

1 Introduction
The working draft of the human genome released in June of 2000 yielded a comprehensive view of the 3-billion-base sequence of the human genetic-blueprint. One of the most striking features observed was the less than anticipated number of genes (estimated by various computational predictions to be around 30 000–40 000) encoded by a very small fraction (∼1–2%) of the vast genome. Consequently to this, a number of processes, including alternate splicing and domain sharing, have been invoked to explain the intricate complexity and diversity of human gene expression. However, the precise number of genes and true mechanisms of transcriptional diversity remain unknown at the current time. The first step in elucidating this calls for a systematic cataloging of all of the human genes and their various coding forms. This has resulted in a number of full-length (FL) cDNA sequencing initiatives for human [1] and mouse [2,3].
In order to carry out very large-scale cDNA sequencing, we developed a strategy, concatenation cDNA sequencing (CCS), for simultaneous batch sequencing of cDNA clones largely through shotgun reads [4]. This strategy was subsequently applied toward the batch sequencing of 69 human brain cDNA clones as one shotgun library [5]. The method was then modified to that shown in Fig. 1. Using this approach cDNA inserts are isolated by restriction enzyme digestion followed by gel purification. Equal molar amounts of the DNA from 50–100 cDNA inserts are concatenated through ligation, followed by the construction of a shotgun library from the ligation product and random shotgun sequencing. This strategy has been generally very successful and enables many of the cDNA clones in a pool to be finished through random shotgun reads.
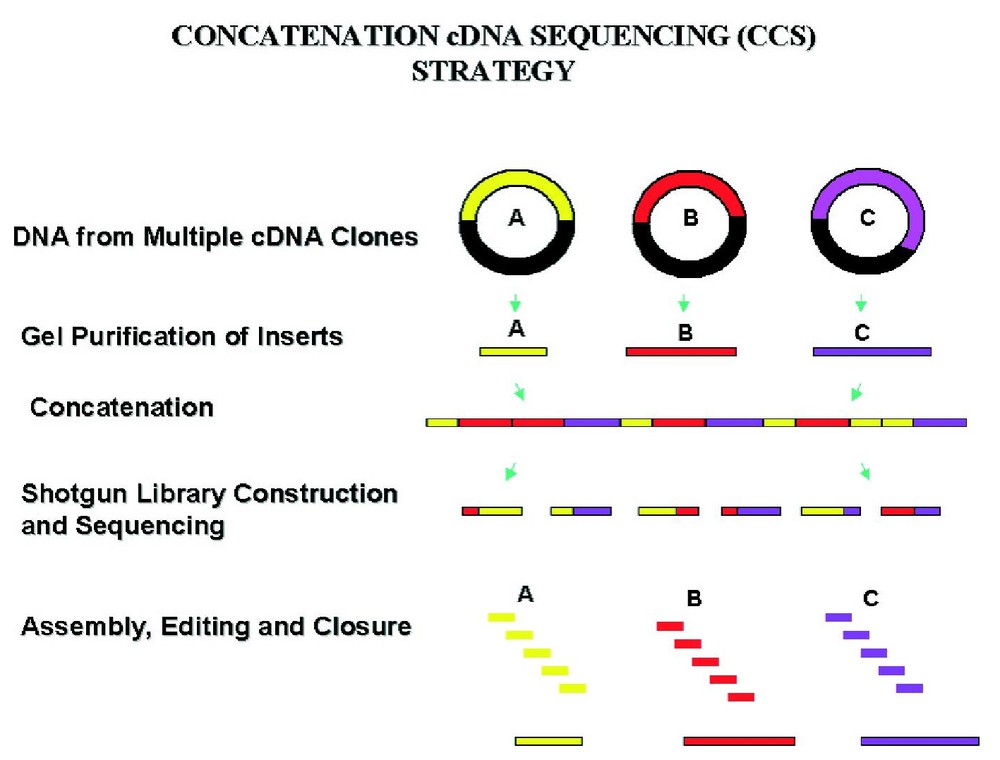
A representation of the concatenation cDNA sequencing strategy.
