

1 Introduction
The demographic history of populations is thought to depend on the interplay between the history of the environment and the ability of species to respond to its changes. For instance, cold climate in Europe during glacial periods resulted in the southward retreat of many species to southern refugia, whereas the postglacial warming resulted in the northward spread of these species through various expansion patterns, due to an unequal contribution of populations originating from the different Mediterranean peninsulas (Hewitt, 2000). The role of interspecific differences in adaptation and of chance events, thus, limits the generality of the process. Even though a global environmental factor may affect all species of a community, examining a single species is not sufficient to assess the effects of environmental changes on biological systems as a whole. This suggests that adequate studies must rely on groups of comparable species.
While the fate of species during glacial periods in Europe remains relatively simple to describe since biodiversity was swept out from large areas (Provan and Bennett, 2008), the history of tropical African species is more difficult to reconstruct. Biogeographical data on dry forest trees led Aubréville (1949) to suggest that the rain forest had a smaller extension during the last glaciation event. Palynological data led Maley (1987) to put forward the refuge hypothesis, according to which the African rain forest was limited to fragmented areas 20,000 years ago and during former climatic cycles (Maley, 1996). The existence of alternate cycles of savanna and forest extension is well supported by palaeoenvironmental data (Dupont et al., 2000; Lézine et al., 2012). Past vegetation maps can be inferred using simulations (Hély et al., 2009) and a theoretical approach can be applied to the determinants of savanna and forests as alternative biome states (Staver et al., 2012). The consequence of these events on faunas cannot be reconstructed using the same methods. Due to environmental conditions, fossils are less preserved in Africa than in Europe (Hamilton and Taylor, 1991).
Typically, environmental changes in Africa led to local displacement of the dominant ecosystem (savannas vs. forests) or to a reorganization of the spectrum of dominant species within a given biome, forest or savanna (Lézine et al., 2012). Therefore, animal populations responded not only to climatic changes, but also (particularly for primary consumers, such as fruit-feeding insects) to changes in vegetation composition.
Because of the paucity of other sources of data, reconstruction of the history of tropical African species has to rely on the genetic variation estimated from natural populations. The demographic history of a species leaves its footprints in the genome. Therefore, the observed genetic variation can be used to infer the processes which shaped it. In the last thirty years, coalescent theory (Hudson, 1983; Kingman, 1982; Tajima, 1983; Tavaré, 1984) has provided a powerful tool to reconstruct the evolutionary history of populations and species (see, e.g. Charlesworth and Charlesworth, 2010; Hein et al., 2005; Slatkin and Veuille, 2002; Wakeley, 2008). The revolution of the coalescent theory relies mostly on its mathematical simplicity and on the fact that it is possible to reconstruct the history of populations by sampling only a modest number of individuals, rather than all of them. First of all, coalescent theory provides a way to quantify the genetic drift of a population. Genetic drift is a random change of the genetic composition of a population due to the recurrent sampling of allele frequencies over generations. Being a consequence of the finite nature of population numbers, it is a function of the effective population size. More generally, all phenomena involving a change in population size and thus, modifying patterns of genetic variation, including population expansions, genetic bottlenecks (the loss of genetic variation due to a population decrease), migration, can be investigated using predictions of coalescent theory. Mitochondrial DNA has often been a most convenient marker to infer the genetic history of natural populations. Several reasons have contributed to its success: the high copy number of the mitochondrial genome in cells, the uniparental (maternal) inheritance, the lack of recombination and the high mutation rate, compared to nuclear DNA (see Ballard and Whitlock, 2004 for a review). Therefore, investigating mitochondrial DNA variation remains an important step in studying the demography of a species. In order to assess the effect of vegetation dynamics on animal populations, we used data on genetic variation in several species belonging to a single guild of primary consumers. Fruit flies belonging to the family Drosophilidae include species that feed primarily on fruit, fungi and flowers, as well as on a few other minor substrates. Their short generation time (2 weeks for egg-to-adult development) and their ability to disperse over long distances by aerial transport potentially allow their populations to respond quickly to changes in the distribution of plant substrates in the forest.
