

1 Introduction
The complete sequencing of a plant genome is a landmark in characterisation of plant genes 〚1〛. Theoretically, a catalogue of all genes from one plant can be established directly from the sequence and from gene prediction programmes. However, as gene prediction alone is not efficient in determining the precise structure of all genes and for many genes, isolation of a full-length cDNA will be necessary to identify the fine gene structure and presumably several cDNA clones will have to be sequenced to recognise alternative splicing events 〚2〛. Yet, when a catalogue of all genes is established, we are far from understanding their function. During the last few years, many independent approaches have been developed to investigate gene function. One of them relies on constructing saturated mutant collections, in which each gene has a chance to be interrupted or mutated. Insertion mutants have been considered as the best choice, since usually once a phenotype is observed it is possible to characterise the sequence flanking the insertion and to identify the mutated gene. In Arabidopsis, several types of collection have been set up, using either T-DNA or heterologous transposons 〚3–14〛. These collections have been extremely useful to isolate a number of important genes based on the observation of a phenotype 〚6–11〛. However a major problem is to establish the linkage between the visible phenotype and the insertion. In addition, multiple screenings are necessary to identify different mutants. With the availability of sequence information, it became possible to screen the collection using DNA pools prepared from pooled insertion lines by PCR methods, to retrieve lines with an insertion in a specific gene. This technique is particularly useful to isolate insertion mutants in genes belonging to multigene families and for which the observation of a phenotype is unlikely 〚15–22〛. Screening of such DNA pools is time consuming as well and not exportable to many labs. An alternative strategy is to develop a more systematic and random strategy, by determining systematically the flanking sequence of a given insertion (FST for Flanking Sequence Tags). These sequences can be archived in a database that can be consulted by any scientist through a web server. So far a limited number of such results have been reported 〚12, 13, 23〛. Using the T-DNA insertion collection developed at the INRA Versailles (France) 〚5, 18, 24〛, we carried out a pilot study to investigate the feasibility of such an approach. In this paper we report our results on a set of 850 lines selected on a phenotypic basis. This approach was chosen for the pilot study, rather than just random selection, because it was expected to harvest at the same time the information on a number of potentially interesting genes. The results demonstrated that systematic approach was feasible and was a shortcut to identify many genes function and mutants. The systematic analysis of the whole collection is now in progress 〚23〛.
2 Material and methods
2.1 Plant material and growth conditions
The Arabidopsis T-DNA lines used in this analysis were selected from the collection of transformants generated at the INRA Versailles. This collection of mutants was constructed using the previously described pGKB5 vector 〚24〛 and infiltration transformation techniques 〚5, 18〛. Because this was a pilot study, we decided to systematically sequence a set of lines corresponding to putative mutants showing a particular phenotype. These phenotypes, the origin, and the number of lines used in this study are described in Table 1. Except for lines selected on the basis of GUS staining, in which the T-DNA is linked to the phenotype, in all others the linkage between the phenotype and the T-DNA was not yet systematically established. This work is still in progress, and so far, our unpublished results indicate that linkage occurs in about 20% of the lines. Each line has been selected on the basis of a phenotype observed in one of the laboratories and amplified. Resulting sterilised seeds were forwarded to the laboratory in charge of FST determination and germinated on culture medium supplemented with 50 μg ml–1 kanamycin, in a controlled environment at 22 °C under continuous light and grown for 15 days. Then, the first growing leaves were collected and used for DNA extraction.