In this manuscript, we describe subsequent key technical and strategic modifications to the CCS procedure that have enabled the establishment of a high-throughput cDNA sequencing and finishing pipeline at the Baylor HGSC. This pipeline has been used successfully to feed the comprehensive FL-cDNA sequencing initiative by the NIH mammalian gene collection (MGC: http://mgc.nci.nih.gov) program [6], as well as a number of other projects. We have established that the CCS strategy is an efficient alternative to other commonly used cDNA sequencing approaches such as sequential deletion [7], primer walking [8], and transposon insertion [9,10] sequencing.
2 Results
2.1 Factors influencing the efficiency of CCS libraries during the scale up
We found that the most critical aspect impacting the scale up of the procedure was accurate quantitation of individual cDNA inserts to ensure near equal representation within a CCS pool. The efficacy of CCS libraries as measured by the percentage of clones sequenced to completion through shotgun reads (auto-finish rate) was directly impacted by even clone representation. Implementation of the modifications shown in Fig. 2 improved the average auto-finish rate from ∼48 to ∼66% with some CCS libraries finishing 80–90% of the clones through random shotgun reads in the initial round. The first modification involved a significant reduction in the range of cDNA insert sizes from 1 kb to 100 bp within a pool. The second change was a decrease in the average number of clones per pool and therefore the reduction of the average size of the concatenation product from ∼100 kb down to 50–75 kb. An increase in the robustness of the DNA preps was achieved by switching to a commercial kit from LTI-Iinvitrogen modified here for large scale DNA preparation in a 96-well format. Collectively these changes resulted in improvements in CCS library quality, yield and an increase in auto-finish rates from 48 to 57%. In contrast, the gel purification step appeared to be less sensitive, having little effect on library quality, sequencing success and auto-finish percentage (data not shown). Inserts purified using Qiagen columns and Geneclean fared equally efficiently in the concatenation reaction and variations in the time of exposure of long-wave UV during gel excision of cDNA inserts did not appear to significantly impact library quality. Two other factors which did influence the efficacy of CCS libraries included the elimination of the phenol extraction step used to remove the ligase prior to nebulization of the CCS product and an increase in the number of attempted reads per library from 15 reads per kb to 22 reads per kb. These changes resulted in a further increase in the auto-finish rate from 57 to 66%.
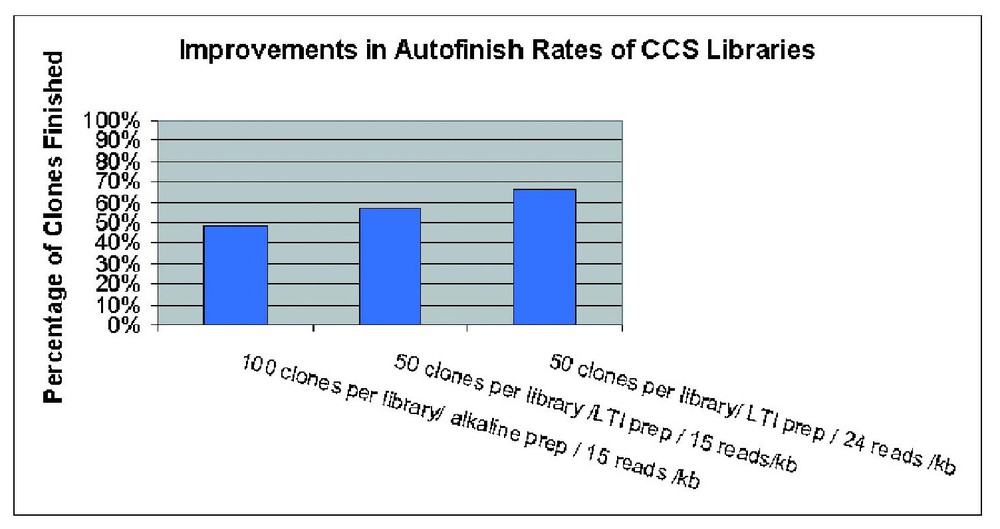
Incremental improvements in efficiency of CCS libraries with changes in the CCS protocol.
