has changed and that the rain forest distribution has fluctuated throughout the Quaternary, there is little direct evidence documenting the extent to which the rain forest was fragmented and where forest refugia occurred. A reconstruction for the LGM proposed by Anhuf et al. (2006; Figs. 1B) suggests that the rain forest was less fragmented than under Maley's hypothesis. However, this reconstruction ignores the impact of CO2 concentration and relies on current rainfall distribution patterns, while rainfall gradients are likely to have been very different during the LGM. Reconstructions based on palaeovegetation modelling coupled to palaeoclimate modelling are also being developed, but are still subject to substantial uncertainty (e.g., Hély et al., 2009). Finally, forest refuge hypotheses usually neglect the heterogeneity of the rain forests and the ecology of their constituent species. However, different rain forest variants occur according to climatic and edaphic factors (e.g., length and intensity of dry and wet seasons, drainage, soil fertility), and azonal vegetation types like gallery or swamp forests can also host many rain forest species. Hence, the size and location of past refuge areas might vary substantially among rain forest species according to their ability to survive in the different forest types.
The paucity of dated phylogenies for African plants limits identification of general patterns of diversification (Plana, 2004). However, preliminary data indicate that the divergence between congeneric species often predates the Quaternary. Thus, genetic signatures of Late Quaternary events are more likely to be detected from analyses of within-species patterns of genetic diversity, rather than from among-species patterns.
3 Types of genetic markers commonly used to infer the phylogeographic history of plant species
Most genetic inferences of the demographic history of species have been based on (often single-locus) DNA sequences or on multilocus genotypic data (using various types of markers such as microsatellites, AFLPs, RAPDs, RFLPs, see Box 1). The advantage of DNA sequences is that the different variants (called haplotypes) can be ordered into lineages (groups of closely related haplotypes) and the geographic distribution of these lineages can be very informative. For example, if two lineages occupy distinct (possibly overlapping) areas, it is likely that the two populations have been isolated for a long time, and their divergence can potentially be dated using the principle of a molecular clock and calibration points (based on fossil evidence and/or the occurrence of endemic species on an island with a maximal known age). The frequency distribution of the different haplotypes can also be informative to infer whether the population has undergone a demographic expansion or bottleneck, or recent range dynamics (e.g., Arenas et al., 2012; Rogers and Harpending, 1992).
Phylogeographic studies in plants have most often relied on plastid DNA (hereafter pDNA, e.g. DNA from the chloroplast genome) because universal molecular markers enable a straightforward application to a wide spectrum of taxa and because the haploid nature of this genome facilitates sequencing compared to a diploid (or polyploid) nuclear genome (for an introduction, see Petit and Vendramin, 2004). In angiosperms, chloroplasts are usually transmitted exclusively maternally, so that their phylogeographic patterns solely reflect the history of seed colonization without being complicated by the effect of long-distance pollen flow. However, the chloroplast genome typically suffers two drawbacks. First, it evolves slowly (low mutation rate) so that it may display little polymorphism within species and hence convey little information on relatively recent events. Second, being non-recombinant, it reflects a single instance of the demographic history and the associated stochastic processes (mutation and genetic drift), while simulations show that the same population history can lead to a wide range of resulting phylogeographic patterns. For example, if a new population was founded in the past, depending on whether the founder individuals bore one or several haplotypes that survived until the present and/or whether new mutations created a lineage unique to this population, very different patterns emerge, and allow the identification of the population founding event, either detectable or not. For this reason, the comparison of phylogeographic patterns at multiple unlinked nuclear loci, which represent different instances of the stochastic processes that structure diversity, is a more reliable method to infer past processes. However, there are still few such studies using multiple markers because markers must be optimized for each species or genus.
As an alternative to DNA sequencing, genetic markers can be used to genotype individuals at several loci at once (e.g., microsatellites, AFLP, RAPD). Generally, these markers do not allow the identification of lineages (i.e. variants, called alleles, are not ordered) but their multilocus nature allows the identification of groups of individuals behaving as populations (i.e. without internal barriers to gene flow) that are often called “genetic clusters” or “gene pools”. The occurrence of different gene pools in allopatry or parapatry indicates that they have been isolated for long enough to become differentiated by genetic drift and/or the accumulation of new mutations. Parapatric gene pools might even show evidence of admixture in their contact areas (i.e. occurrence of individuals with ancestors from both gene pools), in which case genetic data can reveal past population fragmentation that could not be suspected from the current distribution of the species.
Finally, combining several markers is usually insightful because they can reduce the noise due to the stochasticity of genealogical sorting and, furthermore, they can leave signatures of population history at different time depths according to their mutation rates. Similarly, comparing the patterns displayed by multiple sympatric species with similar ecology, the domain of comparative phylogeography, allows highlighting shared patterns driven by past changes in their ecosystem.
4 Expected phylogeographic patterns according to biogeographic scenarios
In this section, we consider biogeographic scenarios that might explain phylogeographic patterns occurring in widespread species at the scale of (1) the whole Guineo-Congolian region, and (2) the putative refuge areas within the sub-centres.
