

1 Introduction
The Quaternary, which includes the Pleistocene and Holocene epochs, started at the onset of the Northern Hemisphere glaciations, approximately 2.58 Mya, and continues to the present-day. It was characterized by a succession of about 50 glacial/interglacial cycles, which are attributed to variation in solar radiation [1]. The decrease of temperature and precipitation during glacial periods had major impacts on the fauna and flora of high and middle latitudes, but also in low latitudes, where widely distributed Pliocene rainforests, which today contain high levels of biotic endemism and richness [2,3], were partly replaced by more open vegetation [4]. A critical question is what was the influence of Pleistocene glaciations on rainforest biodiversity?
The late Jürgen Haffer proposed in 1969 [5] the hypothesis of Pleistocene forest refugia to explain speciation in Amazonian forest birds. He postulated that “during several dry climatic periods of the Pleistocene, […] the Amazonian forest was divided into a number of smaller forests, which were isolated from each other by tracts of open, non-forest vegetation” and “served as ‘refuge areas’ for numerous populations of forest animals, which deviated from one another during these periods of geographic isolation”. This hypothesis has been questioned by palynological data that do not support forest fragmentation of Amazonia during the Last Glacial Maximum (LGM) [6–8]. Despite these criticisms, the hypothesis still continues to attract the attention of many biographers and has been applied to a wide range of plant and animal groups in tropical regions around the world [9–15].
On the African continent, tropical rainforests are currently divided into two blocks: the Upper Guinea Forest in West Africa and the Congo Basin Forest in Central Africa [16–18]. These two blocks are separated in West Africa by the Dahomey Gap, a savannah corridor that extends from central Ghana, through Togo and Benin [19]. During the Pliocene epoch, the climate in Africa was warmer and wetter than today and tropical rainforests covered the Dahomey Gap and extended to higher latitudes [17,20]. In contrast to the Amazonian situation, in Africa there is solid palynological evidence for Pleistocene fragmentation of tropical rainforests [21–24]. In addition, distributional patterns of animals and plants have suggested the existence of several centers of endemism, generally interpreted as forest refugia [16,18,22,25]. However, the number of refugia as well as their location and size remain a matter of debate among specialists [16,22].
Comparative phylogeographic analyses based on molecular data for forest-dwelling organisms provide a good opportunity to test hypotheses of African Pleistocene refuges and to infer aspects of their periodicity and location. Several molecular studies addressing these issues have been published on different biotic groups, including birds [26–29], primates [10,30,31], rodents [32], and plants [12,33–35]. These analyses have produced contrasting results, but in many cases they are not based on assemblages of closely related rainforest dependent taxa (species or subspecies) with geographically overlapping distributions and with broad sampling across different African tropical forests. These criteria are important to test fine-level aspects of African Pleistocene refuge models with molecular data.
Here, we examine the phylogeography of the tribe Scotonycterini, a group of fruit bats (Chiroptera, Pteropodidae) containing four species restricted to tropical rainforests of Africa and characterized by white fur patches on the nose and behind the eyes [36,37]. The tribe was first described by Bergmans [38], who recognized only three species: Scotonycteris zenkeri (Zenker's fruit bat), Casinycteris argynnis (short-palated fruit bat), and C. ophiodon (Pohle's fruit bat). Casinycteris ophiodon was originally placed in the genus Scotonycteris, but a recent study based on morphological and mitochondrial data have indicated that it is best included in the genus Casinycteris [37]. Recently, an additional species, C. campomaanensis (Campo-Ma’an fruit bat), was described from southwestern Cameroon [37].
The geographical distribution of S. zenkeri makes it an ideal candidate to test aspects of the Pleistocene rainforest refugia hypothesis. Indeed, its distribution overlaps with the two large blocks of African tropical rainforests – the Upper Guinea Forest in West Africa and the Congo Basin Forest in Central Africa (Fig. 1) [39]. The other species within this tribe are less widespread: C. ophiodon is also found in both west and Central Africa (Fig. 1) [40], but only known from 16 localities [41], which exclude areas around the Nigeria Delta and central and eastern parts of the Congo Basin; C. argynnis is endemic to the Congo Basin, from southeastern Cameroon and eastern Gabon in the west, across southwestern Central African Republic (CAR), the Republic of the Congo (RC) and all provinces of DRC, except Bas-Congo, to the Mitumba Mountains in eastern DRC (Fig. 1) [42]; and C. campomaanensis known from a single specimen obtained in southwestern Cameroon (Fig. 1) [37]. All four species of Scotonycterini are found in Cameroon (Fig. 1).
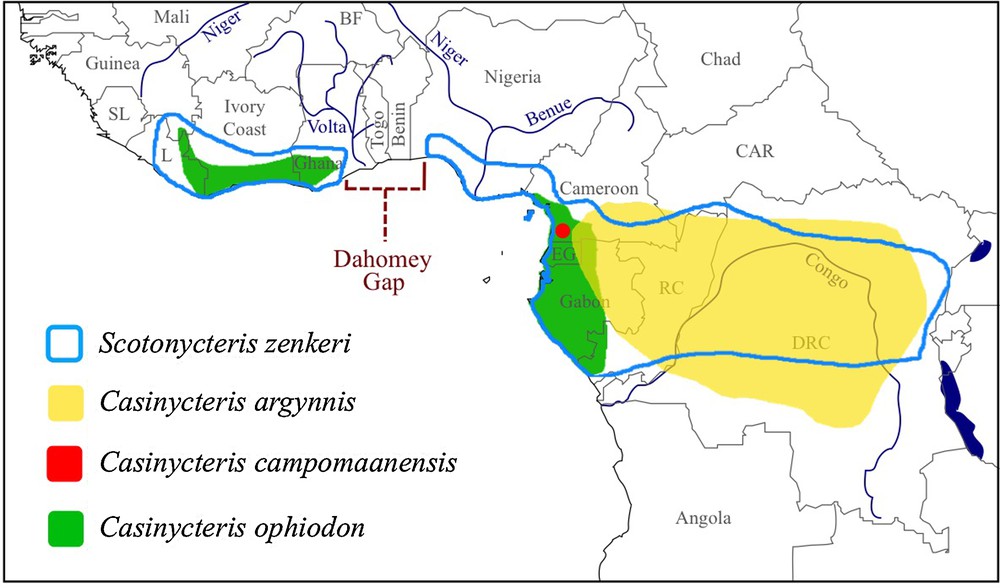
(Color online.) Geographic distribution of the four species of the tribe Scotonycterini. The geographic ranges of Scotonycteris zenkeri, Casinycteris argynnis, and C. ophiodon were modified from the maps provided by the IUCN [39,40,42]. The newly described species C. campomaanensis is known from a single locality [37]. Note that the distribution of S. zenkeri fits perfectly that of African rainforests, with two blocks separated by the Dahomey Gap, Upper Guinea in West Africa and the Congo Basin in Central Africa [16–18].
Herein, the phylogeographical patterns of Scotonycterini were assessed using 105 individuals collected at 37 different localities, including tissue samples from three holotypes, and DNA sequences from the mitochondrial cytochrome b gene (1140 nt) and 12 nuclear introns (9641 nt). These data were used to address the five following questions:
- • How many species are there within the tribe Scotonycterini?
- • Is the monophyly of the genus Casinycteris supported by both mitochondrial and nuclear data?
- • Is there a common phylogeographic pattern shared by the two species distributed in both West Africa and Central Africa (S. zenkeri and C. ophiodon)?
- • Similarly, is there a common phylogeographic pattern in Equatorial Africa for the two species distributed in both western and eastern regions of the Congo Basin forest (S. zenkeri and C. argynnis)?
- • What was the impact of putative rainforest refugia on the structure of genetic diversity?
2 Materials and methods
2.1 Taxonomic sampling
Most of the Scotonycterini samples analyzed in this study were collected by the authors using Ecotone mist-nets (Poland) during field trips to Cameroon (AH), CAR (AH, EN and NN), Ivory Coast (BK and NN), DRC (AH and GCG), Gabon (SMG and XP), and RC (XP). Fruit bat species were identified using the key of Bergmans [38]. In addition, several samples were obtained from specimens housed in the following museums: Musée national d’Histoire naturelle (MNHN; Paris, France), Naturalis Biodiversity Center (NBC; Leiden, Netherlands), Senckenberg Museum Frankfurt (SMF; Frankfurt, Germany), The Field Museum (FMNH; Chicago, USA), and Zoologisches Museum Berlin (ZMB; Berlin, Germany). The holotypes of three species were included in this study: C. campomaanensis (MNHN 2011-637), C. ophiodon (ZMB 50001), and S. zenkeri (ZMB 66533).