2.2 A PCR rescue format for problem clones
Approximately 7% of the cDNA clones from a batch of 6144 were not able to be successfully processed through CCS pipeline. The problems included: (i) a low yield due to inefficient growth; (ii) failed restriction digestion possibly due to the loss of the restriction site because of a mutation; and (iii) insert in the size range of the vector (∼4 kb). The majority of the problem clones we analyzed exhibited growth retardation (∼60%). Using primers directed toward vector sequence, we amplified the cDNA inserts via PCR directly from the bacterial cells or in some cases from the small amounts of DNA isolated from LTI preps. Results from the PCR rescue are shown in Fig. 3. Out of a 136 problem clones we were able to obtain single robust PCR products from 112 (82%). The rescued PCR products were restriction digested, gel purified and pooled into five CCS libraries (Fig. 4C). Approximately 36% of the clones were finished through shotgun reads with another 49% finishing with minimal primer walking, and 15% were subjected to an additional round of CCS due to initial under-representation of reads.

PCR rescue of problematic cDNA clones showing poor growth. A. PCR products obtained from 18 PCR cycles. B. Products obtained from 28 PCR cycles performed on samples that failed to yield products after 18 cycles. C. Concatenated PCR products following restriction digestion and ligation.

The automated data management pipeline for assembly and closure of multiple cDNA sequences in the CCS pool.
2.3 High-throughput finishing of cDNA clones
The finishing strategy was also designed to increase throughput while maintaining high quality (cumulative error bases). The flow chart of the finishing process is shown in Fig. 4. Clones are finished using Consed Autofinish by retaining all of the clones from a single concatenation (CCS) in a single PHRAP assembly. Individual clones are finished as individual contigs by performing electronic digests on the random shotgun reads at the restriction site sequences used to isolate cDNA inserts (Fig. 4). This is achieved by masking sequences on either side of restriction sites. Chimeric reads joined at a restriction site are therefore handled as two separate reads in order to prevent co-assembly of sequences from two independent cDNA clones. EST reads performed on individaul cDNA clones are used to link individual sequence contigs to the respective cDNA clones. Finished sequences are processed by an automated three-step quality control program. The first program calculates the cumulative error for each clone. If the clone passes the MGC standards of <1 error/50 000 bases, a file is generated for the purpose of submitting the sequence. Clones, which fail the error-check are flagged and re-examined in Consed for editing regions of low quality are resolved by primer walking in order to decrease the cumulative error to an acceptable range. The second program, which determines the number of manually edited bases per sequence, is designed to monitor excessive editing of the sequence by the assembler. Excessive manual editing is indicated, the sequence is re-checked in Consed by an independent human assembler to confirm the edits. The third and final checkpoint is achieved through an EST BLAST step, which confirms that the cDNA sequence matches an EST sequence generated from the same clone by an independent source (MGC).
Clones that do not meet the finishing standards but have good representation in terms of shotgun reads are processed by an automated primer design process to resolve both gaps and low quality regions. Primer walks are performed in a 96-well format. Clones which are sparsely represented by shotgun reads (∼10%) in the assembly are returned for a second round of CCS with the expectation that they may be sequenced more efficiently with a different combination of clones. In general, the second round CCS libraries yield an auto-finish rate of approximately 45%, with another 30% requiring 1–2 primer walks, and the same number remaining in the under-represented back to CCS category. Clones, which fail to finish after three rounds of CCS are processed by PCR rescue.
2.4 An annotation tool for the batch analysis of cDNA clones
We carried out an informatic analysis of 5000 human cDNA clones sequenced to completion by the MGC. The results were used to refine the parameters for setting up an automated cDNA analysis tool for the Baylor-HGSC cDNA sequencing and finishing pipeline. From the results shown in Fig. 5, it appears that the extent of the cDNA sequence which is aligned on the unfinished genome (percent clone coverage), and the number of mismatches allowed are important parameters to be considered when aligning the cDNA sequence with the genome. This graph shows the majority of clones will align with their genomic source at 98% clone coverage and 1–6 mismatches per clone. Relaxing the stringency further to 90% clone coverage and >20 mismatches per clone appear to allow the identification of related sequences (pseudogenes, gene families etc.) associated with a specific cDNA. The vast majority of true genes also appear to be aligning the genome in multiple blocks representing exons. The small percentage of cDNA clones aligning the genome as a single block merit further investigation to distinguish pseudogene hits from single exon gene hits.