4.1 Genetic differentiation among sub-centres
UG, LG and C have been recognized because they host a significant portion of endemic plant species (White, 1983). However, there are also many species occurring in two or three of these sub-centres, or even beyond sub-centres (e.g., in the transition zones towards the Sudanian or Zambezian centres of diversity). Three hypotheses might explain these large distributions:
- • H1: species have been widespread during a period of maximal forest extension and persisted in isolation in two or three sub-centres of endemism when the forest became fragmented. Populations from different sub-centres should then display substantial genetic differentiation, leading to a correlation between geographic patterns of differentiation at the within-species and among-species levels;
- • H2: species survived in a single sub-centre of endemism during the Last Glacial Maximum and re-colonised other sub-centres postglacially. Therefore, either populations should be little differentiated among sub-centres (if genetic drift was limited during re-colonisation), or re-colonized areas should harbour a lower genetic diversity than the region of origin, no endemic alleles and a genetic signature of past population size reduction;
- • H3: species have wide enough ecological amplitude to persist even between the sub-centres of endemism when the forest was most fragmented, and might maintain gene flow across sub-centres, their distribution being limited by other factors than the ones determining the rain forest biome. Hence, populations should be little differentiated or display differentiation patterns not coinciding with the limits of sub-centres.
4.2 Genetic differentiation within sub-centres (endemic alleles/gene pools vs. putative rain forest refuges)
Maley's (1996) version of the refuge hypothesis (hereafter H4, Fig. 1B) predicts the following pattern:
- • H4: the Guineo-Congolian forest was highly fragmented during glacial maxima, persisting in a few rather small areas, so that the forests surrounding these refugia must be of recent (Holocene) origin. While mutations could have accumulated new allelic variants within the old forests of the refuge areas, most rare alleles might have been lost during the re-colonisation process. Hence, different refuge areas should be well differentiated and display distinct endemic alleles, while recent forests should carry mostly widespread alleles. If the differentiation between forest refugia was strong enough to have no or few shared alleles, the origin of re-colonised areas could be traced back, and the secondary contacts between colonisation fronts should be characterized by the co-occurrence of differentiated genetic lineages or gene pools. Finally, the patterns exhibited by different species characteristic of rain forests should be similar.
Different alternative hypotheses can be proposed:
- • H5: rain forest was restricted to a few refugia, but their position was different from the ones proposed (e.g. reconstruction by Anhuf et al., 2006; Fig. 1B). In this case, the same expectations as H4 would remain, but for a new set of refuge locations;
- • H6: forest fragmentation was minimal or took the form of a network of refugia (along rivers and/or in mountains) where most species survived. The maintenance of extensive gene flow would have caused little differentiation across space and no spatial heterogeneity of genetic diversity or allelic endemism;
- • H7: distribution of many species became fragmented in response to climate changes but with contrasted responses according to species depending on their life history traits and the habitat types in which they could survive (in other words, H4, H5 and H6 could simultaneously apply to different sets of species). Here, evidence of population fragmentation should appear in many species, but different species would display different patterns, although a correlation could appear among species having similar life history traits or ecological requirements.
It should be noted that diversity patterns that can currently be observed within species depend on the mutation rate of the markers used and the strength of bottleneck events and other stochastic processes, so that the genetic signals do not necessarily allow discriminating among scenarios.
5 A review of observed phylogeographic patterns in Guineo-Congolian rain forest trees
This review is based on phylogeographic patterns documented for 17 tree species in published articles, recent PhD theses and a few unpublished ongoing studies (Table 1). We focused on species occurring in lowland Guineo-Congolian rain forests, ignoring species occurring only in (sub)montane forests (e.g., Prunus africana; Kadu et al., 2011), as well as phylogenetic studies focusing on interspecies patterns. Most of the studies had a fairly good sampling in LG with typically at least 50 and often > 100 individuals sampled per species, but a poor or no sampling at all in UG and C. Sampling deficiency is due to the difficulty of access to most of the Congolian region while, in UG, it partly reflects the degree of degradation of UG forests (some species are now difficult to find), and partly the fact that the few laboratories that have contributed to the accumulation of phylogeographic data have focused their work on LG. Nevertheless, the sampling of genetic data covered two or three sub-centres of endemism for nine species, allowing a comparison of their phylogeographic patterns at the scale of the whole Guineo-Congolian forest. At the regional scale, available data allow a comparison of patterns occurring within the LG for 16 species. In most studies, genetic markers consisted of pDNA sequences and/or nuclear microsatellites, and a few studies were based only on RAPDs, AFLPs, or RFLP of nuclear genes (Table 1).
