

1 Introduction
Alternative splicing is one of the most important mechanisms for regulating gene function and has been implicated in many physiological phenomena including sex determination [1], sound recognition [2], neuronal path finding [3] and apoptosis [4]. Due to the apparent low number of genes in the human genome [5,6], it has been suggested that alternative splicing may be of critical relevance in expanding the human genetic repertoire, generating the complexity required for human development and homeostasis [7,8].
Variation of exon/intron structure can occur in several different ways [9,10]. Exons can be spliced out from the pre-mRNA or skipped, resulting in alternative exon usage (AEU) patterns. Alternatively, introns can be retained in the mature mRNA or cryptic donor and acceptor splicing sites can be used, generating altered exons. At the protein level, alternative splicing can have profound effects, including synthesis of a truncated protein due to the presence of a premature stop codon. Changes in splicing have been shown to determine the ligand binding activity of cell adhesion molecules and to affect the activity of transcription factors [9]. The dscam gene in Drosophila encodes an axon guidance molecule and is considered an example of the diversification a gene can attain through alternative splicing. It is estimated that through the combinatorial use of three different groups of exons, around 38 000 different protein products can be generated from the dscam gene [11].
Large sets of human gene sequences have been analyzed with the aim of estimating the degree of alternative splicing [12–15]. More recently, Modrek et al. [16] analyzed alternative splicing in the EST data from UniGene using a genome-based approach. Alternative splicing has been estimated to occur in at least 1/3 of all human genes.
In this report, we present a comparison of a large-scale analysis of AEU within the human transcriptome by the genome mapping of Open Reading Frame ESTs (ORESTES) generated by the Human Cancer Genome Project [17] and sequences generated by other techniques (5′ and 3′ ESTs). ORESTES are expressed sequence tags generated by low-stringency PCR and map preferentially in the central part of the messages. There is also a bias towards low abundance transcripts in the set of sequences generated by ORESTES [17,18].
It is expected that the ORESTES dataset would be enriched with rare splicing variants that affect the structure of the corresponding protein. In this report we present data that support this notion. We found that clusters that represent genes of low expression, as quantified by Serial Analysis of Gene Expression (SAGE) [19,20], contain more ORESTES than other ESTs. In addition, less ORESTES are required to detect splicing variants and the ORESTES set is shown to be more effective in detecting AEU events within the coding region of the corresponding genes.
2 Results
2.1 Representation of AEU
A critical issue in studies involving alternative splicing is the representation of variants. In general, alternative splicing patterns are depicted by multiple alignments of transcripts. These alignments, however, are usually long and do not provide a concise, schematic view of transcript variability. The design of a topological structure graph represents an alternative that allows the use of graph theories for the study of alternative splicing patterns but remains relatively complex. We suggest a different simple form of representing the exon usage for all transcripts derived from a gene that takes the form of a binary matrix where each column represents an exon and each row represents a sequence in a given cluster (see Methods). A matrix representing a hypothetical cluster is shown in Fig. 1 where a given cell in the matrix is numbered 1 if the corresponding exon is represented in the corresponding sequence and the cell is numbered 0 if the exon is skipped.
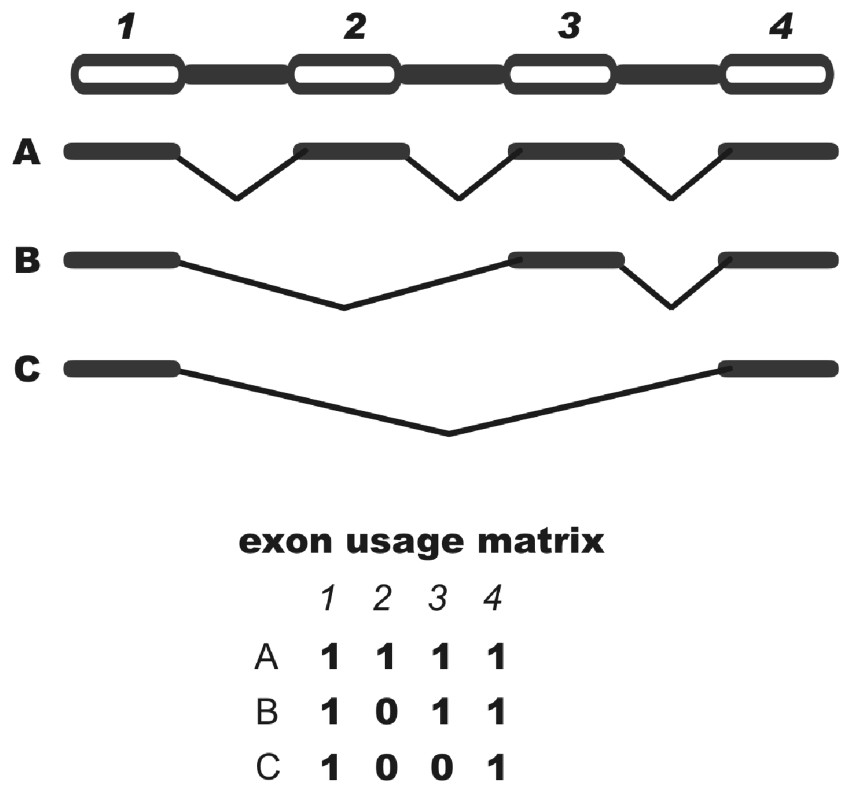
Schematic view of a binary matrix. Exons are represented by columns while sequences are represented by rows. When present, an exon is annotated as 1 and when absent as 0. Exon skipping events can be detected by searching for 10+1.
