

Abridged version
1 Introduction
In the Aquitaine Basin, groundwater constitutes most of the water resources. The Infra-Molassic Sands Aquifer is used by several economical sectors, like drinking water, irrigation, thermal and hydrothermal and gas storage in the aquifer.
Different studies, especially in hydrogeology and geology, have been done to improve the aquifer management [6,12,18]. This work is an approach of the geochemistry aquifer behaviour and of its consistency with the geologic and hydrogeological data. Here, we focus on the sulphate concentration in the groundwater, which shows an important variability, with an augmentation in the flux direction. In particular, we will use (34S/32S) and (18O/16O) ratios of dissolved sulphate to determine the origin sulphur in solution.
2 Geographical and geological situation
The studied area is located in the southern part of the Aquitaine basin in southwestern France. It is limited to the south by the outcrops near Pau, to the north by the Barbotan area, where the aquifer reaches the ground surface, and to the west partly by the Audignon anticline.
This Eocene aquifer, composed by sandy Tertiary sediments, presents a high permeability and a thickness of several tenths of metres to a hundred metres. It is covered by molassic sediments, which form an aquiclude with low permeability and a thickness up to some hundred metres.
3 Results and interpretations
Isotope measures have been done on two samples of gypsum (Table 1) from molasse of the aquifer top. The obtained values (‰ CDT and ‰ SMOW) are coherent with sulphate from evaporites, according to numerous authors [7,8].
Composition isotopique en soufre 34 et oxygène 18 dans les sulfates dissous et dans l'hydrogène sulfuré et apports en sulfates des différentes sources, calculées selon le modèle.
Sulphur-34 and oxygen-18 isotopic composition in dissolved sulphates and in sulphur hydrogen and sulphate contribution of each source, computed according to the model.
| N° | Forages | [SO42−] | [H2S] | δ34SSO4 | δ18OSO4 | δ34SH2S | SO4 précipitation | SO4 gypse | SO4 pyrite |
| (mg L−1) | (mg L−1) | (‰ CDT) | (‰ SMOW) | (‰ CDT) | (mg L−1) | (mg L−1) | (mg L−1) | ||
| 1 | Bordes 3 | 14,43 | <LD | 4,87 | 5,13 | 14,43 | 0,00 | 0,00 | |
| 2 | Blagnac | 131,00 | 0,10 | 13,23 | 15,23 | 7,86 | 2,00 | 129,00 | 0,00 |
| 3 | Lalbarède | 58,38 | 0,02 | 13,06 | 17,23 | 10,78 | 2,00 | 56,38 | 0,00 |
| 4 | Lectoure | 651,80 | <LD | 12,23 | 16,13 | 2,00 | 649,80 | 0,00 | |
| 5 | Gondrin | 27,67 | <LD | 11,56 | 15,40 | 2,00 | 25,67 | 0,00 | |
| 6 | Castéra-Verduzan | 79,20 | <LD | 10,32 | 15,15 | 2,00 | 73,94 | 3,26 | |
| 7 | Garlin | 4,12 | 0,13 | 20,40 | 12,85 | −28,05 | 2,00 | 2,12 | 0,00 |
| 8 | Dému | 7,75 | 0,32 | 18,93 | 12,84 | −16,33 | 2,00 | 5,75 | 0,00 |