Résumé des patrons phylogéographiques observés chez les espèces d’arbres des forêts pluviales Guinéo-Congolaises de plaine. Les espèces sont ordonnées en fonction de l’étendue de leur distribution géographique.
| Species | Family | Distribution in Africaa | Life formb | Functional typec | Genetic markersd | Referencese | Phylogeographic patterns | |||
| Differentiation among sub-centers (UG, LG, C)f | Putative LG refugia with endemic lineages or gene poolsg | Number of distinct gene pools in LG | Different gene pools in LG across 0° to 3°N latitude | |||||||
| Erythrophleum suaveolens | Fabaceae | GCp | T | LD | SSR, pDNA | 1, 2 | YES (UG-LG) | None | 2 | YES |
| Milicia excelsa | Moraceae | GCp | T | LDP | SSR, pDNA | 3, 4 | YES (UG-LG-C) | None | 2 or 3 | YES |
| Coffea canephora | Rubiaceae | GCp | ST | ST | SSR, RFLP | 5, 6 | YES (UG-LG-C) | nd | 2? | nd |
| Santiria trimera (small leaflets) | Burseraceae | GC | T | ST | SSR, pDNA | 7, 8, 9 | YES (UG-LG) | 1 + 2 | 3 | YES? |
| Symphonia globulifera | Clusiaceae | GC | T | ST | SSR, pDNA | 10 | YES (UG-LG-C) | 1 | 3 | NO |
| Pericopsis elata | Fabaceae | GC | T | LDP | SSR | 11 | YES (LG-C) | None | 1 | nr |
| Afrostyrax lepidophyllus | Huaceae | GC | T | ST | pDNA | 7 | YES (LG-C) | 1 | nd | nd |
| Greenwayodendron suaveolens subsp.suaveolens var. suaveolens | Annonaceae | LG + C | T | ST | SSR, pDNA | 7, 12, 13 | YES (LG-C) | 1 + 2 | 3 | YES |
| Scorodophloeus zenkeri | Fabaceae | LG + C | T | ST | SSR, pDNA | 7, 13 | nd | 2, 6 | 4 | YES |
| Afrostyrax kamerunensis | Huaceae | LG + C | ST | ST | pDNA | 7 | nd | 2 + 3 | nd | nd |
| Distemonanthus benthamianus | Fabaceae | UG + LG | T | LDP | SSR | 14 | nd | None | 3 | YES |
| Pentadesma butyracea | Clusiaceae | UG + LG | T | ST | SSR, pDNA | 15 | YES (UG-LG) | nd | nd | nd |
| Erythrophleum ivorense | Fabaceae | UG + cLG | T | LDP | SSR, pDNA | 1, 2 | YES (UG-LG) | None | 2 | YES |
| Baillonella toxisperma | Sapotaceae | UG + LG | T | LD | SSR, pDNA | 16 | nd | None | 3 | YES |
| Irvingia gabonensis | Irvingiaceae | LG | T | ST | RAPD, RFLP, pDNA | 17, 18 | nr | 1 | 2 | YES? |
| Leonardoxa africana | Fabaceae | LG | ST | ST | pDNA, AFLP, SSR | 19, 20, 21 | nr | 1, 2? | 4? | nd |
| Aucoumea klaineana | Burseraceae | sLG | T | LDP | SSR, pDNA | 22, 23 | nr | 3, 4, 5? | 4 | nr |
a UG: Upper Guinea; LG: Lower Guinea; C: Congolia; sLG: southern LG; cLG: coastal LG; GC: Guineo-Congolian (UG + LG + C); GCp: Guineo-Congolian and its periphery.
b T: medium to large tree; S: shrub or small tree.
c ST: shade tolerant; LD: long-living light demanding; LDP: long-living light demanding and pioneer of open habitat.
d RAPD: random amplification of polymorphic DNA; SSR: simple sequence repeats (microsatellites); RFLP: restriction fragment length polymorphism; AFLP: amplified fragment length polymorphism; pDNA: plastid DNA markers or sequences (chloroplast genome).
e References: (1) Duminil et al. (2010); (2) J. Duminil & O.J. Hardy (unpublished); (3) Daïnou et al. (2010); (4) Daïnou (2012); (5) Gomez et al. (2009); (6) Musoli et al. (2009); (7) Dauby (2012); (8) Koffi et al. (2011); (9) Koffi (2011); (10) Budde et al. (2013); (11) C. Micheneau & O.J. Hardy (unpublished); (12) Dauby et al. (2010); (13) R. Piñeiro & O.J. Hardy (unpublished); (14) Debout et al. (2011); (15) Ewédjé (2012); (16) Ndiade-Bourobou (2011); (17) Lowe et al. (2000); (18) Lowe et al. (2010); (19) Brouat et al. (2001); (20) Brouat et al. (2004); (21) Léotard (2007); (22) Muloko-Ntoutoume et al. (2000); (23) Born et al. (2011).
f UG: Upper Guinea; LG: Lower Guinea; C: Congolia.