Drosophilids present a great and unique ecological and morphological diversity among muscomorphan flies, as shown by the large number of species (> 4000). Drosophilids have succeeded in colonizing almost every possible terrestrial habitat, except the polar regions (Markow and O’Grady, 2006). Almost one-eighth of this rich fauna is found in the Afrotropical region, covering a wide range of habitats, including rainforests, savannas, mountains, oceanic islands, and others, and ecological niches, for example, as consumers of fruits, unripe figs, flowers, fungi, bee nests, and eggs of blackflies and dragonflies (Lachaise and Tsacas, 1983; Yassin and David, 2010). The particular paleogeographical and paleoenvironmental histories of tropical Africa, such as periods of connectivity and disjunction with Eurasia and episodes of expansion and contraction of savannas, certainly influenced and shaped the diversification of the drosophilid fauna (Yassin and David, 2010). Restricted accessibility has, however, limited field investigations of the role of environmental changes during the Quaternary in shaping the genetic structure of populations of Afrotropical drosophilids.
In tropical Africa, molecular population genetics studies have been carried out on three species of the Drosophila melanogaster species subgroup, which includes nine species (Lachaise et al., 2004). Two species originating from Africa, D. melanogaster and D. simulans, have become commensals of humans (Begun and Aquadro, 1993; Benassi et al., 1993; Lachaise and Silvain, 2004; Lachaise et al., 1988; Veuille et al., 1998, 2004). The study of the African populations of both species showed evidence of ancient population structuring in Africa (Aulard et al., 2002; Baudry et al., 2004; Benassi and Veuille, 1995; Lemeunier et al., 1994; Pool and Aquadro, 2006) and of expansion in eastern Africa (Baudry et al., 2006; Li and Stephan, 2006). The third species of the melanogaster subgroup, D. teissieri, is geographically limited to rain forests from the African mainland, where it is structured into populations that are morphologically differentiated, based on the male genitalia (Lachaise et al., 1981). The genetic structuring of its populations was interpreted in the framework of the history of Pleistocene rain forest fragmentation events (Cobb et al., 2000).
The genus Zaprionus is a substantial component of the Afrotropical drosophilid fauna. Recent phylogenetic studies showed that Afrotropical species belonging to the subgenus Zaprionus form a monophyletic clade that probably colonized Africa from Asia in the Middle or Late Miocene (Yassin et David, 2010; Yassin et al., 2008a, 2010), an age which has also been suggested for the ancestor of the Drosophila melanogaster species subgroup (Lachaise et al., 2004; Obbard et al., 2012). While the melanogaster subgroup of Drosophila includes only nine species, the Zaprionus subgenus includes 50 species, most of which live in the tropical rain forest. These species may, thus, provide crucial information on the history of this ecosystem. A molecular taxonomic study of African species of Zaprionus is underway (Suwalski et al. in preparation). The current study provides an important body of data on mitochondrial DNA variation about some species that are ecologically dominant in a set of different environments: forests, open areas, lowlands and highlands. We analyze these data below in an attempt to detect and characterize the signature of the main demographic events having affected the history of these populations in western and central Africa.
2 Materials and methods
2.1 Sample collection
The sample includes nine Zaprionus species for which we had 10 or more individuals per geographical location (Table 1): Z. africanus, Z. indianus, Z. aff. proximus, Z. aff. vittiger, Z. davidi, Z. taronus, Z. tuberculatus, Z. sepsoides, Z. vittiger. They were collected in Senegal, Guinea, Cameroon and Gabon (Fig. 1) between 2005 and 2010 using banana traps or by sweeping insect nets over different substrates and over the vegetation. The taxonomic determination of three species remains to be confirmed. They were identified by barcoding (Hebert et al., 2003) and using the Internal Transcribed Spacer (ITS) nuclear sequence (Eickbush and Eickbush, 2007). They could be new species. Wild-caught samples were kept in 70% ethanol until DNA was extracted from single males, using a Qiagen DNeasy Blood and Tissue Kit (Qiagen GmbH, Hilden, Germany), following the manufacturer's instructions. Males were used in order to avoid genetic contamination by mates in fertilized females.
Origin of samples, population variation estimates and neutrality tests.