Five forest regions were sampled for this study (see Fig. 2): Upper Guinea (Liberia and Ivory Coast), Cameroon, western Equatorial Africa (EG, Gabon, and RC), the southwest part of CAR, and the eastern part of DRC. Names and geographical coordinates of all sampled localities are provided in Appendix A. The number of individuals sequenced per species is one for C. campomaanensis, two for C. ophiodon, 46 for C. argynnis, and 56 for S. zenkeri (see Fig. 2 for details).
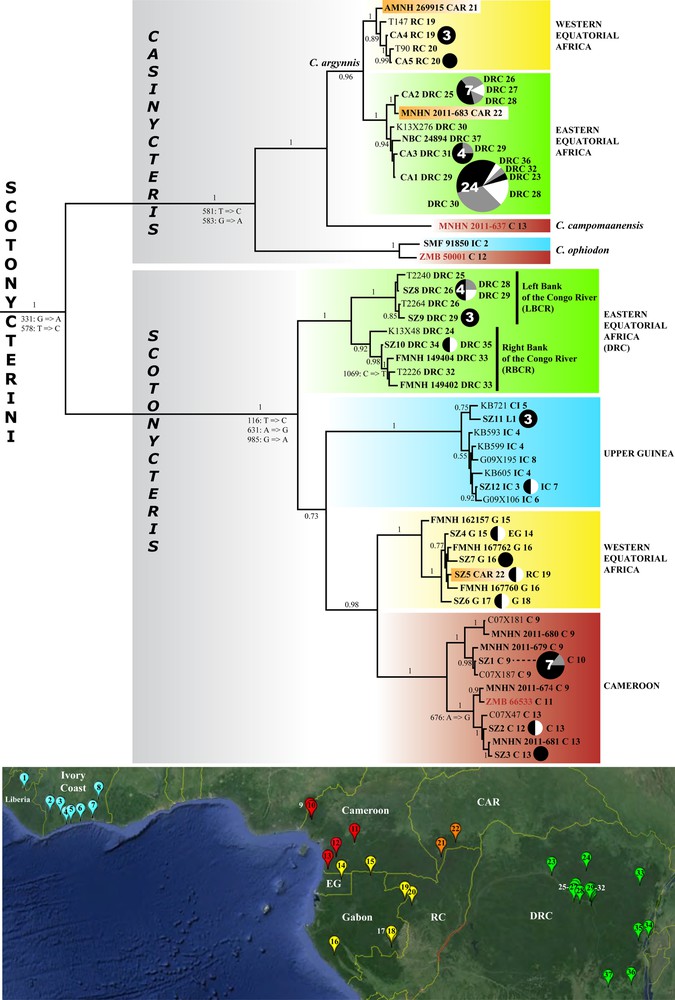
Comparative phylogeography within the tribe Scotonycterini based on complete Cytb gene sequences. The Bayesian tree was reconstructed using 46 outgroup taxa, which are not indicated on the figure (see Appendix B for outgroup), and the 49 haplotypes identified in the 105 Cytb sequences of Scotonycterini. The values at the branches represent posterior probabilities (see § Materials and methods for details). The non-homoplastic substitutions detected in the Cytb alignment of 151 taxa are also indicated at the nodes. At terminal branches, all sample codes indicated in bold are associated with museum specimens (AMNH: American Museum of Natural History [GenBank: JN398197]; FMNH: Field Museum of Natural History; MNHN: Muséum national d’Histoire naturelle; NBC: Naturalis Biodiversity Center; SMF: Senckenberg Museum Frankfurt; ZMB: Zoologisches Museum Berlin). The holotypes of Scotonycteris zenkeri, Casinycteris campomaanensis, and C. ophiodon are highlighted in red. Circle graphs were used for haplotypes shared by several individuals: CA1–CA5 for C. argynnis, SZ1–SZ12 for S. zenkeri (see list in Appendix C). The circle size is proportional to the number of sequences (values > 2 are indicated inside the circle). Black circles are used when all individuals were collected in the same geographic locality. Black, white and grey parts are proportional to the number of haplotypes sequenced in each geographic locality (named according to the initial[s] of the country and a number corresponding to the position on the map below the tree). Geographic localities sampled for this study are indicated on the map. The image was extracted from Google Earth (US Dept of State Geographer; ©2014 Google; Image Landsat; Data SIO, NOAA, U.S. Navy, NGA, GEBCO). Five forest regions were highlighted using different colors: (i) Upper Guinea in blue, including Liberia [L] and Ivory Coast [IC], (ii) Cameroon [C] in red, (iii) western Equatorial Africa in yellow, composed of Equatorial Guinea [EG], Gabon [G], and Republic of the Congo [RC], (iv) Central African Republic [CAR] in orange, and (v) Democratic Republic of the Congo [DRC] in green. For interpretation of the references to color in this figure caption, the reader is referred to the web version of the article.
2.2 DNA extraction, amplification, and sequencing
Total DNA was extracted from muscle or patagium samples using QIAGEN DNeasy Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. The complete mitochondrial cytochrome b (Cytb) gene was amplified and sequenced using the oligonucleotide primers detailed in Hassanin [37]. In addition, 12 nuclear introns from 11 different genes were also amplified and sequenced using published primers. They included FGB intron 8 [43] and 11 introns recently developed for studying Laurasiatherian mammals [44]: CCAR1 intron 6, DHX29 intron 20, DIS3 intron 19, EXOSC9 intron 4, HDAC1 intron 6, HDAC2 intron 4, PABPN1 introns 2 and 6, RIOK3 intron 6, TUFM intron 9, and ZFYVE27 intron 6.
DNA was also extracted from museum specimens, some of them being preserved in alcohol more than 100 years (ZMB 50004, ZMB 54390, and ZMB 66533). For these old samples, DNA degradation was anticipated and explains limitations in amplifying long PCR fragments (> 300–400 nt). Therefore, seven sets of primers were used to amplify and sequence overlapping fragments of the Cytb gene, as previously done for the holotype of C. ophiodon (see details in Hassanin [37]).
The polymerase chain reactions and DNA sequencing were carried out as detailed by Hassanin [37]. Sequences were edited and assembled using Sequencher 5.1 (Gene Codes Corporation). Heterozygous sites in the nuclear loci (double peaks) were coded using the IUPAC code. Sequences generated for this study were deposited in the EMBL/DDBJ/GenBank databases under accession numbers KP305915-KP306513.
2.3 Mitochondrial analyses of Cytb sequences
The Cytb dataset represents a total alignment of 1140 nucleotides and 151 taxa. The 105 Cytb sequences of Scotonycterini were compared to those available in the nucleotide databases for species of the family Pteropodidae (42 additional sequences). The four outgroup species used to root the pteropodid tree, i.e., Artibeus jamaicensis, Hipposideros armiger, Megaderma lyra, and Rhinolophus luctus, were chosen on the basis of previous molecular studies [37,45,46].
DNA sequences were aligned on Se-Al v2.0a11 [47]. The best-fitting model of sequence evolution was selected under jModelTest 2.1.4 [48] using the Akaike information criterion. Bayesian analyses were then conducted using the selected GTR + I + G model on MrBayes v3.2.1 [49]. The posterior probabilities (PP) were calculated using four independent Markov chains run for 10,000,000 Metropolis-coupled MCMC generations, with tree sampling every 1000 generations, and a burn-in of 25%. Pairwise distances were calculated with PAUP version 4b10 [50] using Kimura's two-parameter (K2P) model to allow comparisons with previous molecular studies [37,51].
2.4 Multigene analyses
Analyzing only mitochondrial data to determine a species phylogeny can be highly misleading because of various evolutionary processes, such as mitochondrial introgression, female philopatry, lineage sorting and selection [43,52,53]. To avoid erroneous conclusions, the phylogeny and phylogeography of Scotonycterini were therefore also investigated by analyzing 12 nuclear introns (representing 11 autosomal genes) for 48 taxa. Four pteropodid outgroup species were used to root the trees: Cynopterus sphinx, Eidolon helvum, Epomops franqueti, and Rousettus aegyptiacus. For each gene, DNA sequences were aligned with MUSCLE [54]. All introns were analyzed separately, excepting the introns 2 and 6 of PABPN1, which were used in combination.