g Lower Guinean putative refuge areas (see Fig. 2): 1 = area surrounding the Cameroon volcanic line, 2 = Ngovayang and surrounding massifs, 3 = Monts de Cristal, 4 = Massif du Chaillu, 5 = Monts Doudou, 6 = Mayombe
5.1 Patterns of differentiation among sub-centres
With no exception, all the species occurring in at least two sub-centres of endemism displayed a clear genetic differentiation between sub-centres (distinct gene pools occur in distinct sub-centres; Table 1). This pattern is reported for seven species between LG and UG (or DG) and for six species between LG and C. Thus, at this scale, patterns of differentiation within species seem congruent with patterns of differentiation among species, supporting the hypothesis (H1) that widespread species have maintained isolated populations in different centres of endemism during periods of forest contraction, and did not colonize recently the whole GC region from a single source. However, each sub-centre of endemism could host more than one gene pool (well documented for LG in at least eight species, see below and Table 1), and the geographic limits between parapatric gene pools do not necessarily follow the limits between sub-centres of endemism. For example, the limit between the most adjacent West African and Central African gene pools is situated in Nigeria for Milicia excelsa (Daïnou, 2012), and near the forest–savannah limit in northern Cameroon for Erythrophleum suaveolens (Fig. 2), rather than at the height of the Dahomey Gap. In fact, these two species are not restricted to the Guineo-Congolian forest as they also occur in gallery forests or in dry woodland (for M. excelsa) at the periphery of the Guineo-Congolian region, so that their phylogeographic patterns support H3 and they might not reflect the history of the rain forest cover.
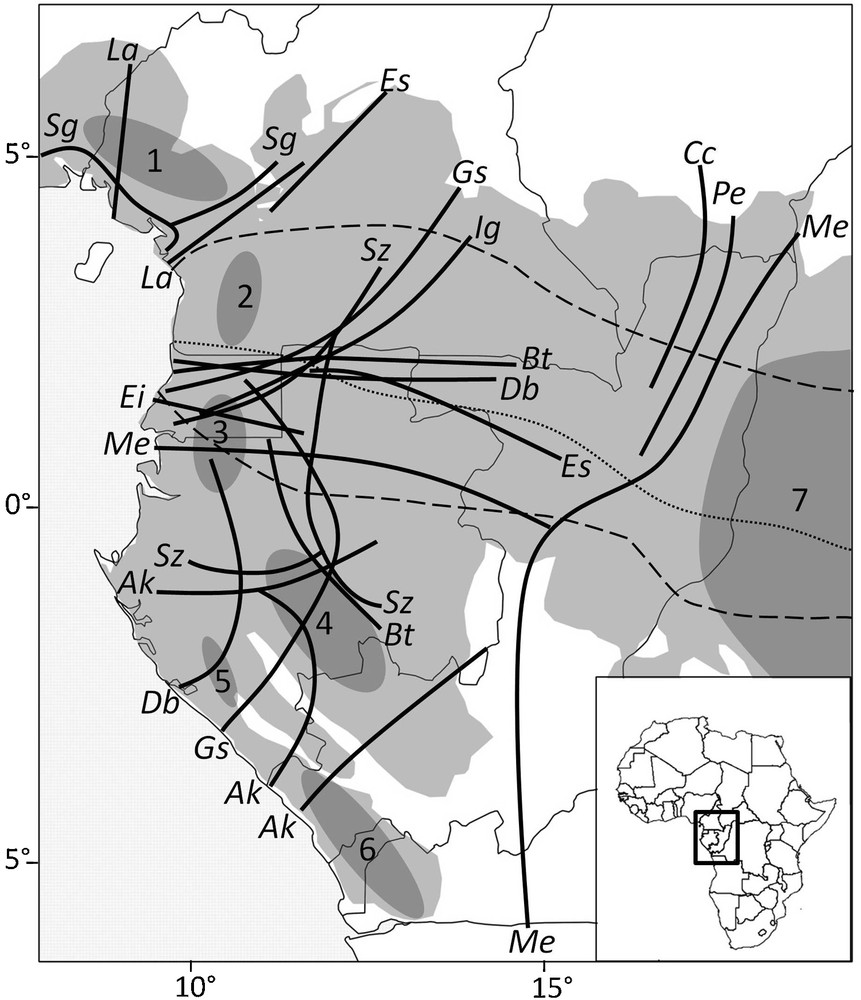
Approximate position of genetic discontinuities found within 13 rain forest tree species in Lower Guinea. Putative forest refugia during the Last Glacial maximum are depicted as grey shaded areas (modified after Maley, 1996). Discontinuities were identified as the geographic limits between parapatric gene pools identified using nuclear microsatellites, except for Ig (RAPD data) and La (limits between parapatric clades of the chloroplast genome). Most species were not sampled on the eastern side of the map (except Cc, Me and Pe) so that the low density of discontinuities there might be an artifact. The two stippled lines delimit approximately the equatorial climate with two wet and two drier seasons, the latter being of equal intensity (in terms of rainfall) along the climatic hinge (dotted line); the boreal climate to the north is characterized by one dry and sunny season from December to February, while the austral climate to the South is characterized by one dry but cloudy (hence less harsh) season from June to August. Species: Ak = Aucoumea klaineana, Bt = Baillonella toxisperma, Cc = Coffea canephora, Db = Distemonanthus benthamianus, Ei = Erythrophleum ivorense, Es = Erythrophleum suaveolens, Gs = Greenwayodendron suaveolens subsp.suaveolens var. suaveolens, Ig = Irvingia gabonensis, La = Leonardoxa africana, Me = Milicia excelsa, Pe = Pericopsis elata, Sg = Symphonia globulifera, Sz = Scorodophleus zenkerii. Putative refuge areas (modified from Maley, 1996): 1 = area surrounding the Cameroonian volcanic line, 2 = Ngovayang and surrounding massifs, 3 = Monts de Cristal, 4 = Massif du Chaillu, 5 = Monts Doudou, 6 = Mayombe, 7 = fluvial refuge of the Congo river.