Selection of the Arabidopsis T-DNA lines.
| Series | Origin | Selected phenotype | Number of lines |
| E, F, L, P, Q, S | CNRS Clermont-Ferrand | GUS activity in seeds and siliques | 417 |
| B, M, K, N | CNRS Perpignan | Putative embryo-defective mutations | 230 |
| A | INRA Antibes | GUS activity induced by plant parasitic nematodes | 77 |
| C | INRA Versailles | Gametophytic, sporophytic mutations | 51 |
| R | CEA Cadarache | GUS activity in roots | 51 |
| D, G, H, T | others | 24 | |
| 850 |
2.2 Genomic DNA isolation and characterisation of T-DNA flanking regions
The Dneasy plant DNA extraction kit (QIAGEN) was used to isolate DNA from 100 mg of 15 day-old seedlings. Approximately 500 ng of DNA was double-digested by EcoRV/DraI or ScaI/SspI to create blunt-ended fragments. Genomic sequences flanking the T-DNA LB and RB were isolated by PCR walking. The detailed method (ligation of adaptors, nested primers and PCR conditions) has been described 〚17〛. The following minor modifications were used. After the second step of reactions, PCR products were electrophoresed on a 1% agarose gel, bands larger than 200 bp were recovered and purified using a QIAquick gel extraction kit (QIAGEN) and used for direct sequencing using ABI Bigdye terminator cycle sequencing kit and an ABI 377 automatic DNA sequencer (Applied Biosystem). Fragments were sequenced from both ends.
2.3 Sequence analysis
A first step of sequence analysis consists in trimming all uncertain sequences. Then, each sequence is systematically aligned with the complete sequence of the T-DNA and the adjacent sequences of pGKB5. The sequences that corresponded only to T-DNA or vector and those without recognisable T-DNA signal sequence (LB or RB) were discarded. Finally, the T-DNA sequence was removed in order to obtain the plant FST, which is usually in the range of 200–300 bp. The resulting sequences were analysed with Sequencher software (Gene Codes Corp., Ann Arbor, Mi, USA), and subjected to BLAST 〚25〛 searches using Genbank, Atdb Arabidopsis sequences, dbEST, Swissprot and non-redundant protein databases. For BAC clones, we used gene prediction analysis supplied by Genbank and periodically updated. Chromosomal location was determined using physical and genetical mapping data from TAIR, the Arabidopsis Information Resource (http://www.arabidopsis.org).
3 Results
3.1 FST number and quality
From 850 lines analysed, 510 have produced at least a single exploitable plant sequence: 98 produced two FSTs, in opposite orientation, corresponding to both sides of an insertion at a single locus, 56 produced two FSTs, corresponding to different loci, and two lines yielded three FSTs, corresponding to three insertions each. These results indicate that it is possible to isolate different FSTs in lines carrying more than one T-DNA insert. T-DNA or vector sequence was obtained for 130 lines. It was impossible, in our conditions, to obtain FST sequence in 210 lines. This failure can result from complex T-DNA insertion, truncation of the T-DNA border resulting in loss of primer sequence, or absence of restriction site in the immediate vicinity of T-DNA.
A total of 851 sequences was obtained. Among them, 72% are flanking the left signal sequence (LB) of T-DNA because it was observed that this sequence was usually better preserved than the right one. It was also observed that 36.5% of the amplified fragments corresponded to T-DNA or vector sequence when the primer targets the right end signal, instead of 10.3 when a left border primer is used. This difference might be due to the fact that the right signal sequence is frequently engaged in RB/RB inverted repeats. As a result, 701 authentic plant FST sequences were observed.
3.2 Characterisation of FST sequences
Sequences were cleaned from the T-DNA and adapter sequences and they were compared to sequences in Genbank and dbEST. They were also compared to protein sequences in public databases. A match with the Arabidopsis genome sequence was observed for 97.5% of the FSTs (684 sequences), in spite of some ecotype differences (the sequenced genome is Columbia ecotype, whereas the insertion line collection is in the WS ecotype). The missing 2.5% can correspond to rare unavailable genomic sequences, or to sequences too much rearranged to be recognisable. Only 16% of the FSTs give a hit with an Arabidopsis EST. This apparently surprising observation can be explained by the small size of the FSTs (usually less than 400 bp), the small size of ESTs, and by the distribution of FSTs in all parts of the genome and not only in exons.