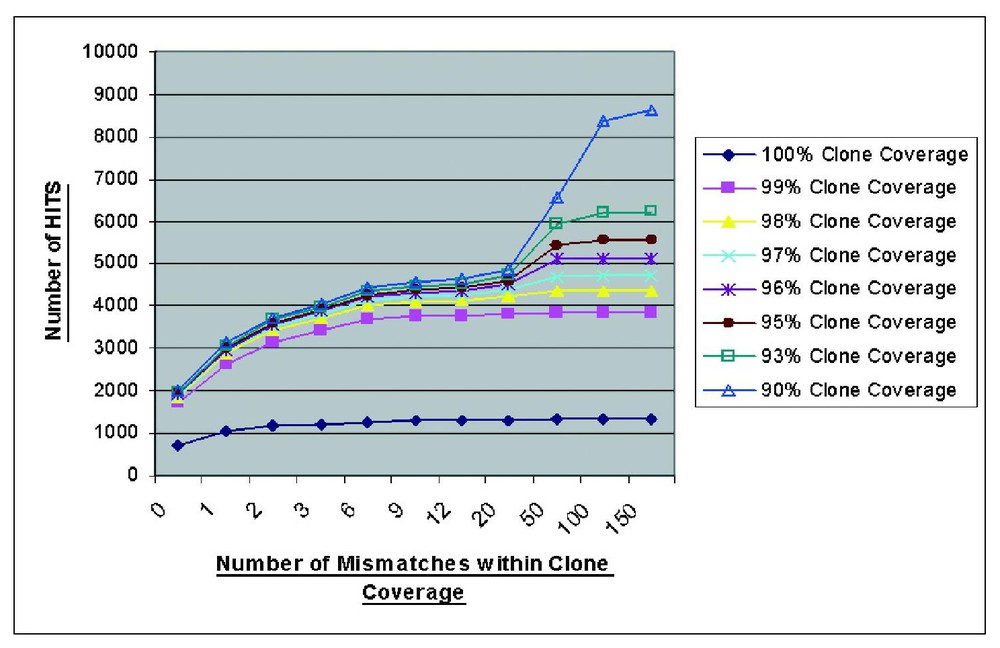
Results from a BLAT analysis of ∼5000 MGC sequences in relation to the genome. Criteria developed from this graph were used to design the annotation pipeline.
2.5 A cDNA sequencing and annotation pipeline
Based on these observations, a cDNA analysis and annotation pipeline has been established and analyze finished cDNA clones in large batches using BLAT analysis against the latest version of the genome build. This pipeline includes several filters, including a stringent first pass filter to identify true gene hits (⩾98% clone coverage, ⩽1 mismatch, multiple block alignment and >50 bp between blocks (or exons)). A less-stringent second pass filter using the criteria ⩾90% clones coverage and >20 mismatches will be used to identify related sequences in the genome with a view to identifying pseudogenes and/or multigene families associated with a specific cDNA clone. Single block hits and cDNAs with multiple hits at high stringency will be analyzed in greater detail searching the top five alignments to distinguish single exon genes from pseudogenes and recently duplicated gene families.
3 Discussion
In this manuscript we present a modifications to improve the previous format for the Baylor-HGSC high-throughput full-insert cDNA sequencing and characterization pipeline. The results and methods presented here represent significant improvements in the volume and accuracy of the prototype CCS pipeline presented at the original Transcriptome 2000 meeting. The pipeline has also evolved to allow characterization of the cDNA clones in terms of their true genomic source, related sequences including pseudogenes and multigene families, various structural and functional homologues and gene structure information including exon/intron structure, best ORF, 5′ and 3′ UTRs and poly A signals. Other features of interest such as GC content, simple repeat distribution and content can easily be extracted.