Positions approximatives des discontinuités génétiques identifiées chez 13 espèces d’arbres des forêts pluviales de basse Guinée. Les refuges forestiers hypothétiques durant le dernier maximum glaciaire sont représentés par les aires grisées (modifié à partir de Maley, 1996). Les discontinuités correspondent aux limites géographiques entre pools géniques parapatriques, identifiés à l’aide de microsatellites nucléaires, excepté pour Ig (données RAPD) et La (limites entre clades du génome chloroplastique). La plupart des espèces n’ayant pas été échantillonnées dans la partie est de la carte (excepté Cc, Me et Pe), le peu de discontinuités rapportées dans cette zone pourrait être un artéfact. Les deux lignes en traits délimitent approximativement le climat équatorial avec deux saisons humides et deux saisons plus sèches, ces dernières étant d’intensité égale (en terme de pluviométrie) le long de la charnière climatique (ligne pointillée); au nord, le climat boréal se caractérise par une saison sèche et ensoleillée de décembre à février, alors qu’au sud, le climat austral se caractérise par une saison sèche et nuageuse (donc moins rude) de juin à août.
A current difficulty in assessing whether the demarcation between the LG and UG (i.e. the DG) also corresponds to a discontinuity within-species is that sampling in southern Nigeria is lacking for most species (except M. excelsa, Daïnou, 2012, and Irvingia gabonensis, Lowe et al., 2001). The same problem occurs at the limit between LG and C, but here sampling is also largely insufficient for the whole C.
5.2 Patterns of allelic endemism and genetic differentiation in Lower Guinea
Analyses of patterns of allelic or haplotypic variation in relation to putative LG forest refugia (following Maley, 1996) gave mixed results. On the one hand, several species display genetic variants endemic to a putative refuge. This is particularly the case for the putative refugia around the Cameroon Volcanic line (CVL Fig. 2), where endemic variants (pDNA haplotypes, SSR gene pools, RFLP alleles) were found for at least five species, and four species displayed variants endemic to at least one of the other LG putative refugia (Table 1). For example, Santiria trimera (Koffi et al., 2011, considering the morphotype with small leaflets which probably constitutes a well-defined species, Koffi et al., 2010) displayed localized pDNA haplotypes only in putative refugia from Cameroon and had widespread haplotypes elsewhere. On the other hand, six other species did not display such a pattern. The case of E. suaveolens is unique because it displays a clear pattern of a geographic gradient of rare pDNA haplotypes (much higher in eastern Gabon than in eastern Cameroon) unrelated to putative forest refugia (Duminil et al., 2010), but this gradient does not appear when the species is analysed with nuclear microsatellites (J. Duminil, unpublished). Moreover, although species like Greenwayodendron suaveolens subsp. suaveolens var. suaveolens (Dauby et al., 2010) or Symphonia globulifera (Budde et al., 2013) have rare endemic pDNA haplotypes (and a gene pool, in the case of S. globulifera) associated with the CVL, other endemic variants occur in different populations not necessarily close to putative refugia.
These trends support hypothesis H7, that different tree species have had quite different demographic histories. Nevertheless, the CVL appears to have played an important role in the genetic diversification of many species, at least more often than other LG putative refugia, possibly supporting the hypothesis that the location of the main refugia should be reconsidered (H5).
In fact, the range of sampling schemes, types of markers and analyses used in the different studies, and differences in successional status of the species, do not facilitate an objective comparison among species. As a consequence, the visual interpretation of patterns is prone to much subjectivity, guided by preconceptions about the main drivers of biodiversity. Hence, it is likely that new insights could be obtained from a spatial analysis of the gradients of endemism of genetic variants in multiple species starting from the original data, a study beyond the scope of this review.
5.3 Genetic discontinuities in Lower Guinea
Comparing the distribution of genetic discontinuities (parapatric gene pools based on multilocus nuclear markers or parapatric clades based on DNA sequences) among species (Fig. 2) is easier than comparing their spatial patterns of diversity, because the geographic limits between gene pools can be drawn approximately. In LG, most species displayed several gene pools (Table 1), generally with parapatric or allopatric distribution (except in Coffea canephora, where three gene pools exhibited a large contact zone, whose range might have been obscured by human introduction and naturalization, Gomez et al., 2009). The occurrence of distinct gene pools indicates a lack of gene flow between them, at least in the recent past. Hence, gene pools showing a parapatric distribution are suggestive of a past-range fragmentation leading to genetic differentiation between gene pools, followed by the re-colonisation of the intervening space and partial admixture of gene pools. This is precisely what is expected to occur under the refuge hypothesis driven by climatic oscillations (H4 and H5). However, it must be kept in mind that other factors might be the cause of a gene flow barrier, such as physical barriers (e.g., mountains, large rivers), pre-zygotic barriers (e.g., phenological delay in flowering time between gene pools, see below), or post-zygotic barriers (e.g., incompatibilities between gene pools).