| 9 | Lamazère | 25,90 | <LD | 17,50 | 14,93 | 2,00 | 23,90 | 0,00 | |
| 10 | Nogaro 2 | 10,01 | 0,45 | 8,76 | 12,61 | −20,17 | 2,00 | 7,48 | 0,53 |
| 11 | Pléhaut | 51,98 | <LD | 9,76 | 15,28 | 2,00 | 47,07 | 2,91 | |
| 12 | EF1 | 4,40 | <LD | 35,80 | 14,30 | 2,00 | 2,40 | 0,00 | |
| 13 | ELB2 | 11,07 | 0,90 | 33,18 | 14,14 | −10,55 | 2,00 | 9,07 | 0,00 |
| 14 | LUG 47 | 19,61 | 0,08 | −3,77 | 8,33 | ||||
| 15 | LUG 57 | 20,47 | 0,08 | −3,21 | 9,19 | −15,48 | 2,00 | 9,24 | 9,23 |
| 16 | IZA 2 | 18,66 | <LD | −0,40 | 9,49 | ||||
| 17 | IZA 4 | 19,67 | <LD | −1,98 | 10,69 | ||||
| 18 | IZA 5 | 14,68 | 0,49 | −1,63 | 9,11 | −24,05 | 2,00 | 6,91 | 5,77 |
| 19 | Geaune 2 | 16,05 | <LD | −3,20 | 8,30 | 2,00 | 6,91 | 7,14 | |
| 20 | Lotus 2 | 53,47 | 0,03 | −15,80 | 3,95 | −32,25 | 2,00 | 5,49 | 45,98 |
| 21 | B 102 | 40,01 | 0,08 | −20,12 | 4,41 | 4,28 | 2,00 | 0,00 | 38,01 |
| Gypse (Pléhaut) | 11,87 | 15,87 | |||||||
| Gypse (Beaucaire) | 13,57 | 13,87 |
The from dissolved sulphates allows proposing four zones (Fig. 1):
- – near the Pyrenees, in the outcrop zone, the measured values are close to those of dissolved sulphates in precipitation [14];
- – in zone B, from the Montagne Noire to the centre of the basin, values are close to those measured in gypsum from molasse; we will note some values slightly more important on some points (7 to 9), located in the southern part of the basin;
- – in zone C, having a SW–NE direction, δ34S values are negative, with a diminution from the south to the north, whereas δ18O variations follow the same evolution;
- – in zone D, at the limit of the aquifer, we can observe a strong enrichment in δ34S (more than‰ CDT).
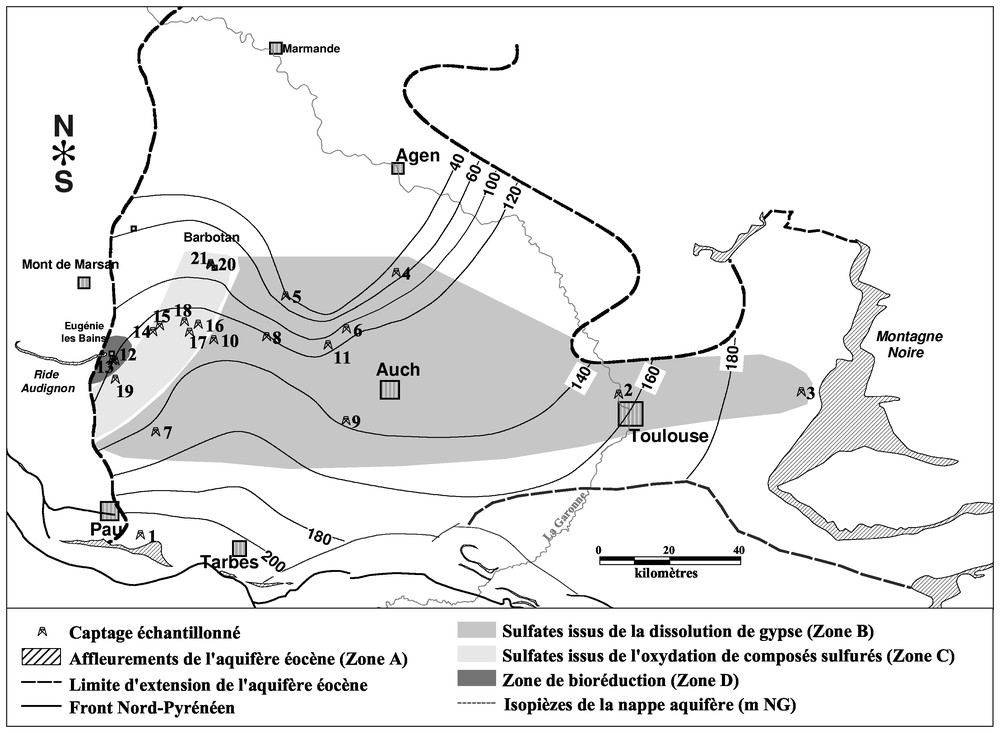
Représentation géographique des résultats isotopiques obtenus sur le soufre 34.
Geographic representation of isotopic results on sulphur-34.
4 Origin of dissolved sulphates
These variations give some clues on the origin of dissolved sulphates.
Values at the outcrops confirm that a part of sulphur composition comes from precipitation and that some mixes occur with unconfined aquifer.
To the North and to the East, sulphates seem to come from gypsum dissolution. The water age and the replacement rate of the aquifer confirm the absence of soluble mineral in the matrix. So, the isotope signature suggests the need to take into consideration exchanges with aquitards. Gypsum has been described by many authors at the aquifer top [2,4]. A progressive and continuous transfer of mineralised solution from the molasse to the aquifer could be the origin of the variations of sulphate concentration. The vertical transfers, by leakage and/or diffusion processes, are consistent with hydrogeological data: the topographic layer is behind the piezometric layer, inducing transport of matter from the bottom aquitard to the aquifer.