Table 2 gives the distribution of FST tags according to the fraction of the genome in which the insertion occurs. 112 (16.4%) are in regions annotated as exons, in agreement with the percentage of hits with EST. A majority of FSTs corresponds to sequences in the immediate vicinity of an ORF (immediate vicinity was arbitrarily defined as a region of 1200 bp upstream of the putative initiation codon and 600 downstream of the stop codon and roughly represents promoter and terminator regions) or in intergenic regions. However, because the lines have not been randomly selected, this distribution is not homogenous among the samples; for instance, the proportion of FSTs corresponding to exon is lower in samples that have been selected on the basis of GUS staining (12%) than in samples selected on the basis of a morphological phenotype (20%). Altogether a total of 482 genes (including promoter, exon, intron and terminator regions) have been tagged by an FST. It should be reminded that these values rely on the present annotation of the Arabidopsis genome, which is not completely accurate 〚2〛 and that some minor changes can occur when the annotation is improved. Approximately 60% of the FST tag a gene for which a putative function can be predicted.
Distribution of FST sequences in Arabidopsis genome.
| Localisation | FST number | FST (%) |
| Exon | 112 | 16.4 |
| Intron | 84 | 12.3 |
| Vicinity of an ORF | 286 | 41.8 |
| Intergenic region | 186 | 27.2 |
| Repeated DNA region | 8 | 1.2 |
| rDNA | 1 | 0.1 |
| Unknown | 7 | 1 |
| 684 | 100 |
The list and accession numbers of the genes tagged by an FST generated in this study cannot be given in this paper, due to page limitation. A partial list is available on the Perpignan laboratory website (http://gamay.univ-perp.fr/∼delseny/). All sequence data, except those concerning a very limited number of genes under further investigation, are being transferred to Flagdb 〚23〛 available through Genoplante-info (http://flagdb-genoplante-info.infobiogen.fr/projects/fst).
Corresponding lines can be requested from the INRA Versailles at the following address: publiclines@versailles.inra.fr.
3.3 Analysis of the junction between T-DNA and plant sequence
The availability of the Arabidopsis and of the T-DNA sequences allows one to investigate rearrangements at the junction between the two types of sequences. As indicated above, a couple of FSTs could be obtained on both sides of the T-DNA for 98 lines. In 50 cases, the couples are LB/RB, but in 46 the couples are LB/LB and in 2 they are RB/RB, corresponding most likely to the insertion of two in-tandem inverted T-DNA at the same locus. In the lines for which the FSTs are available on both sides of the insertion, it is possible to compare the FSTs with the genomic sequence in the non-transgenic plant. This type of comparison revealed that a portion of plant DNA sequence is frequently deleted following insertion of T-DNA. The distribution of the size of the deletion is illustrated in Fig. 1. The majority of the deletions ranges between 0 and 60 bp. A similar observation can be made on the T-DNA sequence. Sequence analysis revealed that 93.1% of the LB and 85.5% of the RB signals are truncated over less than 40 nucleotides. Truncation is usually larger on the right than on the left side. The distribution of truncation sizes in LB and RB in T-DNA is shown in Fig. 2. In a limited number of FSTs, a short sequence with no homology to the Arabidopsis genome is observed at the junction between plant genomic sequence and T-DNA. Such short sequence (usually less than 30 bp) likely corresponds to filler DNA resulting from reparation of breakage necessary for insertion of T-DNA into the genomic sequence.