Mapping the approximate limits between parapatric gene pools (Fig. 2) reveals some congruencies among species. The most striking feature is that at least eight species (among nine with appropriate distribution and data, Table 1) show a north–south genetic discontinuity located between 0° and 3° latitude north. On their western side, these discontinuities are located in or near Equatorial Guinea, but the lack of samples from this country prevents accurate delimitation. On their eastern side, these discontinuities are located close to the border between Gabon and Cameroon for three species (Distemonanthus benthamianus, Baillonnella toxisperma, E. suaveolens), while for three other species (G. suaveolens, I. gabonensis, Scorodophloeus zenkeri) they shift northwards, separating southeastern Cameroon from the rest of the country (Fig. 2). Hence, one may make a distinction between northern and southern LG (hereafter, nLG and sLG). In nLG, the other limits between gene pools tend to be concentrated on each side of the CVL. In sLG, the limits for Aucoumea klaineana seem to occur between the putative forest refugia, but this is not the case for four other species (B. toxisperma, D. benthamianus, G. suaveolens, S. zenkeri), which display an east–west discontinuity in Gabon (Fig. 2).
The coincidental distinction between nLG and sLG gene pools (Fig. 2) was first observed for four species by Debout et al. (2011), and is confirmed here for a total of eight species, making it the most congruent phylogeographic feature observed among species. The degree of admixture between gene pools in the contact area varies substantially among species. E. suaveolens, E. ivorense and M. excelsa display a 100–200-km-wide band where most individuals appear admixed while the limit between gene pools seems sharper in D. benthamianus and especially B. toxisperma, for which genetic data indicate that the divergence between the northern and southern gene pools must be ancient (Ndiade-Bourobou, 2011).
It is worth noting that the limit between nLG and sLG gene pools is also congruent with the distribution range limits of some tree species such as A. klaineana and Scyphocephalium mannii (northern limit) or Triplochiton scleroxylon and Terminalia superba (southern limit; Nicolas, 1977). A phytogeographical delimitation between nLG and sLG has never been underlined, nor sought, until recently. Indeed, Gonmadjé (2012) reported a clear floristic differentiation between nLG and sLG using ordination analyses of LG forest tree inventories, giving support for a congruent biodiversity pattern at the intra-specific and inter-species levels. For animal species, a differentiation between nLG and sLG has also been reported, for example in gorillas (Anthony et al., 2007). Interestingly, the genetic discontinuities between nLG and sLG coincides approximately with the climatic hinge (Fig. 2), the latitudinal area characterized by two dry seasons of similar strength and making the transition between the boreal and austral climatic regimes where one dry season becomes more and more important during the boreal and austral summer, respectively, as one moves away from the equator (Suchel, 1990). This congruence suggests that the nLG, sLG genetic discontinuities might be related to climatic seasonality.
The congruent genetic limits around the CVL tend to confirm the interesting phytogeographic history of this area, already known for its richness in endemic species. It gives support to the hypothesis that the rain forest survived in this area during glacial maxima. As a forest refuge, it is however unclear whether it would have contributed much to the recolonization of adjacent areas. For southern Nigeria, data are available only for I. gabonensis that shows no differentiation between Nigerian and West Cameroonian populations (Lowe et al., 2000, 2010), compatible with a re-colonisation from the CVL. In southern Cameroon, there is genetic evidence for a southward expansion of at least one subspecies of Leonardoxa africana (subsp africana) along the coastal forest (Léotard, 2007), while phylogenetic data support an origin of the species in the CVL (Brouat et al., 2004). By contrast, S. globulifera displays a gene pool endemic to the CVL with little evidence of geographic expansion, and the occurrence of scattered endemic pDNA haplotypes over most of its range in LG rather suggests that the species has survived in multiple refuge areas and possibly also along river floodplains in most of its current range (Budde et al., 2013). It must be noted that a genetic discontinuity coinciding with the CVL does not necessarily imply a role as a forest refuge because the CVL could also act as a barrier to gene flow causing differentiation. In this case, however, we would not except to find endemic alleles or gene pools.
Finally, it is worth noting that the major rivers of the region do not coincide with genetic discontinuities. Thus rivers play no, or only a minor, role as gene flow barrier for most tree species, which is contrary to inferences of their role for many animal species (e.g., Anthony et al., 2007; Gonder and Disotell, 2006; Nicolas et al., 2011).
6 Origin of the north–south differentiation in Lower Guinea
Assuming that the gene pools detected result from ancient population fragmentation followed by recolonization, the most parsimonious explanation of historical vegetation dynamics is that rain forest disappeared from the area surrounding the climatic hinge, before being re-colonised from populations situated in the south and/or in the north. This first explanation would explain the congruent limits between gene pools for species having survived on both sides of the climatic hinge, and the congruent northern or southern distribution limits for species having survived on one side only. It is a version of the refuge hypothesis recognizing two main refugia in LG (or three if the CVL is an additional refugium), but the position of these refugia is not clear. To our knowledge, however, this hypothesis is not consistent with models of the historic climate, which do not predict a particularly dry climate around the climatic hinge during glacial maxima (e.g., Anhuf et al., 2006; Hély et al., 2009). Alternative explanations are based on current climate.