The best-fitting models of sequence evolution were selected under jModelTest 2 [48] for each marker and the two concatenations (nuDNA and supermatrix). Using the Akaike information criterion, the best-fitting models were HKY for CCAR1 and DIS3, HKY + G for EXOSC9, HDAC1, TUFM, and ZFYVE27, HKY + I for HDAC2, GTR for DHX29 and RIOK3, GTR + G for FGB, PABPN1, and the nuDNA concatenation, and GTR + I + G for Cytb, and the supermatrix. Gaps were treated as missing data, but indels (insertions/deletions) with unambiguous position in the DNA alignment were coded as additional binary characters by using 1 and 0 symbols for insertion and deletion, respectively.
Bayesian inferences were performed on each of the 12 independent markers, on the nuDNA dataset (11 loci; 9641 nt + 128 indels = 9769 characters) and on the supermatrix (10,781 nt + 128 indels = 10,909 characters), with the options detailed in the section “Mitochondrial analyses of Cytb sequences”. For SuperTRI analyses (see below), we set the minimum partition frequency (mcmcp Minpartfreq parameter) to 0.01. For the two concatenated datasets (nuDNA and supermatrix), we applied a partitioned approach using the models selected by jmodeltest for each of the markers.
The SuperTRI method [55] was conducted to test for repeated phylogenetic signals in the 12 independent markers. The lists of bipartitions obtained from the Bayesian analyses of the 12 independent markers (11 autosomal genes and mitochondrial Cytb gene; Appendix E) were transformed into a weighted binary matrix for supertree construction using SuperTRI v57 [55] (Python script available at http://www.normalesup.org/∼bli/Programs/programs.html). Each binary character corresponded to a node, which was weighted according to its frequency of occurrence in one of the 12 lists of bipartitions. This way, the SuperTRI method accounts for both principal and secondary signals, as all phylogenetic hypotheses found during the Bayesian analyses are represented in the weighted binary matrix used for supertree construction. The reliability of the nodes was assessed using three measures: supertree bootstrap percentages (SBP) were obtained from PAUP* after 1000 BP replicates of the matrix of 5321 binary characters generated by SuperTRI v57, and the mean posterior probabilities (MPP) and reproducibility indices (Rep) were directly calculated on SuperTRI v57. The SBP, MPP and Rep values were reported on the Bayesian tree found with the nuDNA dataset of 9769 characters (Fig. 3). If a node was recovered with high SBP, MPP, and Rep values (SBP > 50; MPP > 0.5; Rep > 0.5), we concluded that the signal was robust and reliable, i.e., supported by more than half of the genetic markers. If a node was recovered with low Rep values (< 0.17), we concluded that the signal was not reliable, i.e., supported by less than two markers.
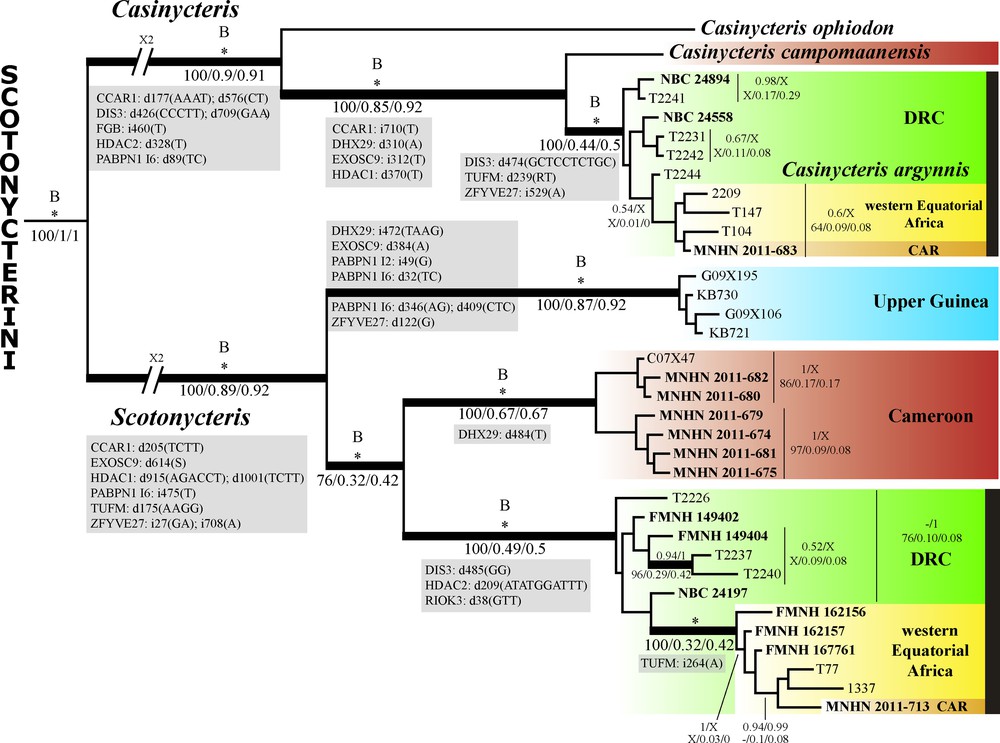
(Color online.) Nuclear phylogeny of the tribe Scotonycterini as inferred from the concatenation of twelve nuclear introns, representing eleven physically unlinked genes and 9769 characters. For each node, the two values above indicate posterior probabilities calculated either from the concatenation of the 12 nuclear introns (PPnu at the left of the slash) or from the supermatrix combining nuclear and mitochondrial genes (10,909 characters; PPtot at the right of the slash). The asterisk (*) indicates that the node was supported by maximal values of robustness (PPnu = 1/PPtot = 1). The nodes labeled with the letter “B” were found to be highly supported (PP = 1) in the *BEAST analysis, for which three species were assigned to the genus Scotonycteris (Appendix H). The three values below were obtained from the SuperTRI analyses of the 12 physically unlinked markers, i.e., the mitochondrial Cytb gene, and the eleven autosomal genes (CCAR1, DHX29, DIS3, EXOSC9, FGB, HDAC1, HDAC2, PABPN1, RIOK3, TUFM, and ZFYVE27): from left to right, Supertree Bootstrap percentage (SBP), Mean posterior probability (MPP), and Reproducibility index (Rep). The symbol “–” indicates that the node was not found in the analysis, and that no alternative hypothesis was supported by PP > 0.5. The letter “X” indicates that the node was not found in the analysis, and that an alternative hypothesis was supported by PP > 0.5. The position and nature of all diagnostic indels (i: insertion; d: deletion) shared by at least two taxa in the DNA alignments of nuclear genes are highlighted in grey.
A species tree was also reconstructed from our multilocus dataset (48 taxa, 12 loci) using *BEAST v.2.0 [56] (software package available at http://beast.bio.ed.ac.uk). Three geographic regions corresponding to three putative species were considered for the genus Scotonycteris based on the results of nuDNA, supermatrix and SuperTRI analyses: Upper Guinea, Cameroon, and Equatorial Africa. For each locus, we applied the model of evolution selected under jModelTest (see above). The species tree was estimated using a Yule speciation prior and 5 × 108 generations, with tree sampling every 5000 generations, and a burn-in of 10%. Adequacy of chain mixing and MCMC chain convergence were assessed using the ESS values in Tracer v.1.6. The trees were visualized with DensiTree v2.1.11.
2.5 Molecular dating analyses
Divergence times for major phylogenetic clades (time to the most recent common ancestor, TMRCA) were estimated using the Bayesian approach implemented in BEAST v.2.0 [56], and a Cytb alignment of 1140 nt and 49 taxa, including the 48 taxa used in the multigene analyses + the holotype of C. ophiodon. As no calibration point (fossil record or biogeographic event) is sufficiently accurate for the Scotonycterini, divergence times were estimated using a uniform rate of 0.02 nucleotide substitutions per site per lineage per Mya with a lower boundary of 0.01 and an upper boundary of 0.025. These values were chosen in agreement with divergence rates previously estimated for different groups of mammals [57], including bats [58]. We applied a GTR + I + G model of evolution (based on jModelTest) and a relaxed-clock model with uncorrelated lognormal distribution for substitution rate. Node ages were estimated using a Yule speciation prior and 109 generations, with tree sampling every 10,000 generations, and a burn-in of 10%.