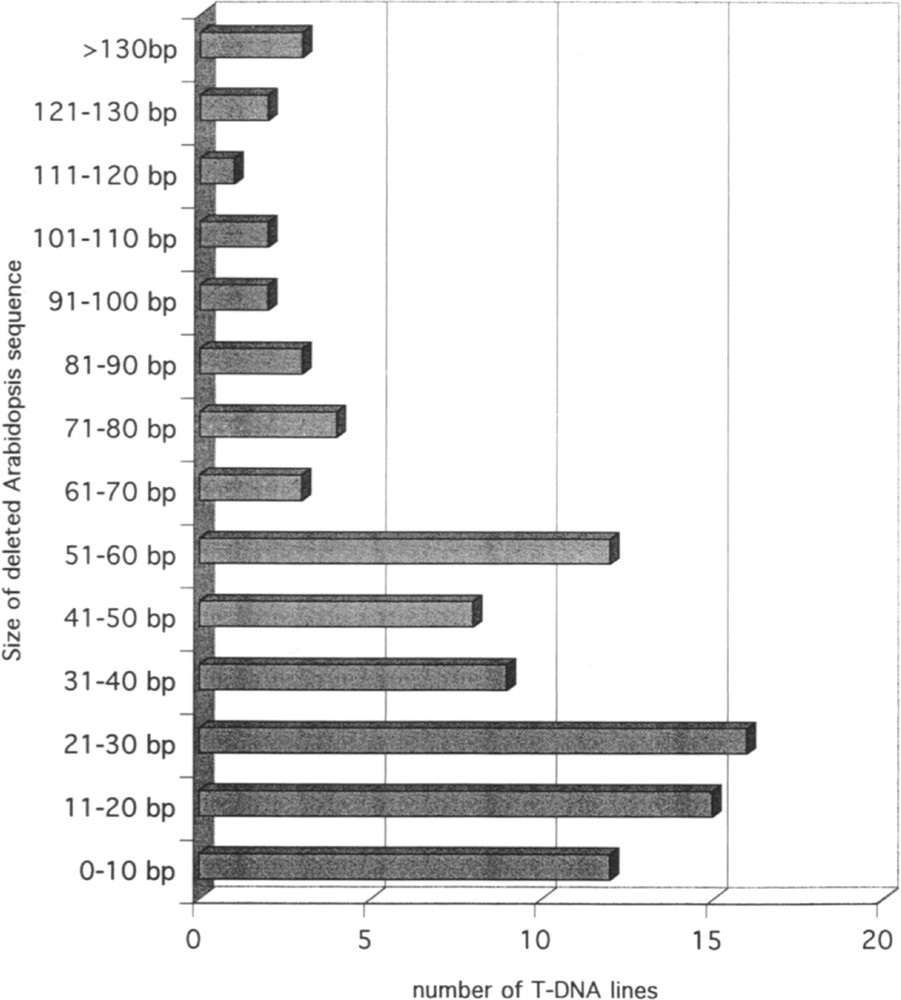
Size distribution of Arabidopsis DNA fragments deleted by T-DNA integration. The histogram based on the analysis of 98 lines gives the number of lines for each range of deletion size between the two borders of the insertion.
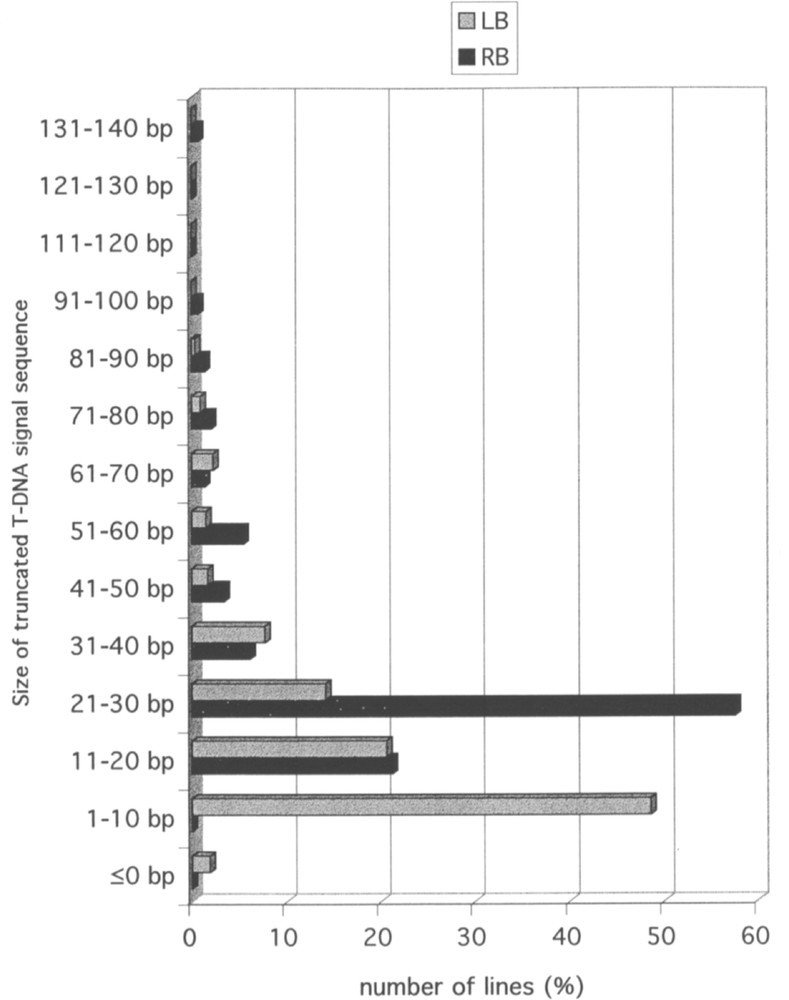
Size distribution of truncations on right (RB) and left (LB) T-DNA signal sequences. The histogram based on the analysis of 673 FSTs gives the percentage of lines for each range of truncation size (<140 bp) on the right (RB) and left (LB) borders.