| Species | Sampling site | Country | Collector (year) | N | S | h | H d | π | θ S | D | F s |
| Z. africanus | Mt Kupe | Cameroon | D. Lachaise and M. Veuille (2005) L. Brazier and M. Veuille (2007) |
21 | 38 | 20 | 0.995 | 0.013 | 0.019 | –1.35 | –13.78* |
| Z. indianus | Mt Kupe | Cameroon | D. Lachaise and M. Veuille (2005) L. Brazier and M. Veuille (2007) |
16 | 31 | 15 | 0.992 | 0.012 | 0.017 | –1.15 | –8.10* |
| – | Libreville | Gabon | M. Ikogou (2009) | 10 | 23 | 8 | 0.933 | 0.012 | 0.015 | –0.93 | –1.29 |
| – | La Lope | Gabon | M. Ikogou (2010) | 19 | 39 | 19 | 1 | 0.018 | 0.020 | –0.57 | –11.70* |
| – | Mbiyeh | Cameroon | M. Veuille (2008) | 14 | 35 | 14 | 1 | 0.017 | 0.020 | –0.66 | –6.92* |
| – | Gouan | Guinea | C. Denys (2008) | 12 | 35 | 12 | 1 | 0.018 | 0.021 | –0.60 | –4.92* |
| – | Salemata | Senegal | M. Veuille (2009) | 28 | 44 | 27 | 0.997 | 0.015 | 0.021 | –0.99 | –24.09* |
| Pooled | 99 | 83 | 90 | 0.996 | 0.016 | 0.029 | –1.53* | –24.66* | |||
| Z. aff. proximus | Mt Kupe | Cameroon | D. Lachaise and M. Veuille (2005) | ||||||||
| M. Veuille (2010) | 23 | 9 | 8 | 0.581 | 0.002 | 0.004 | –1.78* | –4.07* | |||
| – | Mboumi | Gabon | M. Veuille (2009) | 19 | 17 | 15 | 0.959 | 0.004 | 0.009 | –2.08* | –14.09* |
| Pooled | 42 | 21 | 20 | 0.796 | 0.003 | 0.009 | –2.20* | –19.48* | |||
| Z. aff. vittiger | Mt Oku | Cameroon | M. Veuille (2010) | 22 | 13 | 10 | 0.84 | 0.003 | 0.007 | –1.93* | –5.27 |
| Z. davidi | Mboumi | Gabon | M. Veuille (2009) | 60 | 22 | 25 | 0.781 | 0.003 | 0.009 | –2.00* | –25.45* |
| Z. taronus | Mt Fako | Cameroon | L. Brazier and M. Veuille (2008) | 15 | 8 | 6 | 0.648 | 0.003 | 0.005 | –1.06 | –1.18 |
| – | Mt Kupe | Cameroon | D. Lachaise and M. Veuille (2005) | 15 | 7 | 7 | 0.657 | 0.002 | 0.004 | –1.69* | –3.79* |
| – | Gouan | Guinea | C. Denys (2008) | 14 | 7 | 6 | 0.604 | 0.003 | 0.004 | –1.28 | –1.87 |
| Pooled | 44 | 18 | 16 | 0.856 | 0.003 | 0.007 | –1.78* | –9.97* | |||
| Z. tuberculatus | Mt Kupe | Cameroon | D. Lachaise and M. Veuille (2005) L. Brazier and M. Veuille (2007) |
11 | 4 | 5 | 0.709 | 0.002 | 0.003 | –1.32 | –2.42* |
| – | Kovifem/Mbiyeh | Cameroon | M. Veuille (2010) | 10 | 5 | 6 | 0.778 | 0.002 | 0.003 | –1.74* | –3.88* |
| – | Gouan | Guinea | C. Denys (2008) | 11 | 7 | 7 | 0.818 | 0.002 | 0.004 | –1.90* | –4.46* |
| Pooled | 32 | 12 | 14 | 0.75 | 0.0019 | 0.005 | –2.09* | –13.98* | |||
| Z. sepsoides | Mt Kupe | Cameroon | D. Lachaise and M. Veuille (2005) L. Brazier and M. Veuille (2007) M. Veuille (2010) |
27 | 4 | 4 | 0.214 | 0.001 | 0.002 | –1.89* | –2.51* |
| – | Mboumi | Gabon | M. Veuille (2009) | 25 | 1 | 2 | 0.153 | 0.000 | 0.000 | –0.70 | –0.28 |
| – | Crystal Mts | Gabon | A. Suwalski and M. Veuille (2010) | 10 | 1 | 2 | 0.200 | 0.000 | 0.001 | –1.11 | –0.34 |
| – | Sibang/Libreville | Gabon | M. Ikogou (2009) | 20 | 2 | 3 | 0.195 | 0.000 | 0.001 | –1.51* | –1.86* |
| Pooled | 82 | 8 | 8 | 0.186 | 0.000 | 0.003 | –2.16* | –10.09* | |||
| Z. vittiger | Mt Fako | Cameroon | M. Veuille (2010) | 12 | 1 | 2 | 0.167 | 0.000 | 0.001 | –1.14 | –0.48 |
| – | Mbiyeh | Cameroon | M. Veuille (2010) | 40 | 2 | 3 | 0.099 | 0.000 | 0.001 | –1.49* | –2.73* |
| – | Mt Oku | Cameroon | M. Veuille (2010) | 29 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Pooled | 81 | 2 | 3 | 0.073 | 0.000 | 0.000 | –1.31* | –2.98* |
* Significance at P ≤ 0.05.