2.6 Estimates of gene flow between geographic populations of Scotonycteris
All the 11 diploid nuclear loci described above were sequenced for 22 individuals of Scotonycteris. DNA sequences were phased using SeqPHASE [59] and PHASE version 2.1 [60,61]. Each unique indel, regardless of length, was recoded as a SNP (T for insertion and C for deletion). Then, we used the program Migrate-n version 3.6.4 [62] to estimate bidirectional migration rates and effective population sizes between four geographic populations of Scotonycteris: Cameroon (14 haplotypes), eastern DRC (10 haplotypes), western Equatorial Africa (12 haplotypes), and Upper Guinea (8 haplotypes). The analyses were conducted using a static heating scheme with four parallel chains with temperature values of 1, 1.5, 3, and 106 and a swapping interval of 10. In the full analysis, ten long chains were run with 10,000 genealogies discarded as burn-in followed by 107 steps recorded every 100 generations, resulting in a total of 106 sampled genealogies. We used exponential priors with window distribution for population sizes (0 < θ < 0.1) and migration rates (0 < M < 1000). Convergence was determined by the consistency of estimates across two separate runs performed with different starting points (random genealogies and different parameter priors). We also calculated Bayes factors as described in Beerli & Palczewski [63] for comparing the probabilities of four gene flow models between two populations, western Equatorial Africa and eastern DRC.
3 Results
3.1 Phylogeography of Scotonycterini based on Cytb analyses
The Bayesian tree reconstructed under MrBayes and using the alignment of mitochondrial Cytb sequences (1140 nt and 151 taxa; Fig. 2) shows a high support for the monophyly of the tribe Scotonycterini (PP = 1), and that of all taxa previously described within this group, including the genus Casinycteris (PP = 1), and the species S. zenkeri (PP = 1), C. argynnis (PP = 0.96), and C. ophiodon (PP = 1). Within Casinycteris, C. argynnis and C. campomaanensis were found to be sister-species (PP = 1).
Within S. zenkeri, we found a strong geographic structure, with four divergent geographic clades (4.9 < K2P distances < 8.7%, Table 1) supported by maximal values of PP: Cameroon, Upper Guinea (Liberia and Ivory Coast), western Equatorial Africa (EG, Gabon, RC, and CAR), and eastern DRC. Nucleotide K2P distances (DK2P) between members of the same geographic clade range from 0 to 3.2% in Cameroon, 0 to 1.1% in Upper Guinea, 0 to 1.4% in western Equatorial Africa, and 0 to 3.2% in eastern DRC (Table 1). The analyses supported an association between the two clades found in western Central Africa, i.e., Cameroon + western Equatorial Africa (PP = 0.98). Other deep relationships within S. zenkeri were not stable and robust. Indeed, the tree reconstructed with MrBayes showed a weak support for a sister-group relationship between Upper Guinea and the clade of Cameroon + western Equatorial Africa (PP = 0.73), whereas the chronogram reconstructed with BEAST rather favored the grouping of Upper Guinea with eastern DRC (PP = 0.42; Appendix G).
Pairwise nucleotide K2P distances (in %) calculated from Cytb (1140 nt, values above the diagonal) and nuDNA alignments (9641 nt, values below the diagonal).
| Taxaa | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 1. C. argynnis | 2.1/0.08 | 4.3–5.3 | 7.3–8.0 | 13.6–14.8 | 12.4–13.4 | 10.7–11.6 | 12.1–13.2 |
| 2. C. campomaanensis | 0.15–0.20 | NA/NA | 8.3–8.4 | 14.5–14.9 | 13.6–14.3 | 12.0–12.6 | 13.5–13.9 |
| 3. C. ophiodon | 0.78–0.85 | 0.83 | 1.3/NA | 13.9–15.1 | 13.4–14.0 | 11.5–12.7 | 13.8–14.6 |
| 4. S.z. Cameroon | 1.24–1.41 | 1.36–1.41 | 1.45–1.50 | 3.1/0.06 | 4.9–5.9 | 6.5–8.3 | 7.7–8.7 |
| 5. S.z. western EA | 1.35–1.58 | 1.48–1.56 | 1.56–1.63 | 0.45–0.59 | 1.4/0.09 | 5.3–7.2 | 6.3–7.9 |
| 6. S.z. eastern DRC | 1.30–1.44 | 1.39–1.44 | 1.47–1.55 | 0.38–0.52 | 0.08–0.22 | 3.2/0.13 | 5.7–7.2 |
| 7. S.z. Upper Guinea | 1.34–1.47 | 1.43–1.48 | 1.51–1.58 | 0.55–0.64 | 0.62–0.75 | 0.56–0.69 | 1.1/0.03 |
The two individuals of C. ophiodon, collected in Ivory Coast (SMF 91850) and Cameroon (holotype ZMB 50001), were found to share similar Cytb sequences, with only 1.3% nucleotide divergence. Within C. argynnis, the Cytb analyses showed the existence of two geographic groups: the first includes individuals from western Equatorial Africa, i.e., Gabon, RC, and the extreme western region of CAR (Dzanga Sangha; locality 21) (PP = 1); and the second contains individuals collected from Ngotto (southern CAR; locality 22) and eastern DRC (PP = 1). Nucleotide K2P distances between members of the same geographic lineage range from 0 to 0.6% in the first group, and from 0 to 0.7% in the second group. Members of these two groups show nucleotide divergences between 1.4 and 2.1% (Appendix D). The smallest nucleotide interspecific K2P distances were found between C. argynnis and C. campomaanensis (from 4.3% to 5.3%), and all other distances between species of Casinycteris were higher than 7.3% (Table 1).
3.2 Multigene analyses of Scotonycterini
The relationships within the tribe Scotonycterini were further studied by sequencing 12 nuclear introns of 48 taxa. The nuclear tree reconstructed from the total alignment of 9641 nt and 128 indels is presented in Fig. 3. The nucleotide K2P distances calculated from nuDNA alignments (9641 nt) are detailed in Table 1. For all nodes of the nuclear tree supported by PP of at least 0.5, we also indicated the results of the supermatrix (Cytb + nuDNA; 10,781 nt + 128 indels) and SuperTRI analyses, as well as the diagnostic indels found in the nuclear alignments. The Bayesian trees obtained from the analyses of the 12 independent markers (11 autosomal genes and the mitochondrial Cytb gene) are presented in Appendix E. The list of non-homoplastic substitutions found in the alignment of the 12 nuclear introns for major clades of Scotonycterini is presented in Appendix F.
The analyses of nuclear markers confirmed most nodes supported by mitochondrial Cytb sequences: the monophyly of Scotonycterini, Scotonycteris, Casinycteris, and C. argynnis, and the sister-group relationship between C. argynnis and C. campomaanensis. All these nodes were highly supported by nuDNA (PP = 1), supermatrix (PP = 1), SuperTRI (SBP = 100; 0.44 < MPP < 1; 0.5 < Rep < 1), and *BEAST analyses (PP = 1), each of them can be diagnosed by at least three exclusive indels (see in Fig. 3).
Three geographic clades of S. zenkeri, previously identified with mtDNA data, were also found to be monophyletic: Cameroon, Upper Guinea, and western Equatorial Africa, and further supported by nuDNA (PP = 1), supermatrix (PP = 1), and SuperTRI (SBP = 100; 0.32 < MPP < 1; 0.42 < Rep < 1) analyses, and each group can be diagnosed by at least one exclusive indel (see in Fig. 3).
Although highly supported by the supermatrix analysis (PP = 1), three mtDNA clades were not found monophyletic in the nuDNA tree or in the separate analyses of the 12 nuclear markers: the clade grouping individuals of S. zenkeri from eastern DRC (SBP = 76; MPP = 0.10; Rep = 0.08), the western group of C. argynnis (SBP = 53; MPP = 0.11; Rep = 0.08), and the eastern group of C. argynnis (SBP = not found; MPP = 0.09; Rep = 0.08). No alternative hypotheses were highly supported by the nuclear data.
Within S. zenkeri, we detected a robust topological conflict between mtDNA and nuDNA analyses. In the Cytb tree, the clade of western Equatorial Africa was found to be the sister-group to the Cameroon clade (PP = 0.98), whereas it was grouped with individuals from eastern DRC in the nuclear tree (PP = 1). The node grouping western Equatorial Africa and eastern DRC was also retrieved with high support in the supermatrix (PP = 1) and SuperTRI (SBP = 100; MPP = 0.49; Rep = 0.5) analyses. Members of this group can be diagnosed by three deletions found in three independent genes, “GG” at position 485 of DIS3, “ATATGGATTT” at position 209 of HDAC2, and “GTT” at position 38 of RIOK3.