There are advantages of such matrices over other forms of representation of splicing variants: (i) their interpretation is straightforward, (ii) they are a starting point for many statistical analyses, (iii) they are suitable for cluster analysis, with the goal of identifying all unique forms of exon usage and (iv) they are easy to parse and therefore appropriate for large-scale computational studies. A shortcoming of this matrix approach, at least in its present form, is that the identification of other types of alternative splicing (e.g., the use of cryptic splicing sites) is not possible. However, AEU corresponds to approximately half of the cases of alternative splicing [16].
2.2 Detection of splicing variants
A binary matrix was generated for every cDNA cluster mapped in the human genome (see Methods for a description of the genome-based clustering strategy). This clustering strategy generated a non-redundant set of clusters containing all mapped cDNAs. A relational database was created in which the source of each cDNA was annotated. A list of all genes and the corresponding matrices are available at http://www.ludwig.org.br/AEU.
Alternative splicing events in different parts of the same gene may not be independent. In this analysis, however, we combined all events independently even when no single cDNA supported the full exon/intron structure. This may have inflated the total number of splicing variants found without affecting the number of known genes that express more than one splicing variant. Detection of splicing variants is described in the Methods section. Each variant represents a set of redundant sequences.
We analyzed cDNA clusters containing at least one cDNA sequence derived from a known human gene (called here full-insert cDNAs), corresponding to a non-redundant set of 17 553 such clusters. Eight hundred and sixteen (816) clusters contained only full-insert cDNAs. The remaining 16 737 clusters and their corresponding matrices, containing ORESTES and other ESTs, were used for the analyses described. We considered ORESTES and other ESTs separately as presented in Table 1.
Overall statistics for the comparative exon usage analysis
| Other ESTs | ORESTES | |
| clusters w/full-insert cDNAs | 16 737 | 13 384 |
| clusters w/full-insert cDNAs and | ||
| with >2 variants | 8133 | 2989 |
| AEU events in 370 000 sequences | ||
| normalized by length | 7463 (5′) | 4808 |
| 3666 (3′) |
The difference in the number of clusters with more than two variants can be explained by the fact that the total number of ORESTES sequences is about six-fold lower than that of all other ESTs. The numbers of AEU events counted in a non-redundant set of variants sampled from an equal number of sequences of ORESTES, 5′ and 3′ ESTs, normalized by length, are shown in Table 1. More events were found for 5′ ESTs, followed by ORESTES.
2.3 Distribution of AEU events within transcripts
Since ORESTES are preferentially mapped in the central part of a transcript, it is expected that most of the AEU events represented by these sequences would affect the structure of the corresponding protein. We found that 85% of AEU events mapped by ORESTES variants are located within the coding region. This frequency is statistically significantly higher (P<0.001) than that observed employing conventional ESTs only (77%) (Table 2). When we compared ORESTES to 5′ and 3′ ESTs separately, we found that ORESTES are more likely to report an event affecting the respective protein (75% for 5′ ESTs, P<0.001 and 82% for 3′ ESTs, P=0.001).
Number of AEU events in the coding region (CDS) and in the untranslated region (UTR) of full-insert cDNAs mapped by ORESTES and other ESTs
| Other ESTs | ORESTES | |
| events in the CDS | 9840 (77%) | 2152 (85%) |
| events in the UTR | 3035 (23%) | 374 (15%) |
2.4 Variants represented exclusively by ORESTES sequences
We found that several variants were exclusively represented by either ORESTES or conventional ESTs. Since the dataset of conventional ESTs is much larger than that of the ORESTES, we performed a comparison of all ORESTES variants with a random pool of conventional ESTs containing the same number of variants as the total set of ORESTES (17 745). In this simulation, about 40% of the variants were found to be exclusively represented by a single type of expressed sequence, indicating that ORESTES contribute to transcriptome coverage by detecting novel variants. However, a more quantitative assessment of the ORESTES contribution depends on the availability of more sequence data.