Map of collecting sites in western and central Africa; mountains are shown as shaded areas.
We PCR-amplified a 546 bp fragment of the mitochondrial DNA cytochrome c oxidase 1 gene (COI) using primers Lep F1 (5′-WTTCWACCAATCATAAAGATATTGG-3′) and Lep R1 (5′-TAAACTTCTGGRTGTCCAAAAAATCA-3′), redesigned from those used by Hebert et al. (2004). Each PCR mix (30 μL) contained 3 μL of Fermentas Dream Taq Buffer, 200 μM of each dNTPs, 0.5 μM of each primer, 0.9 u of Fermentas Dream Taq DNA polymerase and 1 μL of DNA. Amplification included an initial denaturation step at 94 °C for 5 min, 10 cycles (94 °C for 30 s, 45 °C for 1 min and 30 s, 72 °C for 1 min), followed by 35 cycles (94 °C for 30 s, 51 °C for 1 min and 30 s, 72 °C for 1 min) and a final elongation step at 72 °C for 5 min. DNA sequencing was performed by Sanger Sequencing using a 96-capillary ABI 3730×l DNA sequencer (Applied Biosystems).
2.2 Sequence analysis
Sequence quality was checked using CodonCode Aligner 2.0.6 (CodonCode Corporation, Centerville, MA, USA) and data were analyzed using MEGA 5.05 (Tamura et al., 2011). Intra-population molecular variation was characterized using DnaSP 5.10 (Librado and Rozas, 2009) by the number of segregating sites (S), the number of haplotypes (h), haplotype diversity (Hd) and nucleotide diversity (using Tajima's π and Watterson's θs). Two species, Z. vittiger and Z. sepsoides, were monomorphic and were removed from further analyses. Departure from neutrality was assessed using Tajima's D (Tajima, 1989) and Fu's Fs (Fu, 1997) and their significance was computed from 1000 coalescent simulations using Arlequin 3.5 (Excoffier and Lischer, 2010). Tajima's D is based on the difference between two coalescent estimators of the effective population size, which under neutrality is distributed around zero. Departure from neutrality skews this difference towards positive values in case of a genetic bottleneck, and towards negative values in case of a population expansion (Tajima, 1989). Fu's Fs is based on the expected number of alleles under a certain value of the effective population size, and it assumes values close to zero under stationarity (Fu, 1997). Population expansions skew this statistic towards negative values; bottlenecks skew it towards positive values.
In species represented by more than one population, we computed the pairwise Φst and its significance (from 1000 permutations) using Arlequin 3.5 (Excoffier and Lischer, 2010).
Parameters of the instantaneous demographic expansion model (Rogers and Harpending, 1992) were estimated by means of DnaSP 5.10 using the distribution of pairwise differences between chromosomes (the mismatch distribution). Comparing this distribution to predictions of the coalescent theory can be used to detect changes in the effective population size (Schneider and Excoffier, 1999). Expansion times were scaled in calendar units assuming a mutation rate of 5.7 × 10−8 per site per year (Tamura, 1992) and 10 generations per year. We discuss these assumptions in the discussion section below.
The variation of the effective population size over time was reconstructed by means of the Extended Bayesian Skyline Plot (EBSP, Heled and Drummond, 2008) as implemented in Beast 1.7 (Drummond et al., 2012). Beast 1.7 is a software using Monte Carlo Markov Chain (MCMC), a simulated random walk through parameter space for the purpose of sampling from the posterior probability distribution of interest (Gilks et al., 1996). Each analysis was run until the posterior parameters converged (between 10,000,000 and 50,000,000 iterations) with a 10% burn-in and a thinning of 1000. We assumed the HKY + γ model of sequence evolution.