3.3 Time tree of Scotonycterini
Our molecular estimates of divergence times based on the Cytb sequences (Table 2, Appendix G) suggest that the two existing genera of Scotonycterini diverged around 5.5 Mya, and have then co-diversified during the Early Pleistocene, with the identification of two major concomitant events: the first one, dated at around 2.7 Mya, corresponds to the separation of C. ophiodon from the two other species of Casinycteris, and to the divergence of western Central African populations of S. zenkeri (Cameroon + western Equatorial Africa) from other populations of S. zenkeri (Upper Guinea and eastern DRC); the second one, dated at around 1.6 Mya, corresponds to the split between C. argynnis and C. campomaanensis, and to the split within S. zenkeri between Cameroon and western Equatorial Africa.
Molecular estimates of divergence times.
| Taxa | PPB | Mean ages | 95% intervals | Geologic period |
| Tribe Scotonycterini | 1 | 5.51 | 3.14–8.99 | L. Miocene/E. Pliocene |
| Genus Casinycteris | 1 | 2.72 | 1.46–4.52 | L. Pliocene/E. Pleistocene |
| C. argynnis + C. campomaanensis | 1 | 1.61 | 0.79–2.73 | E. Pleistocene |
| C. argynnis | 1 | 0.65 | 0.29–1.13 | M. Pleistocene |
| C. ophiodon | 1 | 0.42 | 0.15–0.78 | M. Pleistocene |
| C. argynnis eastern clade | 1 | 0.17 | 0.08–0.42 | M. Pleistocene |
| C. argynnis western clade | 1 | 0.23 | 0.04–0.35 | M. Pleistocene |
| Genus Scotonycteris | 1 | 2.66 | 1.50–4.30 | E. Pleistocene |
| Scotonycteris UG + DRCa | 0.42 | 2.30 | 1.26–3.80 | E. Pleistocene |
| Scotonycteris C + wEA | 1 | 1.63 | 0.88–2.69 | E. Pleistocene |
| Scotonycteris DRC | 1 | 1.00 | 0.44–1.74 | E. Pleistocene |
| Scotonycteris Cameroon | 1 | 0.73 | 0.36–1.24 | M. Pleistocene |
| Scotonycteris wEA | 1 | 0.48 | 0.20–0.86 | M. Pleistocene |
| Scotonycteris UG | 1 | 0.29 | 0.10–0.54 | M. Pleistocene |
a Not supported in the Bayesian tree reconstructed with MrBayes (see Fig. 2).
4 Discussion
4.1 How many species exist within the tribe Scotonycterini?
The tribe Scotonycterini defined by Bergmans [38] included three species: Scotonycteris zenkeri, S. ophiodon, and Casinycteris argynnis. In a recent paper, Hassanin [37] described a new species, C. campomaanensis, which was the sister-group to C. argynnis, and placed S. ophiodon in the genus Casinycteris. Accordingly, Scotonycteris is monospecific with S. zenkeri, whereas the genus Casinycteris contains three species (C. argynnis, C. campomaanensis and C. ophiodon).
In the present study, several specimens from known localities were sequenced for each of the four species of Scotonycterini; the exception being C. campomaanensis, which is known only from the holotype. Our results are in strong agreement with the classification of Hassanin [37], as all analyses based on mtDNA, nuDNA, supermatrix, SuperTRI, and *BEAST approaches resulted in high support for the monophyly of Scotonycteris, Casinycteris, and C. argynnis, as well as the sister-group relationship between C. argynnis and C. campomaanensis. Each of these groups can be diagnosed by a minimum of three indels in at least three independent nuclear introns (Fig. 3).
Our analyses of mitochondrial sequences have also revealed strong phylogeographic structure within S. zenkeri, with the existence of four clades corresponding, from west to east, to: Upper Guinea (Liberia and Ivory Coast), Cameroon, western Equatorial Africa (EG, Gabon, RC, and southwestern CAR), and eastern DRC (Fig. 2). The mtDNA distances calculated between these four geographic clades are slightly higher than those found between the three species of the genus Casinycteris, i.e., 4.9-8.7% versus 4.3-8.4% (Table 1). Such results suggest that S. zenkeri may contain four cryptic species. Three of the four geographic mtDNA clades of Scotonycteris were recovered monophyletic based on the nuclear marker analyses, i.e., Upper Guinea, Cameroon, and western Equatorial Africa. The nuDNA distances calculated between these three clades were between 0.38% and 0.75% (Table 1), which is in the range of interspecific variation in the fruit bat genera Casinycteris (C. argynnis/C. campomaanensis = 0.15–0.20%; C. ophiodon/other species of Casinycteris = 0.78–0.85%), Megaloglossus (M. woermanni/M. azagnyi = 0.32%) and Myonycteris (M. torquata/M. angolensis/M. leptodon/M. relicta = 0.21–0.51%) (data not shown). As both mtDNA and nuDNA data indicate that these three geographic populations of Scotonycteris have been reproductively isolated for a considerable period (between 1.6 and 2.7 Mya according to our molecular estimates; Table 2), we consider that they should be treated as different species (see systematic descriptions below).
The taxonomic status of Scotonycteris specimens collected in eastern DRC is more problematic owing to a strong conflict between the mtDNA and nuDNA analyses. In the mtDNA tree, the specimens from eastern DRC grouped together in the same clade, which was found to be highly divergent from other groups, i.e., Upper Guinea and Cameroon + western Equatorial Africa (5.3–8.3%; Table 1). In the nuDNA tree, the specimens from eastern DRC do not constitute a monophyletic assemblage, because of an inclusive position of the western Equatorial African clade. These incongruent patterns may be explained by secondary contact(s) between populations from eastern DRC and western Equatorial Africa. Our Migrate-n estimates of gene flow between the four geographic populations of Scotonycteris (Cameroon, eastern DRC, Upper Guinea, and western Equatorial Africa) showed a significant migration rate from western Equatorial Africa to eastern DRC (Nem = 0.501), whereas all other migration rates were found close to zero (0.009–0.020) (Appendix I). According to our scenario, females from eastern DRC and western Equatorial Africa have remained isolated since the end of the Pliocene (2.7 Mya), whereas males were able to disperse, at least occasionally during interglacial periods, resulting in male-mediated nuclear gene flow between these distant populations, apparently from western Equatorial Africa to eastern DRC. This hypothesis is supported by the fact that sex differences in site fidelity, with female natal philopatry and male dispersal, are commonly encountered in mammals, especially bats [52,64,65]. From the taxonomic point of view, we conclude therefore that populations from western Equatorial Africa and eastern DRC are not genetically isolated and still belong to the same species.
4.2 Systematics of Scotonycteris
On the basis of our mtDNA and nuDNA data, which showed the existence of three genetically isolated geographic clades, we propose to divide S. zenkeri into three distinct species. In the new classification, S. zenkeri is restricted to populations from Cameroon, the region the type specimen was collected, whereas populations from Upper Guinea and Equatorial Africa are treated as two different species. The status of animals from Nigeria and Bioko Island need to be assessed based on molecular data, since Bergmans [36] indicated that geographical barriers, i.e., the Dahomey Gap and Niger Delta for the Nigerian population, and a distance of 32 km of open sea for the Bioko Island population, have isolated theses populations from others assigned to Scotonycteris.
GenusScotonycteris Matschie, 1894: Sitzungsberichte der Gesellschaft Naturforschender Freunde zu Berlin, p. 200.
Type species. Scotonycteris zenkeri Matschie, 1894.
Distribution. African rainforests, from Guinea and Liberia to eastern DRC.
Description. A small fruit bat with a forearm length (FA) between 45 and 56.5 mm and body mass between 15.5 and 27 g ([36]; this study). The dorsal pelage is dense, soft, and woolly. The general coloration is brown to rusty brown, with hairs being three-colored: dark brown basally, pale medially, and rusty brown proximally. The ventrum pelage is shorter and paler than the dorsum, with the throat, breast, and belly being whitish. Flanks are brown. The pelage of the head is brown with two kinds of white markings, including an oblong patch extending from the frontal (between the eyes) to the rostrum, and a small spot at the outer corner of the eyes. The lips are bordered with whitish hairs. There is no white patch at the base of the ears. The wing membranes are brown and the finger-joints are not yellowish. The plagiopatagium is attached to the first toe. There is no visible tail. There is a sexual dimorphism in body size, females being slightly larger than males (FA: 5% longer; body mass: 10% heavier). The soft palate is composed of two series of transverse ridges: the first includes four thick, prominent interdental ridges, the first three undivided, the fourth sometimes medially divided; the second series contains 6–9 thin, irregular and serrate postdental ridges.