A second explanation is that the inversion of seasons on each side of the climatic hinge induces a delay of approximately six month in flowering phenology, preventing gene flow by pollen between the northern and southern gene pools. However, the phenological transition across the climatic hinge might well be gradual (data are currently lacking), in which case no barrier to gene flow would be expected. Moreover, even species with long distance seed dispersal capability, such as B. toxisperma (Ndiade-Bourobou et al., 2010), display a clear north–south differentiation. In this case, it would be unlikely that a phenology lag could create a barrier to seed dispersal, unless seeds dispersed over long distance do not find adequate establishment conditions if the climatic transition is abrupt (see next explanation). Finally, this explanation cannot elucidate the congruent species distribution limits.
A third explanation relies on constraints for the establishment of populations crossing the climatic hinge. For example, the limiting factor inhibiting the northern expansion of A. klaineana seems to be related to the lack of precipitation during the germination period (Born, 2007), a problem dependent on the mechanisms determining the phenology of the reproductive stage. In addition, the boreal and austral climates in LG are not symmetric because the dry season is sunny in the north and cloudy in the south, so that the latter is much less harsh for plants suffering from water stress (Gonmadjé, 2012; Suchel, 1990). This might explain the distribution limits of species depending on their ability of coping with water stress, and may explain the distribution limits between gene pools if they display adaptive differences with respect to water stress. Major climatic gradients in LG also occur along an east–west axis, such as the annual rainfall, which decreases from the coast to the interior of Cameroon and Gabon. This rainfall gradient might well explain the longitudinal distribution limits of Erythrophleum ivorense and E. suaveolens (Duminil et al., 2010). It may also explain the east–west genetic discontinuities displayed by four species in Gabon (B. toxisperma, D. benthamianus, G. suaveolens, S. zenkeri). However, further investigations are needed to test whether gene pools limits really follow environmental gradient.
In conclusion, the origin of the genetic differentiation around the climatic hinge remains rather mysterious, although the different hypotheses are not mutually exclusive and could reinforce each other. This question certainly deserves attention for a multidisciplinary approach involving finer-scale genetic analyses including adaptive traits, phenological surveys, palaeoenvironmental reconstruction with climate modelling, and species or gene pool distribution modelling.
7 Perspectives for future research
In this review, only geographic patterns of genetic diversity within species were considered. However, molecular genetic data can potentially provide much more information, such as detecting past demographic changes (population expansion or bottleneck), estimating the timing of population (or species) divergence or the timing of past demographic changes, or detecting signs of selection. While these types of inferences have been successfully applied in other contexts (e.g., for Guineo-Congolian animals: Anthony et al., 2007; Nicolas et al., 2006), few investigations have been made on Guineo-Congolian plants and usually with limited success (but see Budde et al., 2013; Daïnou et al., 2010; Duminil et al., 2010). Of particular interest is the estimation of the divergence time between gene pools, the timing of secondary contact between differentiated gene pools, and the timing of population demographic changes. Indeed, dating past fragmentation or expansion events is essential to assess both whether congruent phylogeographic patterns among species can be related to shared historical events and whether these have been driven by known past environmental events (e.g., glacial cycles).
Inferring divergence time typically relies on the principle of a molecular clock, where the accumulation of mutations (and/or of genetic drift) differentiating populations allows the setting of a time scale. Calibrating an absolute timescale is however difficult, especially in plants for relatively recent events (e.g., less than a few million years) for two main reasons. First, the mutation rates of plant genomes are relatively low compared to the commonly used animal mitochondrial genome (Petit and Vendramin, 2004). A low mutation rate is advantageous to infer remote events (e.g., the divergence time between plant families), but not for recent events (e.g., the divergence time between gene pools within species). Second, finding adequate calibration points for plants is often difficult. These calibration points can consist in dated plant fossils presenting characters typical of a particular plant clade, providing a minimal age for the origin of this clade and thus for the divergence with its sister clade. However, appropriate fossilized plant tissue is rare. Another type of calibration point is provided by species endemic of a volcanic island of known age and which can unambiguously be considered as derived from a sister plant species occurring on the adjacent continent. The age of the island then provides a maximal age for the divergence between these sister species.
Future research could thus be oriented towards finding new calibration points. One potential is studying the phylogeny of the still barely known flora of the islands of the Gulf of Guinea (e.g. São Tomé, Prìncipe), to provide new examples of species sister to continental Guineo-Congolian species. On the molecular genetics side, much progress is expected from the advent of the so called “next generation sequencing” technologies which are providing a tremendous increase in DNA sequencing throughput (Davey et al., 2011). Hence, exploiting these technologies on Guineo-Congolian plants should soon largely compensate for the low rate of molecular evolution of the plant genomes, and projects aiming at sequencing the whole chloroplast genome in many samples are already ongoing.