2.5 Rare transcript variants are represented within the ORESTES dataset
The normalization capacity of the ORESTES technology [17] and the fact that most of the splicing variants comprise rare transcripts lead to the supposition that the ORESTES dataset should be enriched with rare transcripts.
Virtual SAGE tags were generated for the 16 737 full-insert cDNAs (mRNA from known genes) [20] from the clusters containing ORESTES and other ESTs and their frequencies in all SAGE libraries were counted [20]. Although SAGE is an excellent tool for gene expression analysis, low abundance genes may be under-represented. Tags for 13% of all full-insert cDNAs were not present in any SAGE library. These lowly expressed genes were represented by 10 536 ORESTES (out of 100 000 sequences). The same clusters were represented by a lower number of other ESTs (8591 out of 100 000 EST sequences).
Fig. 2 shows a region of a plot of the normalized number of cDNA sequences as a function of SAGE tag frequency. Lower SAGE tag frequencies correlate with a higher number of ORESTES than conventional ESTs.
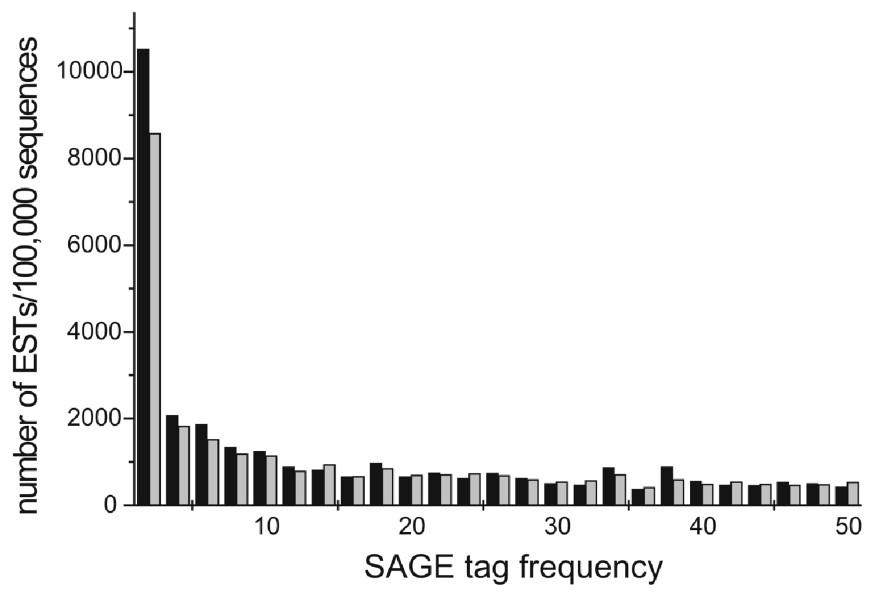
Normalized number of expressed sequences in clusters as a function of SAGE tag frequency (in classes of 2). Low expression clusters are more populated by ORESTES than by other ESTs. Black bars: ORESTES, grey bars: other ESTS. Only SAGE tag frequencies up to 50 were plotted for the sake of clarity.
Velculescu and collaborators [21] observed that genes represented by ⩽5 SAGE tags corresponded to 127 342 transcripts accounting for 25% of the total mRNA mass (low expression) while genes represented by ⩾500 SAGE tags corresponded to 55 transcripts representing 18% of total mRNA mass (high expression). In our database, clusters that presented SAGE tag frequencies ⩽5 contain a total of 14 485 ORESTES (out of 100 000 sequences) and only 11 947 conventional ESTs (out of 100 000) (Table 3). This difference (P⩽0.001) supports the concept that ORESTES map to rare transcript variants more efficiently than other ESTs.
3 Discussion
The accumulation of both cDNA and genomic sequence data has become sufficiently large so to allow its utilization in large-scale analyses of alternative splicing in the human genome. However, one limitation of the EST data is the intrinsic bias towards the 3′ end of the transcripts. Since these regions of the transcripts have a lower frequency of introns [22] and alternative splicing [12], these ESTs are not expected to be enriched with splicing variants. One alternative is the collection of ORESTES sequences, which are biased toward the central region of messages [18]. Another feature of the ORESTES sequences is a bias towards less abundant transcripts [17,18]. Based on these two features it is reasonable to suppose that the ORESTES set contains a higher number of splicing variants when compared to ESTs from different sources. We have already characterized a variant of the nabc1 gene represented exclusively by an ORESTES that is down-regulated in colon tumors [23].
In this report we presented a genome-wide analysis of alternative exon usage in the human transcriptome using the collection of ORESTES generated within the FAPESP/LICR-Human Cancer Genome Project [17]. Since around half of all splicing variants presents AEU [16] it is reasonable to suppose that a large-scale analysis of this type of alternative splicing is important and informative. Our approach has been centered on the generation of a binary matrix built for every cDNA cluster obtained through a genome-based clustering strategy. It is well known that problems may exist in large-scale bioinformatics based studies of alternative splicing [24]. The use of both cDNA and genome data excludes artifacts from our analysis that are intrinsic to approaches that are based solely on cDNA data.