Molecularly, the genus Scotonycteris can be diagnosed by three non-homoplastic substitutions in the Cytb alignment of 151 taxa (positions 116: T→C; 631: A→G, 985: G→A), 24 non-homoplastic substitutions in the nuclear alignments of 48 taxa (see details in Appendix F), and eight non-homoplastic indels detected in the introns of six independent nuclear genes, i.e., CCAR1 (d205: TCTT), EXOSC9 (d614: S), HDAC1 (d915: AGACCT; d1001: TCTT), PABPN1 (intron 6, i475: T), TUFM (d175: AAGG), and ZFYVE27 (i27: GA; i708: A).
Scotonycteris zenkeri Matschie, 1894: Sitzungsberichte der Gesellschaft Naturforschender Freunde zu Berlin, p. 202.
Holotype. ZMB 66533: adult ♀, body in alcohol, skull extracted. The specimen was collected by G.A. Zenker. A partial sequence of the mitochondrial Cytb gene (460 nt) has been deposited in the EMBL/GenBank/DDBJ nucleotide databases.
Type locality. Cameroon, Centre Region, Yaoundé, 3.87°N, 11.52°E.
Referred specimens. Ten additional voucher specimens conserved in alcohol were sequenced for this study. They were collected in three different geographic localities of Cameroon: village of Nkoélon-Mvini (South Region, 2.40°N, 10.04°E) for MNHN 2011-681 (♀) and MNHN 2011-682 (♂); Korup National Park (Southwest Region, 5.01°N, 8.86°E) for ♂♂ MNHN 2011-674, 2011-678, 2011-679, 2011-680 and for ♀♀ MNHN 2011-675, 2011-676, 2011-677; and Bez (South Region, 3.10°N, 10.50°E) for ZMB 54390.
Etymology. The specific epithet is in honor of Georg August Zenker, who collected the holotype. The English common name is “Zenker's fruit bat”. The French common name is “Scotonyctère de Zenker”.
Distribution.Scotonycteris zenkeri is known from Cameroon. Additional molecular data are needed to assess the taxonomic status of populations from Nigeria.
Description. Morphologically, S. zenkeri is not distinguishable from the other two species of Scotonycteris, although its body size tends to be smaller, with a mean FA of 49.4 mm in females (n = 11) and 47.7 mm in males (n = 5). Genetic distances between S. zenkeri and other members of the genus were between 4.9 and 8.7% with Cytb sequences (n = 56; 1140 nt) and between 0.38 and 0.64% with the nuclear dataset (n = 23; 9641 nt). Molecularly, S. zenkeri can be diagnosed by one deletion in DHX29 (d484: T) and 10 non-homoplastic substitutions detected in five independent nuclear genes, including CCAR1 (pos. 542: C→T), DHX29 (pos. 336: T→C, 684: T→C, 687: C→T), DIS3 (pos. 259: C→T, 659: G→A, 762: G→A), EXOSC9 (pos. 368: G→A, 739: T→C), RIOK3 (pos. 181: A→G).
Scotonycteris occidentalis Hayman, 1947 Annals and Magazines of Natural History, series 11, 13, p. 503.
Scotonycteris zenkeri occidentalis Hayman, 1947. Originally described as a subspecies of S. zenkeri Matschie, 1894, hereby raised to the level of species.
Holotype. BMNH 1946.898: adult ♀, skin and skull. The specimen was collected by G.S. Cansdale.
Type locality. Ghana, Eastern Region, Oda, 5.92°N, 0.93°W.
Referred specimens. Four voucher specimens conserved in the MNHN were sequenced for this study, collected at two different localities: Liberia, Fape (7.23°N, 9.28°W) for ♂♂ MNHN 2012-545, 2012-546 and ♀ MNHN 2012-547; and Ivory Coast, Azagny National Park (5.24°N, 4.80°W) for ♀ MNHN 2014-565. Several additional sequenced specimens conserved in the University Félix-Houphouët-Boigny (Abidjan) were destroyed during the Ivorian political crisis (2010–2011).
Etymology. The specific epithet refers to its geographic distribution in West Africa, where the holotype was collected and described by Hayman in 1947 [66]. We suggest “Hayman's tear-drop fruit bat” as the English common name and “Scotonyctère de Hayman” as the French common name.
Distribution.Scotonycteris occidentalis is endemic to West Africa, where it is known from Ghana, Ivory Coast, Liberia, and Guinea [36,41]. Additional molecular data are needed to assess the taxonomic status of populations from Nigeria, although they are isolated by the Dahomey Gap.
Description. Morphologically, S. occidentalis cannot be distinguished from the other two species of Scotonycteris, although it has a tendency of being larger. The mean FA is 52.1 mm in females (n = 4) and 50.9 mm in males (n = 6). Our molecular analyses support the elevation of this named subspecies to a full species, S. occidentalis. Genetic distances between S. occidentalis and other species of Scotonycteris are 5.7–8.7% based on Cytb (n = 56; 1140 nt) and 0.55–0.75% with the nuclear dataset (n = 23; 9641 nt). Molecularly, S. occidentalis can be diagnosed by six indels in the introns of four independent nuclear genes, including DHX29 (i472: TAAG), EXOSC9 (d384: A), PABPN1 (intron 2 i49: G, intron 6 d32: TC, d346: AG, d409: CTC), and ZFYVE27 (d122: G), and 22 non-homoplasic substitutions detected in nine independent nuclear genes, including CCAR1 (pos. 217: A→G, 423: C→T, 698: T→C, 790: G→A), DHX29 (pos. 320: G→A), EXOSC9 (pos. 474: G→T, 530: C→T), FGB (pos. 73: G→A, 90: A→G), HDAC1 (pos. 194: G→A, 555: G→A, 1053: A→C), HDAC2 (pos. 160: C→T), PABPN1 (intron 2 pos. 340: T→A; intron 6 488: A→T, 816: T→G), RIOK3 (pos. 26: T→C, 464: G→A), ZFYVE27 (pos. 167: T→G, 239: T→G, 476: C→T, 649: C→T).
Scotonycteris bergmansi sp. nov.
Holotype. MNHN 2011-713 (field number: R08-119), ♀ in alcohol. The specimen was collected on 16 November 2008 by Alexandre Hassanin, Emmanuel Nakouné, Nicolas Nesi, and Carine Ngoagouni. The sequences from the mitochondrial Cytb gene and 12 nuclear introns have been deposited in the EMBL/GenBank/DDBJ nucleotide databases.
Etymology. The specific epithet honors Dr. Wim Bergmans, a Dutch zoologist, for his outstanding contributions in the fields of taxonomy and biogeography of African fruit bats. We suggest “Bergmans's fruit bat” as the English common name and “Scotonyctère de Bergmans” as the French common name.
Type locality. Central African Republic, Mbaéré-Bodingué National Park, Case of Kpoka, 3.90°N, 17.16°E, 430 m above sea level.
Referred specimens. Ten voucher specimens conserved in three different museums, were also sequenced for this study, collected in five different geographic localities: Gabon, Projet OIBT trail (PK 29) (2.08°N, 12.41°E) for specimens FMNH 162156 (♂) and 162157 (♂); Gabon, Aire d’exploitation rationnelle de faune des monts Doudou (2.23°S, 10.39°E) for ♀♀ FMNH 167759, 167760 and ♂♂ FMNH 167761, 167762; Equatorial Guinea, Nkolentangan (1.85°N, 10.8°E) for ZMB 50004; DRC, Epulu (1.42°N, 28.58°E) for FMNH 149402 (♀) and 149404 (♂); and DRC, Irangi (1.83°S, 28.50°E) for specimen NBC 24197 (♂).
Distribution.Scotonycteris bergmansi sp. nov. occurs in Equatorial Africa, where it is known from the rainforests of Gabon, EG, RC, southern CAR, and eastern DRC.
Description.Scotonycteris bergmansi sp. nov. closely resembles S. zenkeri and S. occidentalis in external, cranial, and dental characters. Our molecular analyses support the designation of S. bergmansi as a distinct species. Genetic distances between S. bergmansi and other Scotonycteris spp. are 4.9–8.3% based on Cytb sequences (n = 56; 1140 nt) and 0.38–0.75% with the nuclear dataset (n = 23; 9641 nt). S. bergmansi can be diagnosed by three deletions detected in three independent genes, i.e., DIS3 (d485: GG), HDAC2 (d209: ATATGGATTT), and RIOK3 (d38: GTT), and by 13 non-homoplasic substitutions detected in six independent nuclear genes, DHX29 (pos.561: G→A), DIS3 (pos. 488: C→A, 570: A→C), EXOSC9 (pos. 925: C→T), HDAC1 (pos. 119: T→C, 968: T→C, 1015: C→T, 1016: C→T), HDAC2 (pos. 379: A→T), PABPN1 (intron 2 pos. 494: T→C; intron 6 pos. 160: G→A, 469: A→C, 796: T→C).