3.4 Distribution of FSTs along the chromosomes
For each FST matching the genomic sequence, it was possible to assign a position along the chromosome. A map of FST positions is given in Fig. 3. Although the average distribution of FSTs is roughly the same for all chromosomes (in average 4.3 insertion/Mb for chr. 1, 5.3 for chr. 2, 5.2 for chr. 3, 5.2 for chr. 4 and 5.0 for chr. 5), there are clearly regions without, or with very few, insertions in the centromere regions. Absence of insertion in these regions is not due to insufficient sequence information, since most of these regions are now sequenced. Moreover, they are essentially composed of repetitive sequences that would be easy to recognise. On the other hand, in some regions the insertion density is higher, notably at both ends of chr. 3 and at the bottom of chr. 4.
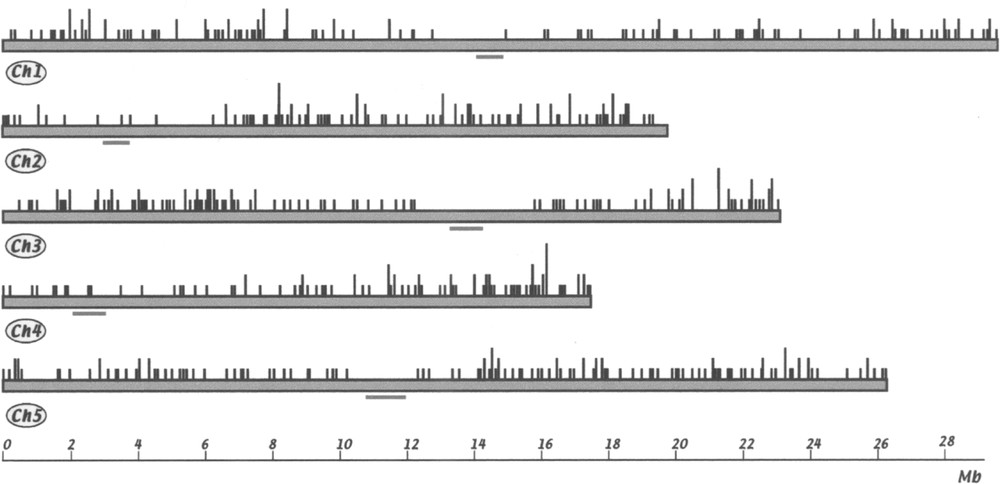
Chromosomal location of FSTs in the Arabidopsis genome. Each chromosome is represented as a bar. Centromeric regions according to 〚1〛 are underlined. The positions of 576 T-DNA insertions are shown. Vertical dark lines represent one to five FSTs located at each chromosome site (BAC or P1 clone).