Approaches such as reciprocal transplantation experiments and DNA sequences analysis of genes potentially under selection (e.g., genes involved in drought resistance) could reveal whether natural selection has had a substantial impact on the phylogeographic patterns observed. In particular, such methods would help assess whether differentiated gene pools are more adapted to the environmental conditions prevailing in their respective distribution ranges. As outlined earlier, northern and southern Lower Guinea differ in the strength of their respective dry seasons so that the hypothesis that differences in drought-resistance occur between northern and southern gene pools is worth testing. This type of research must be conducted on species with well-documented ecological requirements, which is unfortunately another knowledge gap for species of this region. Next-generation sequencing is also promising in this context, as it facilitates the study of functional markers, which can reveal the genetic bases of ecological adaptations (Stapley et al., 2010).
Finally, other priorities for future phylogeographic research of the Guineo-Congolian region is to expand the geographic focus as well as the types of plants studied (life forms, functional groups). The Upper Guinea and especially Congolia have been much less sampled than Lower Guinea, and would benefit from concerted sampling efforts in new projects. Current data from Lower Guinea also highlight areas of particular interest, such as the area surrounding the climatic hinge, which would benefit from further denser sampling, especially on its western side situated within the insufficiently sampled Equatorial Guinea. More sampling efforts in southern Nigeria would also be worth attention, to more precisely identify the location of the differentiation between LG and UG. So far, most phylogeographic research on Guineo-Congolian plants has focused on long-lived, shade-tolerant or light-demanding tree species (Table 1), although studies on lianas and herbs (not reviewed here) have started (Ley and Hardy, 2010). Short-lived pioneer tree species, epiphytes, lianas and herbaceous species might each provide complementary information because differences in generation time, seed or pollen dispersal abilities, light or water requirement and/or sensitivity to disturbances can affect phylogeographic patterns and provide a more complete picture of the vegetation history of these biomes. This is of course also true for non-plant organisms, such as animals or fungi. In this respect, the comparative phylogeographic analysis of organisms forming (near) obligate associations (symbionts, specialized parasitisms or mutualisms, pollinators, seed dispersers) can provide much insight into their evolutionary history because several independent genomes are evolving in parallel (e.g., Nieberding and Olivieri, 2007). This is illustrated by the research carried out on the Guineo-Congolian ant-plants L. africana (e.g. Léotard, 2007) and Barteria spp. (R. Blatrix and D. McKey, pers. comm.).
8 Conclusion
Comparative phylogeography of Guineo-Congolian plants provides an important source of information to reconstruct the history of the Guineo-Congolian forest. This review synthesizes the main patterns observed across a number of studies conducted in the past few years, but a number ongoing and new projects are likely to bring a lot of new data soon. The validation of phylogeographic data to reconstruct past biomes can only be done through a multidisciplinary approach, involving; palaeoenvironmental (including palaeoclimatic) reconstructions, climate modelling and species-niche modelling. By publishing this review in a journal dedicated to geosciences, we hope that it will inspire colleagues from these disciplines to think about how their data and methods can complement phylogeographic approaches. The main phylogeographic patterns detected so far are:
- • species with a wide distribution range are typically differentiated among the sub-centres of species endemism, suggesting repeated or long-lasting fragmentation of the forest at the largest scale;
- • most species display differentiated gene pools within sub-centres of endemism, at least in Lower Guinea, supporting the hypothesis that forest fragmentation also occurred at this scale, though not necessarily according to the most often considered refuge hypothesis (Maley, 1996);
- • the most striking and unsuspected pattern in Lower Guinea is a north–south differentiation across the climatic hinge and suggests that rain forest has disappeared from this area historically and was recently recolonized form northern and/or southern areas. However alternative explanations based on current climates cannot yet be ruled out. Particular attention to what has happened in the vicinity of this climatic hinge should constitute one of the priorities for future research.
Acknowledgments
This work was supported by the ANR-BIODIV program of the Agence nationale pour la recherche (ANR) of France through projects IFORA and C3A, and by the Belgian Fund for Scientific Research (F.R.S-FNRS) through grants FRFC 2.4.577.10 and MIS 4.519.10. We thank Jean Maley and two anonymous reviewers for their useful comments on a previous draft, as well as Amin Dezfuli for providing the climatic data used to delimit the climatic hinge. We warmly thank all the people, institutions, NGO's, and forestry companies who helped us assemble the necessary plant material by providing logistic support and official authorizations. Detailed acknowledgements are already provided in the cited published studies. However, for their repeated support, we wish to mention the following institutions (Cameroon: Herbier National du Cameroun, ministère de la Recherche scientifique et de l’Innovation, université de Yaoundé I; Côte d’Ivoire: CNRA – Centre national de recherche agronomique, université Lorougnon Guédé; France: CIRAD – Centre de coopération internationale en recherche agronomique pour le développement, IRD – Institut de recherche pour le développement; Gabon: CENAREST – Centre national de la recherche scientifique et technologique, CIRMF – Centre international de recherches médicales de Franceville, CNPN – Conseil national des parcs nationaux, ENEF – École nationale des eaux et forêts, Herbier national du Gabon; Uganda: NARO – National Agricultural Research Organisation), conservation agencies (MBG – Missouri Botanical Garden, Nature +, Smithsonian Institution, WCS – Wildlife Conservation Society, WWF – World Wildlife Fund) and logging companies (Millet-Precious Woods Gabon, Pallisco, Rougier, SFID, Wijma).