Based on mitochondrial Cytb sequences, it is possible to distinguish two different subspecies.
Scotonycteris bergmansi bergmansi ssp. nov.
Holotype. MNHN 2011-713 (field number: R08-119).
Type locality. Central African Republic, Mbaéré-Bodingué National Park, Case of Kpoka, 3.90°N, 17.16°E, 430 m above sea level.
Distribution.Scotonycteris bergmansi bergmansi ssp. nov. is found in the rainforests of western Equatorial Africa in the following countries: CAR (South), RC, Gabon, and Equatorial Guinea EG.
Description.Scotonycteris bergmansi bergmansi ssp. nov. is characterized by mitochondrial sequences of the Cytb gene, which are highly divergent form those of the other subspecies (5.3–7.2%). Morphologically, it is slightly smaller than the other subspecies, with a FA of around 49.6 mm in females (n = 5) and 47.3 mm in males (n = 3). Molecularly, the subspecies S. b. bergmansi can be diagnosed by the insertion of a nucleotide A in pos. 264 of TUFM, and by five non-homoplasic substitutions detected in two independent nuclear genes, i.e., EXOSC9 (pos. 276: G→A), and TUFM (pos. 33: T→A, 78: T→G, 186: C→T, 334: C→A). Primus et al. [67] reported a male karyotype from Gabon with 2n = 32.
Scotonycteris bergmansi congoensis spp. nov.
Holotype. MNHN 2014-564 (field number: K13-48), ♀ in alcohol, deposited in the collections of the MNHN.
Type locality. Democratic Republic of the Congo, Sukisa, 2.31°N, 24.98°E, 470 m above sea level.
Distribution.Scotonycteris bergmansi congoensis ssp. nov. is found in the rainforests of eastern DRC.
Description.Scotonycteris bergmansi congoensis ssp. nov. is characterized by mitochondrial sequences of the Cytb gene, which are highly divergent form those of the other subspecies (5.3–7.2%). Morphologically, it is slightly larger than the other subspecies, with a mean FA of around 50.8 mm in females (n = 5) and 48.7 mm in males (n = 6).
4.3 Ecological speciation at the Miocene/Pliocene boundary
Morphologically, the common ancestor of Scotonycterini was certainly characterized by white fur patches on the nose and behind the eyes, as these features are observed in all living species. These pelage markings are generally observed in taxa that roost in dense vegetation, where they function as disruptive coloration to make them less conspicuous to predators [68]. All these points suggest therefore that the origin and evolution of the tribe Scotonycterini have been associated with tropical rainforests of Africa.
The first phase of diversification of Scotonycterini, i.e. the separation between the genera Scotonycteris and Casinycteris, took place around 5.5 Mya. After a brief event of aridity during the Late Miocene, between 6.5 and 6 Mya, more humid climatic conditions prevailed in Africa, and rainforests expanded during the Early Pliocene (5.3–3.6 Mya), covering much larger areas than today (Fig. 4) [20,69]. On the basis of our phylogenetic reconstructions, it can be inferred that the common ancestor of Scotonycteris was a small sized fruit bat, probably similar to the three living species (FA = 46–56 mm in S. zenkeri, S. bergmansi and S. occidentalis; data from present study and Bergmans [36]), whereas the common ancestor of Casinycteris was a medium sized fruit bat (FA = 50–64 mm in C. argynnis, 73 mm in C. campomaanensis, and 75–88 mm in C. ophiodon; data from present study, Bergmans [36] and Hassanin [37]). An initial selective shift towards different body sizes may have permitted the exploitation of new ecological niches, i.e., such as different-sized fruits or fruit trees. We propose therefore that the basal generic division within the tribe Scotonycterini was driven by the expansion of rainforests after 6 Mya, which have offered more favorable opportunities for forest-adapted animals, such as fruit bats, to spread over the continent and to diversify in different ecological niches. A similar scenario involving speciation driven by ecological factors may also account for the divergence between Myonycteris and Megaloglossus, approximately at the same time, at around 5.7 Mya [51]. In that case, the main factor forcing speciation was not variation in body size, but the development of a new dietary strategy in the ancestor of Megaloglossus. Indeed, the two species of Megaloglossus (M. woermanni and M. azagnyi), both obligate nectivores, are today the only African bats possessing a long tongue adapted for lapping nectar from flowers [70].
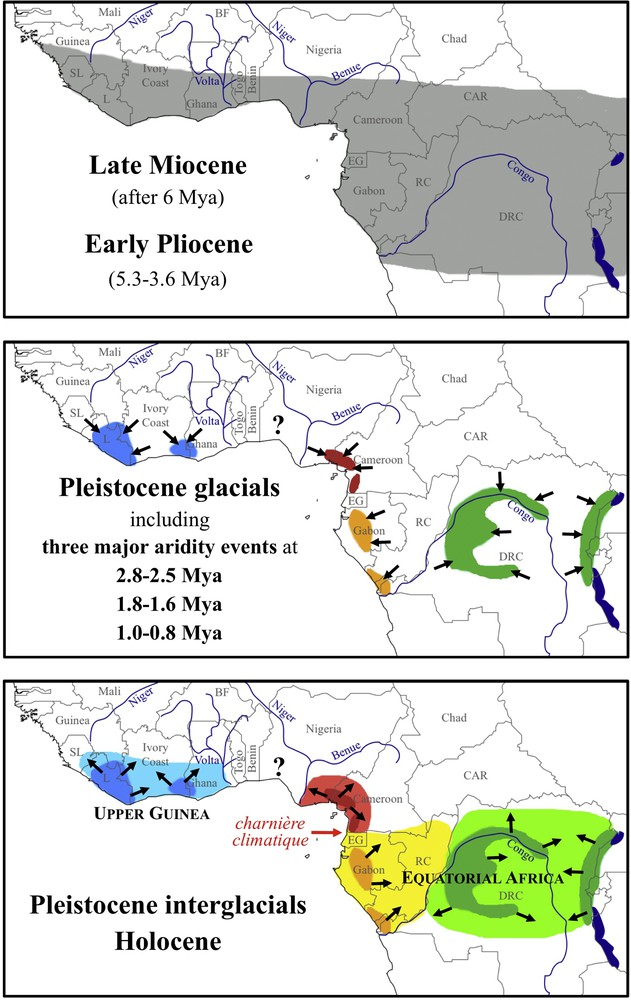
Biogeographic scenario illustrating how Pleistocene aridity events may have shaped the biodiversity of Scotonycteris. During the early Pliocene epoch, rainforests were distributed in most inter-tropical regions of Africa because of favorable climatic conditions corresponding to high levels of humidity. During this period, the common ancestor of Scotonycteris was probably distributed across African rainforests (grey zone). During periods of Pleistocene glaciations, rainforests may have persisted only in a few refugia. The distribution of Scotonycteris was fragmented into at least four main refugia corresponding to Upper Guinea (blue), Cameroon (red), Gabon (orange), and eastern DRC (green). The number and placement of these refugia were interpreted from Maley [22]. During Pleistocene interglacials and the Holocene, rainforests expanded to recover an area of Africa probably similar to modern times. Populations isolated during glacial periods may have dispersed and occurred in sympatry, but may have been reproductively isolated. For interpretation of the references to color in this figure caption, the reader is referred to the web version of the article.
4.4 The role of Pleistocene forest refugia in shaping the biodiversity of Scotonycterini
The onset of glaciation in the Northern Hemisphere took place at the Pliocene/Pleistocene boundary, at around 2.6 Mya, and from that period until today, the Earth's climate has been characterized by a succession of about 50 glacial–interglacial cycles [1]. During glacial periods, the decrease of temperature and precipitation had major impacts on the fauna and flora of high and middle latitudes, but also in low latitudes, where widely distributed rainforests of the Pliocene were partly replaced by more open vegetation [4]. In Africa, deMenocal [71] identified three progressive shifts in climate variability and increasing aridity, which are coincident with the onset and intensification of high-latitude glacial cycles: the first occurred between 2.8 and 2.5 Mya, the second between 1.8 and 1.6 Mya, and the third at around 1.0 Mya. Analyses of African mammal fossils have indicated an increase of arid-adapted taxa at 2.9–2.4 Mya and after 1.8 Mya [72]. Nesi et al. [51] have found that these two phases of aridity, at 2.8–2.5 Mya and 1.8–1.6 Mya, have resulted in speciation by isolation within the tribe Myonycterini. Our present study showed that the two phases of Scotonycterini diversification occurred in parallel in the genera Scotonycteris and Casinycteris, at around 2.7 Mya and 1.6 Mya, respectively (Table 2), suggesting that forest-adapted bats were strongly influenced by these two phases of aridity. During these episodes of drastic climate change, the fragmentation of rainforests was probably more severe than during other Pleistocene glacial periods. In Fig. 4, an evolutionary scenario is presented illustrating how Pleistocene aridity events may have triggered allopatric speciation within the genera Scotonycteris and Casinycteris in three main zones identified as forest refugia, i.e., Upper Guinea, Cameroon and Equatorial Africa.