To the west, δ34S values are negative and here, we can suppose that dissolved sulphates come from reduced sulphur compounds, like pyrite. The existence of this sector seems to be connected with a sub-basin, submitted during Eocene to particular sedimentation conditions, and containing organic matter, observed in different wells.
In the last zone, the high values of δ34S seem to be the consequence of local bioreduction processes. Moreover, these values are in agreement with H2S contents higher than those measured in other groundwaters and lower redox potentials.
5 Modelling
All groundwaters have the same origin, but the chemical composition acquisition seems to be different, depending on the considered sector. For each sampled water, we have described the possible origin of sulphate in solution. A model have been established, based on δ34S values, in which we show that sulphur in solution could come from three origins, which are meteoric waters, gypsum dissolution and reduced sulphur compounds. This model allows quantifying the proportion of each process.
It leans on the δ34S balance equation, which gives the relative importance of the three processes. It has been validated by the δ18O values.
In this step, we confirm that sulphur composition of some waters originate only from gypsum dissolution. For others waters, some sulphur seems to come from reduced sulphur compounds according to two processes: oxidation with water oxygen and/or dissolved oxygen. It seems, according to modelling results, that a proportion of 50% of each process conducts to a satisfying consistency between computed and measured values.
This model is consistent with mineralogical and geologic data and also with hydrogeological data.
In conclusion, isotope tracing of different species of sulphur permits to propose new hypotheses about the behaviour and the flux of the Infra-Molassic Sands Aquifer. Four geochemical basins have been identified, each of them showing a different process affecting sulphate.
The North and the East of the Basin present relatively high sulphate concentrations; the stability of δ34S values, in this zone, confirms a gypsum origin. This hypothesis highlights the leakage process that may happen between aquitard and aquifer.
To the west, negative δ34S values seem to indicate the existence of a sub-basin, isolated from the precedent. The sulphate concentration variation seems to be linked to pyrite or other sulphur compounds oxidation.
We can finally note that bioreduction processes affect the sulphate composition of waters.
1 Introduction
En Aquitaine occidentale, les eaux souterraines représentent une ressource en eau importante. Ceci est particulièrement le cas pour l'aquifère éocène dit des « Sables infra-molassiques », utilisé à la fois pour l'eau potable, l'irrigation, la géothermie et le stockage de gaz en nappe. Actuellement, en vue d'une gestion plus rigoureuse, de nombreuses études, tant géologiques qu'hydrogéologiques, sont menées sur ces aquifères [6,12,18].
Ce travail rend compte d'une approche géochimique du fonctionnement de la nappe aquifère et de la mise en concordance de ces investigations avec les données géologiques et hydrogéologiques. Les variations spatiales de nombreuses espèces ont été étudiées ; il s'avère que les concentrations en ions sulfate montrent, au sein de la nappe des Sables infra-molassiques d'Aquitaine, une grande variabilité, avec des valeurs comprises entre 10 et 75 mg L−1 au sud de la zone et entre 50 et 650 mg L−1 au nord. L'augmentation suit les sens d'écoulement.
On se propose de montrer ici que les rapports d'abondance entre les isotopes du soufre (34S/32S) couplés à ceux de l'oxygène (18O/16O) des sulfates en solution permettent d'attribuer l'origine du soufre à :
- – des phénomènes de drainance descendante, à partir de l'éponte supérieure molassique et capacitive, pour une partie de l'aquifère ;
- – une oxydation chimique de la pyrite, au sein de l'aquifère transmissif, pour une autre partie.
À ces phénomènes se superpose une bioréduction des sulfates dissous, plus ou moins intense selon les secteurs de la nappe considérés.
2 Site d'étude et méthodologie
2.1 Situation géographique et géologique
La nappe étudiée, située dans le Sud-Ouest de la France, est limitée, au sud, par les affleurements des Sables de Baliros, près de Pau, au nord, par la structure de Barbotan, où l'aquifère recoupe la surface, à l'est, par la Montagne noire et, à l'ouest, par la ride d'Audignon (Fig. 1).