4 Discussion
The data obtained in this study and those developed afterwards can be aligned on the Arabidopsis genomic sequence; when the database will be completed it will be easy to identify a mutant line containing an insertion in a given gene just following a BLAST search. With such lines in hands it will be easier to carry out more refined and selective screens than on the whole collection. It also becomes possible to examine in more details the phenotype of lines in which an orphan gene is tagged. This should considerably facilitate functional identification of a number of genes. So far we have characterised an insertion in more than 500 genes in much less time than required by previous techniques. The advantage of carrying this pilot study on lines with phenotypes is that the technique identifies immediately a number of candidate genes, because their function can explain the phenotype and allows one to discard others. This facilitates the choice of the lines to be further studied. Obviously, determination of the FST is not sufficient: the linkage of the phenotype to the T-DNA insertion should be formally established by a segregation analysis and the candidate gene should be validated by transformation and complementation of the mutant or sequencing of alleles.
Very few insertions are strictly preserving the plant genome or the T-DNA structure. Rather, in most of these cases, short deletions are observed in both the T-DNA and in the plant sequence, confirming that insertion most likely occurs through an illegitimate recombination process, which creates small deletions and insertion of some filling sequences 〚26–28〛. Recent studies indicate that in yeast, non-homologous end-joining proteins are required for Agrobacterium T-DNA integration 〚29〛 and that, in planta, the VirE2 protein is a major player 〚30〛. Our study also confirms that part of the vector is inserted at the same time as T-DNA 〚31, 32〛 at a significant frequency (32 sequences out of 851). Sequences adjacent to either LB or RB, as well as more distant vector sequences were observed. Finally, we could observe that several insertions are not simple, even though a single locus is detected by segregation analysis. In 118 lines, we could only amplify T-DNA fragments, suggesting that several T-DNA occur in tandem, either in direct (75 lines) or inverted (40 lines) orientation. In the inverted orientation most structures correspond to two internal RB (92%), in which one of the T-DNA is truncated. Finally three lines have undergone a more complex rearrangement, which prevents any obvious interpretation.
Although our experimental sample is not completely random, our results nevertheless illustrate general trends. It is possible that the frequency of insertion in a gene, or in its immediate vicinity, is higher when a phenotype is observed than in a random line. The fact that the gene coding for the selection marker has to be expressed to select a transgenic line might also introduce an additional bias in evaluating the localisation of insertion sites. However, only a fifth to a quarter of the observable mutations are linked to a T-DNA insertion, thus buffering the effect. An additional buffering effect is the very high gene density in Arabidopsis, with a gene every 4 kb in average, which leaves very little space for insertions outside of the genes or their regulatory regions. Nevertheless, we detected some differences in the distribution of FSTs within genome compartments when the selection was based on the observation of GUS expression.
Sequencing the whole collection of lines for the T-DNA flanking sequence will likely confirm the general trends. A limitation of the systematic approach is that only the simple insertions give rise to an FST and in many case only on one side of the T-DNA. Likely a number of flanking sequences are missed, but this drawback is compensated by the high throughput strategy that delivers a large number of sequences.
As a conclusion, this paper reports one of the very few studies of a large number of T-DNA insertions. It shows that the strategy of systematically analysing the flanking sequence of T-DNA is feasible on a large scale. Indeed high throughput programmes are presently developed by several groups (http://arabidopsis.org/Blast). Recently, the Genoplante consortium has made publicly available a few thousand FSTs and is in progress towards characterising the whole Versailles collection after optimisation of our strategy for high throughput production 〚22, 23〛.
Acknowledgements
The authors wish to acknowledge a joint CNRS/INRA grant supporting this programme. They wish to thank also their colleagues Lise Jouanin, Jean-Claude Davidian, Mondher Bouzayen and Magali Pichon, who together contributed by providing 24 additional putative mutant lines also investigated in this project, as well as Michel Caboche, Alain Lecharny and Loïc Lepiniec for fruitful discussions and exchanges. The help of Benoît Piegu in mining the public databases is gratefully acknowledged.