At 2.7 Mya, in what is today tropical Africa, there was an abrupt decline in tree cover, with the extension of savanna and reduction of closed canopy forest. The greatest effect was in West Africa, where the rainforests retreated to occupy an area similar to that of today [69]. This event is proposed to have separated bat populations from west and central Africa, with S. occidentalis and possibly C. ophiodon in Upper Guinea, and the ancestor of other species in Central Africa. This period also fits with the diversification of the genus Myonycteris into four allopatric species: M. leptodon in Upper Guinea, M. brachycephala on Príncipe and São Tomé islands, M. torquata in Central African forests (from Cameroon, and possibly Nigeria, to DRC and Uganda), and M. relicta in the forests of eastern Africa [51]. This scenario implies that the ancestors of Scotonycteris, Casinycteris, and Myonycteris were widely distributed during the Pliocene epoch associated with the existence of continuous rainforests across tropical Africa, and that speciation events took place through vicariance around 2.7 Mya associated with forest fragmentation. This scenario requires, firstly, that these fruit bat genera have always been rainforest dependent, secondly, that they were not able to fly over long distances outside of rainforests, and thirdly, that the Dahomey Gap was an efficient savannah barrier for forest-adapted animals since the early Pleistocene. These assumptions appear reasonable for small fruit bats of the genera Scotonycteris and Myonycteris, but they seem to be questionable in the case of the larger and presumed stronger flyer C. ophiodon. Indeed, our phylogeographic analyses have shown that C. ophiodon is the only species of Scotonycterini sparsely distributed in both West and Central Africa, and recorded from only 16 localities in Liberia, Ivory Coast, Ghana, Cameroon, and possibly in Republic of the Congo [36,41]. Our molecular data suggest recent gene flow between West Africa and Central Africa, as the animal from Ivory Coast (SMF 91850) and the holotype from Cameroon (ZMB 50001) showed high levels of similarity in the mitochondrial Cytb gene (98.7%, 1140 nt) and in nuclear fragments of three selected autosomal introns, i.e. CCAR1 (100%, 162 nt), DHX29 (100%, 159 nt) and TUFM (99.4%, 154 nt) (data not shown). These results may be explained by a higher dispersal capacity for C. ophiodon, probably due to its larger body size and wingspan [36]. In agreement with our scenario involving vicariant speciation in Upper Guinea and Central Africa at around 2.7 Mya, the divergence of C. ophiodon from the two other species of Casinycteris may be explained by two non-exclusive hypotheses: a smaller body size in the Early Pleistocene ancestor, or a broader extent of the Dahomey gap during the Early Pleistocene.
The second phase of allopatric speciation, dated at around 1.6 Mya, also occurred simultaneously in the genera Scotonycteris and Casinycteris. The period between 1.8 and 1.6 Mya corresponds to an intensification of climate variability and aridity, generating more varied and open habitats in both West and East Africa [72,73]. At that time, we hypothesized that the rainforests of West and Central Africa retreated to occupy only four main refugial zones corresponding to the coastal forests of Upper Guinea, Cameroon, and western Equatorial Africa, and to the equatorial forests of central and eastern DRC (Fig. 4). In both genera of Scotonycterini, this episode of aridity may have separated populations of Central Africa, with C. campomaanensis and S. zenkeri in Cameroon and possibly in eastern Nigeria, and C. argynnis and S. bergmansi in Equatorial Africa. The period may also coincide with the onset or intensification of the “charnière climatique”, a climatic hinge located in southern Cameroon/northern EG and Gabon that today separates boreal and austral climates [74] and that appears to be an important phytogeographic barrier [75].
4.5 Rivers as biogeographic barriers during interglacial periods
In the context of current climate change patterns associated with global warming, understanding the evolution of biodiversity during interglacial periods of the Pleistocene is critical to better protect environment for present and future generations. Although our phylogeographic analyses have provided strong support for the diversification of forest-adapted bat species initiated by rainforest fragmentation during two major arid (glacial) periods, rivers have probably also acted as biogeographic barriers, stopping or limiting the dispersal of forest bat populations during interglacial periods. Three phylogeographic results on the genus Scotonycteris support this hypothesis.
First, the Ntem River, also known as the Campo River, in southern Cameroon and northern Gabon seems to have separated bat populations of S. zenkeri to the north (red in Fig. 4) from that of S. bergmansi distributed to the south (yellow in Fig. 4).
Second, the fact that female dispersal of Scotonycteris may be limited by large rivers, at least during interglacial periods, is also corroborated by the existence of two divergent mtDNA haplogroups detected in Cameroon for S. zenkeri (Fig. 2): one contains only animals from the Korup National Park (KNP), at the border with Nigeria (PP = 1; localities 9 and 10), and another includes specimens collected south of the Sanaga River (localities 11–13), as well as one KNP specimen (PP = 1).
Third, the role of rivers is better characterized in eastern DRC, where two divergent mtDNA haplogroups (2.4–3.2% of divergence in the Cytb gene) were detected in populations of S. bergmansi found on opposite sides of the Congo River (Fig. 2 and Appendix J): the first haplogroup contains specimens collected on the left bank of the Congo River (localities 25, 26, 28 and 29; ≤ 0.5%); the second includes specimens collected on the right bank of the Congo River (localities 24, 32, 33, 34 and 35; ≤ 1.4%). Since several localities of the two haplogroups are separated by less than 30 km (e.g., Yoko [locality 29]–Arboretum of Kisangani [locality 32]), this result shows that the Congo River constitutes a strong biogeographic barrier, at least for philopatric females of S. bergmansi. Flying over this large river is probably too risky for females of this small bat species. The phylogeographic pattern is also compatible with the existence of two forest refugia in eastern DRC, as proposed by Maley [22] (highlighted in green in Fig. 4): one surrounding the Congo River and some of its tributaries, and another in the mountain forests of eastern DRC, close to the Great Lakes Albert, Edward, Kivu, and Tanganyika. Such fine scale geographic structure was not found for C. argynnis (Fig. 2 and Appendix J), indicating that the Congo River is not impassable for this species, which is presumably related to its larger body size.
Acknowledgments
We thank numerous individuals who helped in collecting tissue samples during our field missions: Philippe Blot, André Délicat, Jean-Pierre Hugot, and Eric Leroy in Gabon; Jérôme Fuchs, Philippe Gaubert, Flobert Njiokou, and Anne Ropiquet in Cameroon; Alain Le Faou and Carine Ngoagouni in CAR; Christiane Denys, Mireille Dosso, François Jacquet, and Stéphane Kan Kouassi in Ivory Coast; and Raphaël Colombo, Laurent Daudet, Benjamin Dudu, Tamas Görföl, Keunen Hilde, Ros Kiri Ing, Jean-François Julien, Prescott Musaba, and Vuong Tan Tu in DRC. We are very grateful to collection managers and curators who sent samples, skulls, and pictures used in this study (see methods section for definition of institution acronyms): Nora Lange, Carola Radke, Lisa Kluckert, and Frieder Mayer, Museum für Naturkunde (Berlin, Germany); Emmanuel Gilissen and Erick Verheyen, Royal Museum for Central Africa (Tervuren, Belgium); Katrin Krohmann and Virginie Volpato, Senckenberg Museum Frankfurt (Germany); Lawrence R. Heaney, Julian Kerbis, John Phelps, and William Stanley, FMNH (Chicago, USA); Wendy van Bohemen and Steven van der Mije, NBC (Leiden, The Netherlands); and Anne-Marie Ohler, Jean-Marc Pons, and Anne Previato, MNHN (Paris, France). We also acknowledge the anonymous reviewers for their helpful comments on the manuscript. This work was supported by the MNHN, CNRS, PPF “Taxonomie moléculaire, DNA Barcode and gestion durable des collections”, LabEx BCDiv 2012-2013, and “Consortium national de recherche en génomique”. It is part of agreement No. 2005/67 between the Genoscope and the MNHN on the projects “SpeedID” and “Bibliothèque du Vivant”.