4 Methods
4.1 Processing of cDNA clones for concatenation and preparation of CCS libraries
4.1.1 Insert preparation
Individual cDNA clones are inoculated into 625 μl of TB medium with ampicillin () and grown in 96-well Beckman growth boxes at 37 °C, 300 rpm for 18 h. Culture boxes are inspected for growth and centrifuged to pellet the cells. DNA is prepared in a 96-well format using the Life Technologies kit (LTI-Invitrogen). The cell pellet is gently vortexed and resuspended in 260 μl of lysis buffer containing RNaseA. 310 μl of the wells contents (cell culture plus the lysis solution) are filtered through the Life Tech filter plate. The filter plate is transferred on to the corresponding ethanol-filled receiver plate. The filter/receiver plate assembly is centrifuged at 3600 rpm for 15 min at 4 °C. The filter plate is discarded and 350 μl of 70% ethanol is added to the wells of the receiver plate and centrifuged at 3600 rpm for 5 minutes. The DNA is resuspended in 40 μl of TE and a small sample is test digested to determine insert size. Insert sizes are assessed using Kodak Digital Software. Preparative digests are performed on 36 μl of the DNA sample in a 96-well format. The choice of restriction enzymes is dependent on the clones being processed. EcoRI, NotI, KpnI and SalI were commonly used to separate the MGC inserts.
4.1.2 Pooling and concatenation
Approximately 50–75 cDNA inserts within a size range of 100 bp are pooled and electrophoresed in a 1.25% agarose gel. Pooled inserts are purified using standard Qiagen or Geneclean procedures. Purified insert pools are resuspended in 36.5 μl of HPLC water and ligated in a final volume of 50 μl using 5 μl T4 Ligase Buffer w/ATP (New England Biolabs) and 8.5 μl T4 DNA Ligase at (New England Biolabs). The ligation product is sheared through nebulization using a Quick Spin Nebulizer at 2500–3000 rpm for 40 s at 10 psi. The sheared DNA is end repaired using 20U T4 DNA polymerase (Boehringer) and 12.5U Klenow (Boehringer sequencing grade) by incubation for 10 min at room temperature and 2 h at 16 °C. The resulting product is purified using a Qiagen PCR column and cloned into a pUC18-based vector using the double-adaptor cloning method described by Andersson et al. (1996) [11].
4.1.3 Sequencing
Sequencing is performed using a 1/8th sequencing reaction of Big Dye Terminator Version 3.0. Reactions are performed on Packard mini-track robots in a 384-well format. Sequences are assembled using PHRAP and viewed using Consed Autofinish. Finished clones, which pass the three-step checkpoint program to check for cumulative error, number of manual edits and a confirmatory EST are submitted to MGC.
4.2 PCR rescue procedure
Problem clones are picked from the original 384 well plates into 96 well deep-well growth boxes and grown overnight at 37 °C, 300 rpm for 18 h. Of the total growth culture of 625 μl, 500 μl is used to isolate the DNA insert with the LTI-Invitrogen kit and 125 μl is spun down to produce the cell prep. PCR reactions are performed on DNA (1:70 dilution) or directly on cells (1:25 dilution) using Forward and Reverse sequencing primers from the vector. PCR reactions are set up using 2 μl of sample, 4 pmol of Forward Primer (5′ GTAAACGACGGCCAGT 3′) and 4 pmol of Reverse Primer (5′ CAGGAAACAGCTATGAC 3′), 2.5 mM dNTP and 2.5 U of Amplitaq. PCR is carried out with an initial denaturation step at 94 °C for 7 min, and 17 to 27 cycles of 94 °C for 1 min, 58 °C for 30 s, 69 °C for 3 min. This is followed by a single extension step at 69 °C for 7 minutes. PCR products are examined on a 1% agarose gel. The clones that exhibited sharp, bright single bands are re-arrayed onto 96 well plates. The amplified PCR products are grouped based on size, digested in a pool and concatenated as described above.
Acknowledgements
We wish to acknowledge the contributions of the Baylor–HGSC. Ryan Martin, Hermela Loulseged, Carla Kowis, Anna Sneed, Amit Nanavati, Xiuhua Lu, Seema Nair, Vivienne Yoon, Sarah Hale, Laura Gay, Natalie Walsham. Donna Muzny, Erica Sodergren, David Steffen, Graham Scott are also acknowledged. This work was supported by NCI grants 20X182A, 5 UO1 CA 80200 and DOE grant DE-FG 03-97ER62375.