L'aquifère des Sables infra-molassiques est surmonté d'une éponte molassique (s.l.) tertiaire qui forme un aquiclude à faible perméabilité et d'épaisseur parfois supérieure à plusieurs centaines de mètres. Les sables éocènes, d'une épaisseur de quelques dizaines à parfois une centaine de mètres, présentent une forte perméabilité ; leur profondeur varie en fonction des topographies souterraines.
L'eau de la nappe des Sables infra-molassiques s'écoule, d'une part, des affleurements au sud de Pau vers la structure d'Audignon (le long d'un axe SE–NW) et, d'autre part, selon un axe parallèle à la Garonne, vers le système nord-aquitain (Fig. 1).
2.2 Méthodes
Les prélèvements d'échantillons d'eau sur 21 forages ont été effectués au cours d'une campagne qui a eu lieu de mars à avril 2000. Les concentrations en éléments majeurs ont été déterminées par chromatographie ionique. Les paramètres sensibles, tels que la température, la conductivité, le pH, Eh, l'alcalinité, la concentration des formes réduites du soufre en solution ont été mesurés sur le terrain [1].
La composition isotopique en sulfate dissous est obtenue sur précipité de BaSO4, après filtration à 0,45 μm et précipitation en milieu acide. Pour les eaux sulfurées, un prélèvement sur acétate de zinc a été réalisé. Des cristaux de gypse, provenant de déblais de deux forages, au toit de l'aquifère, ont été également analysés.
Les analyses isotopiques ont été réalisées au laboratoire des isotopes de l'environnement (université de Waterloo au Canada). Les résultats sont présentés dans le Tableau 1, selon la notation classique : δ18OSO4 (‰ SMOW) pour l'oxygène et δ34S (‰ CDT) pour le soufre.
3 Résultats et interprétation
3.1 Variations régionales de S
La valeur moyenne de δ34S mesurée sur deux échantillons (Tableau 1), dans le gypse issu de l'éponte au toit de l'aquifère, est de 12,72‰ CDT. Cette valeur est en accord avec celles citées par plusieurs auteurs pour les évaporites de sulfate [7,8]. La valeur moyenne de δ18OSO4, sur ces deux mêmes échantillons, est de 14,87‰ SMOW.
Les valeurs de δ34S déterminées dans les sulfates des eaux nous permettent de proposer quatre zones (Fig. 1) :
- – une zone A, située en bordure des Pyrénées, dans la zone d'affleurement de l'aquifère (point n° 1), la valeur de δ34S est proche des valeurs rencontrées pour les sulfates dissous dans les précipitations (‰ CDT [14]) ; la valeur de δ18OSO4 est légèrement inférieure aux valeurs admises dans les précipitations (18OSO4<+13,34‰ SMOW [20]) et est probablement due à des mélanges entre la nappe captive et la nappe alluviale située à proximité [15] ;
- – une zone B, qui s'étend des affleurements de la Montagne noire, à l'est, jusqu'au centre du bassin ; les valeurs de δ34S et δ18OSO4 sont proches des valeurs mesurées dans les cristaux de gypse présents dans la molasse ; aux points nos 7 à 9, on note des valeurs de δ34S sensiblement plus élevées (comprises entre 17,5 et 20,4‰ CDT) ; ces points sont situés dans une partie plus méridionale du bassin et pourraient s'expliquer par une légère bioréduction ;
- – une zone C, de forme allongée et de direction SW–NE ; δ34S présente ici des valeurs négatives à très négatives (de −0,4 à −20,1‰ CDT), avec un appauvrissement du sud vers le nord ; les valeurs de δ18OSO4 suivent la même variation, avec un appauvrissement le long du même axe ; ces valeurs traduisent une origine très différente des sulfates dissous par rapport à la zone B, qui serait le siège d'une oxydation de composés sulfurés ;
- – une zone D (à l'ouest), le long de la « terminaison » de l'aquifère, où l'on observe des enrichissements importants en soufre 34 (δ34S>33‰ CDT) ; cet enrichissement pourrait s'expliquer par un phénomène de bioréduction, qui affecterait alors les eaux de la partie occidentale du bassin.
3.2 Origine des sulfates dissous
Les teneurs en isotopes (δ34S et δ18O) issus des sulfates dissous mettent en évidence l'existence de quatre sous-bassins géochimiques distincts.
À proximité des affleurements, les eaux sont marquées par du tritium (11 UT en 1992 [3]) ; les valeurs de δ34S sont comparables aux moyennes mesurées dans les précipitations et sont cohérentes avec celles mesurées dans la partie ouest de l'Aquitaine [5].