3 Results
3.1 Population variation
Out of 21 Zaprionus species identified from their morphology and their molecular barcode in the studied region, nine were represented by population samples, including ten individuals or more and were retained for the molecular variation study. Results are shown in Table 1. A striking contrast appears in the level of variation across species, with three different patterns. Firstly, two species, Z. vittiger (n = 81 individuals in three populations) and Z. sepsoides (n = 82 in four populations) were nearly monomorphic despite a large sample size. The second species is known to be infected by Wolbachia, a commensal intracellular bacterium (Cordaux et al., 2008). The lack of variation in the COI mitochondrial gene probably results from hitchhiking by this bacterium, which is transmitted matrilinearly in oocytes, and is known to spread in host populations through a transmission bias conferring an advantage to its host matrilines. We do not know whether or not a similar argument applies to Z. vittiger. Second, two species, Z. africanus (n = 21 in one population) and Z. indianus (n = 99 in 6 populations) present a very high level of variation, with a value of π = 1.3% in the first species, and π = 1.2–1.8% among populations of the other species. They will be referred to below as the high-variation species. Third, the remaining five species (Z. aff. proximus, Z. aff. vittiger, Z. davidi, Z. taronus, and Z. tuberculatus) represented by a total of ten populations, all show a low level of variation, ranging from π = 0.2% to π = 0.4%, depending on the population. They will be referred to below as the low-variation species. Tajima's D was significant in all low- and high-variation species (but in only seven populations out of ten in low-variation species, and not in a single population of the high-variation species), whereas Fu's Fs was significant in all species and populations for both low- and high-variation species. In all cases, Tajima's D and Fu's Fs were negative, including non-significant cases.
3.2 Differentiation between populations
Significant pairwise Φst values were found in two species, Z. indianus and Z. taronus. Data are shown in Table 2 for these two species. Zaprionus taronus showed high and significant values for all three comparisons. As expected, the two populations from the Cameroon volcanic line, Mt Kupe and Mt Fako, were closer to each other than either was to the Guinean population from Mt Nimba. The fact that all populations were significantly different is evidence of substantial divergence between populations from this species, which lives almost exclusively in montane forests. For instance, the collection sites at Mt Kupe and Mt Fako are only 100 km distant, but they are separated by a broad lowland area. In Z. indianus, populations were not significantly different from each other, despite an extended sampling covering distant areas. A surprising fact is that the Mt Nimba population (Gouan), differed from the Mt Kupe and Libreville populations, but did not differ from the populations that are geographically closer to them, such as Mbiyeh (for Mt Kupe) and Lope (for Libreville). This can be explained by the fact that the Mt Kupe and Libreville populations are much less variable than the other populations whereas Gouan is the most variable of all. This increases significance in the random genetic drift of the smaller populations increases genetic distance in these two pairwise comparisons. We have no explanation for the lower variation of these populations. Libreville is a suburban population, whereas Mt Kupe is a forested montane environment. Both locations are not typical of this species, which is generally found in open areas.
Pairwise estimate of population divergence using molecular Φst.
| Z. indianus | Z. taronus | ||||||
| Kupe | Libreville | Lope | Mbiyeh | Gouan | Gouan | Kupe | |
| Z. indianus | |||||||
| Libreville | 0.005 | ||||||
| Lope | 0.029 | 0.046 | |||||
| Mbiyeh | 0.009 | 0.017 | –0.004 | ||||
| Gouan | 0.075* | 0.076* | –0.001 | –0.009 | |||
| Salemata | 0.013 | 0.037 | –0.006 | –0.012 | 0.002 | ||
| Z. taronus | |||||||
| Kupe | 0.418* | ||||||
| Fako | 0.266* | 0.16* |
* P value ≤ 0.05.
3.3 Inference of past demographic history
Since negative values of Tajima's D and Fu's Fs support a demographic expansion, we checked the plausibility of this interpretation by visual inspection of the mismatch distribution (Fig. 2). In agreement with this, it appeared that the distribution was unimodal for almost all species and also for each population of Z. taronus (data not shown). The modes of the distributions fell into two classes corresponding respectively to the low-variation and the high-variation species. The average mismatch value in the low-variation group was k = 1.57 (range 1.038–1.845). In the high-variation group, the average mismatch was k = 7.75 (6.933–8.567). Thus, the average mismatch in the latter group was about five times higher than in the former.