Version abrégée
1 Introduction
L’obtention de la séquence complète du génome de la plante modèle Arabidopsis thaliana permet, en théorie, de dresser un catalogue des gènes de cette espèce. Cependant, il s’avère que les programmes de prédiction de gène sont insuffisants pour définir avec certitude la structure fine de chacun d’eux et que, même une fois le catalogue dressé, la fonction d’une fraction très significative des gènes reste difficile à établir. L’une des stratégies privilégiées par la génomique fonctionnelle consiste à réaliser des collections saturées de mutants d’insertion, soit à l’aide de l’ADN-T d’Agrobacterium tumefaciens, soit à l’aide de transposons hétérologues. Ces mutants d’insertion se sont révélés particulièrement utiles pour remonter à des gènes d’intérêt identifiés par l’observation d’un phénotype. La disponibilité de la séquence de nombreux gènes permet aussi de rechercher par PCR des lignées présentant une insertion dans un gène donné. Néanmoins, ces deux dernières stratégies sont relativement lourdes à mettre en œuvre et ne renseignent que sur un nombre limité de gènes. C’est pourquoi une troisième stratégie s’est développée, consistant à isoler les séquences flanquant les insertions (FST pour Flanking Sequence Tags) et à les séquencer systématiquement. Disposant de la séquence complète du génome et d’une base de données de FST, il devient aisé de rechercher les lignées qui ont une insertion dans un gène donné. Cet article rapporte les résultats d’une étude pilote menée sur une fraction de la collection de lignées réalisée par les laboratoires de génétique et amélioration des plantes et de biologie cellulaire de l’Inra à Versailles.
2 Nombre et qualité des FST
L’étude a porté sur 850 lignées, sélectionnées dans la collection parce qu’elles présentaient un phénotype (Tableau 1). L’ADN des régions flanquantes a été amplifié et séquencé. Parmi ces lignées, 510 (soit 60%) ont fourni au moins une FST exploitable. Pour 98 d’entre elles, deux FST, en orientation inverse, correspondant au même locus, ont été obtenues ; 56 ont produit deux FST, correspondant à deux loci distincts, tandis que deux lignées ont produit trois FST, correspondant à trois loci différents. Des séquences d’ADN correspondant à des insertions complexes comprenant des ADN-T en tandem ont été obtenues pour 130 lignées. Les 210 autres lignées n’ont donné lieu à aucune séquence exploitable. La longueur de ces FST est généralement de 200 à 300 bp.
3 Caractérisation des FST
La comparaison des séquences FST avec la séquence annotée du génome a permis d’analyser leur distribution entre les différents compartiments du génome : 16,4 % sont situées dans des exons, 12,3 % dans des introns, 41,8 % sont à proximité immédiate des gènes dans une région située soit entre 0 et 1200 bp en amont du codon d’initiation, soit entre 0 et 600 bp en aval du codon stop et 27 % sont dans les régions intergéniques restantes (Tableau 2). Au total, 482 gènes distincts ont ainsi été étiquetés par une insertion ADN-T. À l’exception des lignées repérées par l’expression du gène rapporteur localisé dans l’ADN-T, la liaison entre l’insertion et le phénotype observé reste à établir et ne sera vérifiée que dans 20 % des cas en moyenne.
4 Analyse de la jonction ADN-T/séquence génomique et distribution des FST le long de chromosomes
Pour 98 lignées, deux FST ont pu être obtenues, de part et d’autre de l’insertion d’ADN-T. Quarante-six d’entre elles correspondent à une insertion complexe constituée de plusieurs ADN-T en tandem inversé. Pour la majorité de ces 98 lignées, une délétion de l’ADN génomique est observée (Fig. 1). Des délétions limitées dans les séquences signal droites (RB) et gauches (LB) (Fig. 2) sont également fréquentes pour la plupart de ces lignées.
Les FST sont réparties de façon à peu près homogène tout le long des chromosomes, à l’exception des régions centromériques et péricentromériques (Fig. 3).
Bien que les lignées n’aient pas été complètement choisies au hasard, mais sur la base de l’existence d’un phénotype, cette étude démontre ainsi la faisabilité d’une approche systématique d’isolement et de séquençage de FST et rapporte des informations préliminaires sur la distribution des FST le long des chromosomes.