À l'est et au nord, les sulfates dissous semblent provenir de la dissolution du gypse ; les données des isotopes de l'environnement (18O, 2H, 13C, 14C) disponibles sur la zone d'étude montrent bien une origine météorique des eaux et des temps de résidence calculés qui ne dépassent pas 30 000 ans [3].
Dans ces conditions, on peut admettre que le taux de renouvellement de l'aquifère est important. Les minéraux solubles de l'aquifère (type gypse ou anhydrite), susceptibles d'apporter des sulfates en solution, ont été dissous et balayés. Les analyses minéralogiques réalisées sur des phases solides de l'aquifère confirment que l'ensemble des sédiments de la partie transmissive de l'aquifère a largement été lessivé depuis son dépôt. Cependant, une abondance de gypse a été décrite au toit de l'aquifère par plusieurs auteurs [2,4] : un relargage progressif et continu de solutions minéralisées issues de la drainance des épontes argileuses ou argilo-sableuses, à caractère très capacitif, pourrait être à l'origine des teneurs en sulfate. Ce phénomène de drainance est confirmé par les données hydrogéologiques : la surface topographique étant située au-dessus de la surface piézométrique de la nappe, des transferts verticaux, de la surface vers l'aquifère, sont susceptibles de survenir et d'apporter de la matière dans la nappe.
Certaines valeurs de δ34S sont légèrement supérieures aux valeurs moyennes mesurées dans le gypse (points 7 à 9). Des phénomènes locaux de bioréduction semblent en être à l'origine. Le coefficient de fractionnement α entre les sulfates et les sulfures a été déterminé pour les forages 7 et 8 (pour le point 9, le taux de sulfures s'est avéré trop faible pour être quantifié). Les valeurs de α pour les points 7 et 8 sont respectivement 1,044 et 1,032, coefficients très proches de 1,040, valeur communément admise dans le cas d'une réduction par les bactéries Desulfovibrio desulfuricans [11].
À l'ouest, comme dans le secteur précédent, les eaux ont la même origine météorique. Cependant, les valeurs de δ34S mesurées semblent indiquer que le sulfate en solution pourrait provenir de l'oxydation de composés soufrés réduits (tels que la pyrite) [11]. Cette zone est à corréler à l'existence probable d'un sous-bassin naturel, marqué par des conditions de dépôt de sédiments à dynamique d'évolution particulière (lagunes, zones palustres, etc.), en présence de matière organique. Ce processus est conforté par les descriptions de coupes géologiques de forage, sur lesquelles on peut noter la présence abondante de sulfure de fer, au mur et au sein de l'aquifère, ce qui n'est pas le cas dans les zones A et B.
En bordure de ce « sous-bassin naturel », les eaux des forages nos 12 et 13 (région d'Eugénie-les-Bains), qui présentent de fortes teneurs en soufre 34 pour les sulfates dissous, semblent indiquer d'importants processus locaux de bioréduction [13,16]. Le coefficient de fractionnement α=1,040 confirme ce phénomène. Il est à noter que ces valeurs, singulières par rapport aux valeurs mesurées dans les autres eaux, sont à mettre en parallèle avec des concentrations fortes en soufre réduit ([ΣH2S]=0,8 mg L−1) et des potentiels d'oxydo-réduction faibles.
3.3 Modèle de mélange
L'ensemble des eaux étudiées possède des origines similaires, mais l'acquisition de leur composition chimique semble se faire selon différents processus, en fonction du secteur de la nappe envisagé. D'après les valeurs de δ34S mesurées, la minéralisation en sulfate provient majoritairement de trois origines :
- – minéralisation originelle issue des eaux de recharge, météoriques ;
- – minéralisation issue du drainage de la molasse contenant du gypse ;
- – minéralisation par oxydation de composés sulfurés intra-aquifère.
Nous avons élaboré un modèle visant à montrer que les valeurs en δ34S des sulfates dissous pouvaient être calculées à partir d'une superposition de ces trois processus. Ce modèle permet ainsi de quantifier leur importance relative en un point donné.