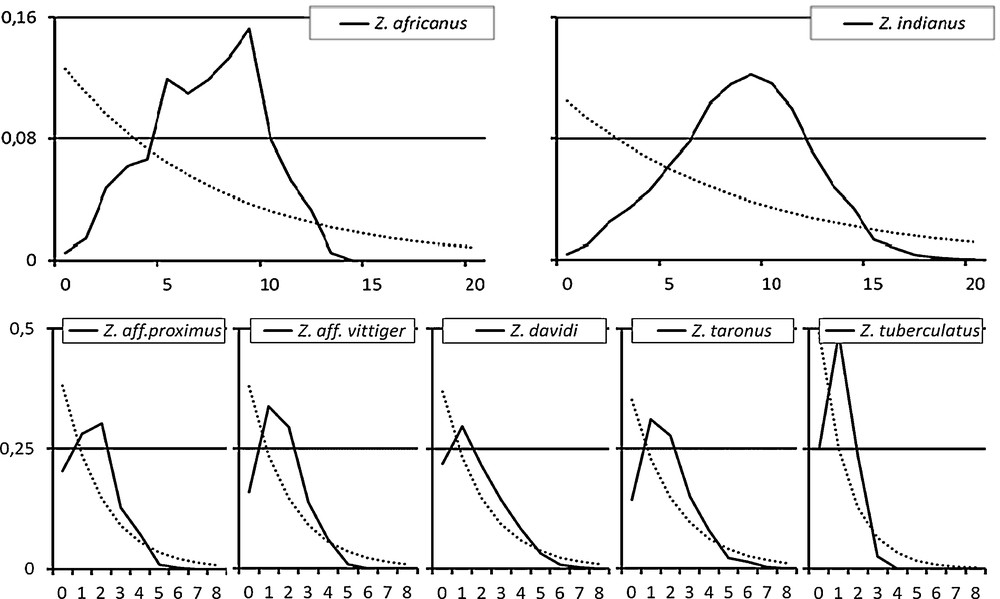
Mismatch distribution in the seven variable Zaprionus species. Continuous line: observed distribution; interrupted line: expected distribution.
For each of these species, the output of the EBSP is shown in Fig. 3, while the parameters inferred by the instantaneous population expansion model are given in Table 3. Using Tamura's molecular clock, the time span since the beginning of population growth was estimated to be 138 kya for Z. indianus and 111 kya for Z. africanus. Even though the uncertainty around these estimates is very large, these values are very close to each other. This estimate corresponds roughly to the onset of the last interglacial. For the low-variation group, the estimates were also very close to each other (average: 25.2 kya, range: 17–30 kya). This period approximately matches the time elapsed since the last glacial maximum (22–26 kya).
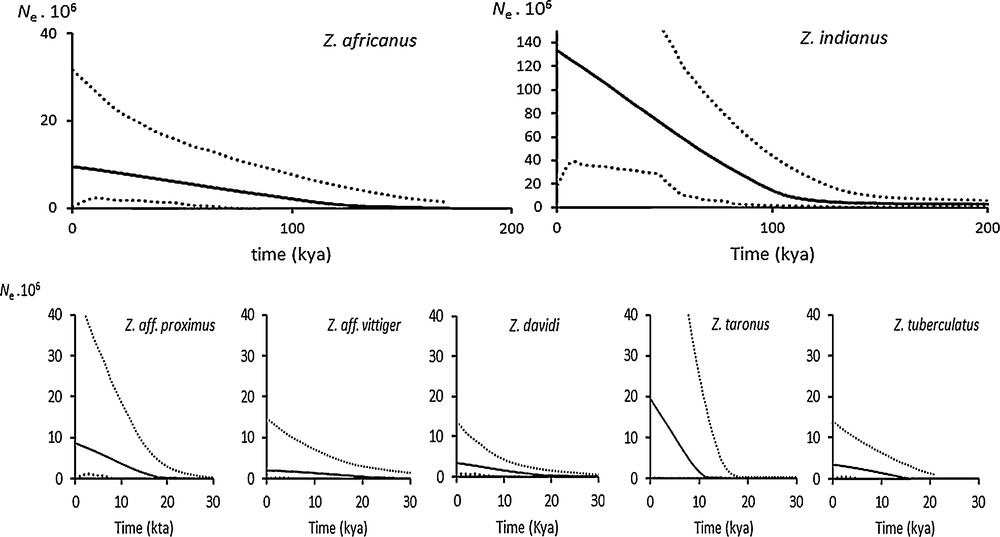
Extended Bayesian skyline plot in the seven variable Zaprionus species. Solid line: median of the posterior distribution; dotted lines: 95% highest posterior density; Ne: effective population size.