The distribution of events along the transcripts for conventional ESTs is in accord with previous work (all types of alternative splicing) [16]. A comparison among ORESTES, 5′ and 3′ ESTs, showed a higher number of AEU events for 5′ ESTs, followed by ORESTES. The higher number of events detected by 5′ ESTs is probably due to a higher rate of alternative splicing in the 5′ half of transcripts, as previously reported [16]. On the other hand, ORESTES probed a higher number of events affecting the encoded protein. Eighty-five percent and 75% of events detected by ORESTES and 5′ ESTs, respectively, affected the coding region. For ORESTES, only 13 and 2% of all splicing events occur within the 5′ and 3′ UTRs, respectively.
Our analysis also suggests that the ORESTES dataset is enriched with rare splicing variants as evaluated by the number of sequences present in clusters represented by ⩽5 SAGE tags when compared to the remaining ESTs. Also, the ORESTES set revealed variants not detected by other ESTs.
In summary, we found that the collection of ORESTES is enriched with rare splicing variants affecting the structure of the encoded protein. This makes this technology a powerful platform for exhaustive screening of the variability found within the human transcriptome.
4 Methods
4.1 Data sources
Complete human genomic sequences (build 29) were obtained from the NCBI. Human ESTs were obtained from human dbEST (July 2002) [25] and mRNA sequences derived from known human genes (called here full-insert cDNAs) were obtained from UniGene (release 153) [26]. The ORESTES collection is almost entirely available in dbEST. A few ORESTES that have not met the quality criteria to be submitted to GenBank were also used in this analysis. A complete set of ORESTES used in this work is available at http://www.ludwig.org.br/AEU.
4.2 Genome mapping of cDNAs
Masking of genomic contigs provided by NCBI was used. MEGABLAST [27] was used to align pairs of genomic and transcribed sequences. Only pairs that aligned over at least 45% of total sequence length and with exons presenting more than 93% identity were considered.
4.3 cDNA clustering
cDNA clusters were generated on the basis of the coordinates of cDNA alignments to human genomic sequences. Two sequences were clustered together if they presented at least one exon presenting a common exon/intron boundary (allowing ±5 bp of difference). Sequences that did not present introns had to overlap at least 100 bp of another sequence in the cluster to be grouped together.
4.4 Alternative splicing
For each cluster, exons were defined by cDNA alignment with the genome using only cDNAs that span at least two exons. Sequences were represented by a binary matrix where each column corresponds to an exon. If the sequence presented a given exon mapped in the cluster, the corresponding position was assigned 1, on the contrary 0. AEU events could be detected by searching for 10+1 in the matrices, where+means at least one absent exon (Fig. 1). Sequences of a given cluster bearing the same exon usage pattern, as represented by the binary matrices, were grouped in one representative variant.
4.5 Analysis of AEU events within transcripts
Reference full-insert mRNAs were elected for each cluster according to the following criteria: (i) should be the longest available, ideally covering the whole cluster and (ii) have annotated coding region coordinates starting at least 100 bp from the beginning of the mRNA. The position of an AEU event (10+1 in the matrices) were considered to be that of the starting coordinate of the exon skipping (in the reference full-insert mRNA).
4.6 Serial analysis of gene expression tag frequency count
Virtual SAGE tags for 16 737 full-insert mRNAs (all clusters containing ORESTES and conventional ESTs) were generated [20] and counted in all SAGE libraries available. The numbers of expressed sequences (ORESTES or other ESTs) were counted in each cluster. Total sequences in clusters of the same SAGE tag frequency were summed and normalized by the total number of sequences available (370 149 ORESTES and 2 160 400 5′ and 3′ ESTs in 16 737 clusters of a total of 17 553). Statistics of data in Table 3 were performed with raw data. Normalized data is presented only for clarity.
4.7 Number of AEU events
Equal numbers of sequences with mean length of 310 nt (the mean of the ORESTES dataset) were randomly picked from separate sets of ORESTES, 5′ and 3′ ESTs. The non-redundant set of variants representing these sequences was determined and the number of AEU events counted. We performed this simulation with increasing numbers of sequences until the total number of the smallest set (370 000 ORESTES) was achieved.
4.8 Statistical tests
Homogeneity χ2 tests were performed to measure significance of difference between classes.
Acknowledgments
This work was supported by fellowships from FAPESP (F.P., M.L., N.J.S) and CNPq (P.A.F.G). Thanks to Maria Vibranovski and Natanja Kirschbaum for careful revision of the manuscript and to Guilherme Marson for generating the figures.