L'équation de bilan s'écrit (Éq. (1)) :
| (1) |
La signature isotopique δ34S des sulfates dissous doit refléter ces trois origines, ce qui se traduit (en considérant comme nuls les fractionnements isotopiques lors de la mise en solution) par :
| (2) |
X est la proportion de sulfate issu des eaux de recharge (X=nSO4 précipitation/nSO4 eau), Y la proportion de sulfate issu de la dissolution de gypse, Z la proportion de sulfate issu de l'oxydation de la pyrite, δ34Sprécipitation la teneur moyenne isotopique des sulfates dissous dans les précipitations. La valeur de +5‰ CDT a été retenue, car elle correspond à la valeur mesurée au point n° 1 (près des affleurements) et est en accord avec la littérature [14]. δ34Sgypse est la valeur mesurée sur les échantillons de gypse analysés (+12,72‰ CDT). δ34Spyrite n'a pas pu être mesuré et une valeur moyenne, issue de la littérature, a été retenue (−20‰ CDT) [9,11].
À partir des Éqs. (1) et (2), les proportions relatives des trois origines possibles de soufre en solution ont pu être établies (Tableau 1).
Ces résultats montrent que les origines du soufre varient très nettement : au forage de Blagnac (n° 2), 100 % des sulfates en solution proviendraient du gypse, alors que 100 % des sulfates dissous du forage B102 de Barbotan (n° 21) seraient issus de composés sulfurés. Pour le forage de Bordes (n° 1), situé à proximité immédiate des affleurements, nous considèrerons que la totalité des ions sulfate proviennent des précipitations, bien que des mélanges d'eaux avec l'aquifère alluvial interviennent [15].
Pour valider les proportions relatives de chaque processus, nous avons utilisé les teneurs en δ18OSO4 des sulfates dissous. Le même type d'équation de bilan peut être proposé (Éq. (3)) :
| (3) |
Les sulfates issus de l'oxydation de la pyrite proviennent de deux types de réactions : oxydation par l'oxygène de l'eau et/ou par de l'oxygène dissous [21].
| (4) |
La comparaison entre les valeurs de δ18O mesuré sur les sulfates dissous et de δ18O calculé à l'aide des Éqs. (3) et (4) est présentée sur la Fig. 2.

Comparaison entre δ18O mesuré sur les sulfates dissous et δ18O calculé à partir des δ34S. Les points sont obtenus avec β=0,5. Les barres d'erreur correspondent à des variations de β entre 0 et 1.
Comparison between δ18O measured on dissolved sulphates and δ18O computed from δ34S values. Points are obtained with β=0.5. The error bars show a 0 to 1 β interval.
Le modèle, correspondant à un mélange d'espèces du soufre issues de différents processus, semble à même de décrire les différences observées dans la composition isotopique du sulfate dissous dans l'ensemble des eaux de la nappe. Il permet de confirmer l'origine commune des eaux et l'évolution de leur composition chimique, en utilisant deux sources de minéralisation :
- – l'éponte, constituée de molasse sablo-argileuse, qui libère graduellement des sulfates, issus de la dissolution du gypse, dans l'aquifère ;
- – l'aquifère, sableux, dans lequel se produit l'oxydation chimique des composés sulfurés, quand ils existent.
Ce modèle géochimique proposé est cohérent avec les données géologiques et minéralogiques, mais aussi avec les données hydrogéologiques.
4 Conclusion
Le marquage isotopique des différentes espèces du soufre nous a permis de présenter de nouvelles hypothèses de comportement et d'écoulement de la nappe des Sables infra-molassiques. Ainsi, quatre principaux sous-bassins géochimiques peuvent être définis. Pour chacun d'entre eux, un processus géochimique différent semble affecter le sulfate.
La partie orientale et méridionale du bassin présente des concentrations en sulfate relativement élevées. La constance des teneurs isotopiques en soufre 34 sur l'ensemble de la zone conforte l'hypothèse selon laquelle les sulfates proviennent bien de la dissolution de gypse contenu dans l'éponte supérieure. Cette hypothèse permet de mettre en évidence les phénomènes de drainance qui sont susceptibles de se produire de l'éponte vers l'aquifère.
À l'ouest de cette zone, un sous-bassin, présentant des valeurs de δ34S négatives, semble relativement isolé du précédent. L'augmentation de la concentration en sulfate semble liée à la mise en solution de composés sulfurés, présents au sein et au mur de l'aquifère, dans cette zone.
Enfin, dans une dernière zone, relativement restreinte, des phénomènes de bioréduction semblent affecter les sulfates. Les valeurs isotopiques trouvées, corrélées à des concentrations élevées en espèces réduites du soufre, mettent en évidence le caractère particulier de cette zone.
Remerciements
Cette étude a pu être réalisée grâce au concours de TotalFinaElf Stockage Gaz France.