Estimates of population expansion parameter in the seven variable Zaprionus species, assuming a neutral mitochondrial DNA mutation rate of 5.7 × 10−8 changes per year.
| Species | Average mismatch (k) |
Time since population expansion (kya) |
| Z. africanus | 6.933 | 111 |
| Z. indianus | 8.567 | 138 |
| Z. aff. proximus | 1.619 | 26 |
| Z. aff. vittiger | 1.628 | 26 |
| Z. davidi | 1.715 | 27 |
| Z. tuberculatus | 1.038 | 17 |
| Z. taronus | 1.845 | 30 |
4 Discussion
Despite limitations inherent in the use of mitochondrial DNA for population genetics studies, we obtained significant results in a sufficiently large number of species to allow us to validate molecular variation in Drosophilidae as a relevant model to document population responses to Quaternary climatic changes in African forests. Below, we will examine in turn the limits of the study and its main conclusions.
4.1 Setting limits to the interpretation of results
Some limits inherent to the methods used need to be mentioned. Firstly, the generation time of drosophilids in the wild is unknown. While the time of development as a function of temperature is well established for several Drosophila species in the laboratory, generation time in the wild is difficult to estimate, since it depends on several factors, including the temperature of development which can be different in lowlands and in highlands, and on the survival of adults in natural conditions. Second, the neutral mutation rate in Drosophila mitochondrial DNA is difficult to evaluate. Several estimates of the mutation rate are available in Drosophila melanogaster. Tamura (1992) estimate of the mitochondrial DNA molecular clock (5.7 × 10−8 per site per year) is based on the assumption from geological evidence that the radiation of the Drosophila genus occurred 40 million years ago. This date could be an underestimate due to a lack of geological record, thus, the neutral mutation rate could be overestimated and lead to underestimated time inferences. Haag-Liautard et al. (2008)’s estimate of the mitochondrial DNA mutation rate (6.2 × 10−8 per site per generation) is based on laboratory mutation accumulation experiments. It includes some proportion of heteroplasmic non-synonymous changes, which could represent mildly deleterious mutations subsisting in experimental lines. Thus this is probably an overestimate of the neutral mutation rate. Obbard et al. (2012)’s estimates of the nuclear molecular clock in Drosophila are based, either on divergence data between species endemic to Hawaiian Islands of known age, or on experimentally derived mutation rates. The authors note that the rate calibrated by Hawaiian Islands is probably an overestimate since it assumes that the divergence of new species started soon after the emergence of a new island. The second estimate, calibrated by the mutation rate, is compatible with geological data on Drosophila radiation. Below, we use Tamura's estimate of the neutral clock, but it should be remembered that the most recent account on the nuclear mutation rate in Drosophila concludes that estimates of divergence dates in Drosophila should still be treated with some caution.
Attention should also be paid to cautiously interpreting mitochondrial DNA variation data. Since mitochondrial DNA is a non-recombining genetic system, any bias in its genetic transmission affects it as a whole. This includes infection by Wolbachia, an intracellular endosymbiotic bacterium. Differential genetic transmission between matrilines affects the transmission of mitochondrial DNA and results in hitchhiking. Hitchicking (Maynard-Smith and Haigh, 1974) results from the simultaneous increase in frequency of advantageous alleles and of neighboring neutral variants, since they do not segregate as independent Mendelian factors. This can result in the “selective sweep” of local variation in whole chromosomal fragments (Kaplan et al., 1989). Selective weeps can be detected by a number of selective neutrality tests (Depaulis et al., 2004), which are also used to detect demographic events (Depaulis et al., 2003; Mousset et al., 2004). Mitochondria and Wolbachia co-occur in matrilines, thus, the endosymbiotic bacterium can induce a selective sweep of variation in the mitochondrion. Interestingly, a Wolbachia strain has been isolated in Z. sepsoides (Cordaux et al., 2008), and its presence can explain the low mitochondrial DNA variation observed for this species in our data. The very low level of mitochondrial DNA variation in Z. vittiger could be explained in the same way, but this species has never been tested for Wolbachia infection. For the other species, the selective sweep hypothesis is less plausible. In this study, the evolutionary history drawn is that of mitochondrial DNA rather than that of the species. Mitochondrial DNA is a very useful genetic system for putting forward new hypotheses on population history, but genetically independent data from nuclear genes should be used to confirm these conclusions.
4.2 Insights into the responses of drosophilid populations to climate changes
The most striking result observed in our study is the long-lasting population expansion of Z. indianus and Z. africanus, the expansion of the former species being associated with a large increase in effective population size, whereas that of the latter species is moderate. Zaprionus indianus is a worldwide distributed species (Yassin et al., 2008b) whereas Z. africanus was recently described as a new species (Yassin and David, 2010) on the basis of mitochondrial analysis and experimental evidence of reproductive isolation of two lines from Gabon and Uganda with the former species. The striking increase in the population size of Z. indianus over a very long period provides new evidence on the remarkable evolutionary success of this species. This is a widely studied invasive species which succeeded in extending its geographical range from Africa to India, the Middle-East, southern Europe, and the Americas over the fifty last years (Chassagnard and Kraaijeveld, 1991; David et al., 2006; Yassin et al., 2008b), revealing its ability to spread into new environments. In tropical Africa, it is more abundant in open environments than in rain forests (Lachaise, 1974; Yassin et al., 2008b), which could explain its ability to invade seasonally dry areas, such as Brazilian savannas (Loh and Bitner-Mathé, 2005) and arid areas, such as oases in the Egyptian desert (Yassin et al., 2009). Our results suggest that the expansion time of both species (about 130 kya according to Tamura (1992)’s molecular clock) is consistent with an expansion beginning in the last interglacial period. Since there is no indication of a bottleneck coinciding with the period of the last glacial maximum (22–26 kya), our findings suggest either that these two species adapted to an environment which remained sufficiently widespread to support large populations, or that this environment was fragmented into numerous patches with large carrying capacities. In line with this, Z. indianus seems to be adapted to a wide range of environments. Our knowledge of the ecology of Z. africanus is limited. The samples used in this study, for which a species identification is confirmed by a molecular barcode, originated from five populations occurring at variable elevations between lowlands and highlands, suggesting that Z. africanus is adapted to a wide spectrum of environments.
The samples of the remaining five species, Z. aff. proximus, Z. aff. vittiger, Z. davidi, Z. taronus and Z. tuberculatus, were all caught in forested environments. The fact that all of these species show evidence of low-variation and of a population expansion beginning with the last glacial maximum remarkably complements the conclusions drawn for the previous two species, Z. africanus and Z. indianus. Thus, the same rationale applies to both cases, with the appropriate changes, while species typical of open environments were expanding during the last glacial period, when savanna was the dominant ecosystem; forest species began expanding after the Last Glacial Maximum. A thorough characterization of the ecological distribution of all five species is underway.
Observations similar to those made on Zaprionus were previously made in the melanogaster subgroup of Drosophila. From nuclear DNA variation, Li and Stephan (2006) estimated the East African population of Drosophila melanogaster to have undergone a population growth having begun 60,000 years ago. Baudry et al. (2006) found evidence of population expansion in several D. simulans populations from eastern Africa, but not from central or southern Africa. Since no reduction of population size during the last glaciation was observed in D. melanogaster, Stephan and Li (2007) suggested that the wild-to-domestic habitat shift of D. melanogaster postulated by Lachaise et al. (1988) occurred before the Last Glacial Maximum thus sheltering non-forest-dwelling populations from a decrease in population size. The fact that no reduction in population size is observed in Z. indianus and Z. africanus rather suggests that the expansion event observed in D. melanogaster was general among species living in open environments.
In the forest species D. teissieri, Cobb et al. (2000) showed that African populations are genetically strongly differentiated, in relation with the history of African forests. Thus, among these three sibling species from the melanogaster subgroup, D. melanogaster and D. simulans are preferentially adapted to open environments, and have become invading species throughout the world, whereas D. teissieri is limited to forest ecosystems. It is striking that, like Zaprionus indianus, the eastern populations of the first two species probably underwent an old population expansion, whereas the last one, which is ecologically closer to Z. taronus, is likewise geographically structured between African forests. These cases could represent two main classes of recurrent conditions, reflecting two different kinds of responses of African drosophilids to recent climate change, depending on their adaptation to the main biomes, forests and savannas.
Acknowledgments
This study was supported by the IFORA ANR (Agence nationale de la recherche) grant to Michel Veuille. Axelle Bouiges is currently supported by the C3A ANR grant to Anne-Marie Lézine. The field work was made possible under authorizations of the Minresi (Cameroon Ministry of Research and Industry) with help from the IRD (Institut de recherche pour le développement) delegation of Yaoundé. We thank Gaston Achoundong, Lionel Brazier, Lise Frézal, Arnaud Suwalski, Xavier Garde, and Philippe Le Gall for their help.