

1 Introduction
In the human genome project, the sequence information of transcripts played an important role supplementing the information of the genome sequence. Expressed sequence tags (EST) [1] were widely used for annotation of the genome sequence and acquisition of the basic transcriptome information, such as identification of genes, their splicing patterns, estimation of the total number of genes and a rough estimate of expression patterns. The IMAGE cDNA clones from which the majority of EST data were acquired served as the indispensable resource for the analysis of the gene structure and its function.
In recent years, more stress was put on the full-length cDNAs. One reason is that EST data are now reaching saturation (about 5 million human ESTs in dbEST for human). The other reason is that the realization of the high degree of incomplete cDNAs in the database, which sometimes hinders the analyses rather than facilitate them. The incomplete cDNAs are generated because of the low processivity of the reverse transcriptase (RT), as well as the breakdown of mRNAs during their isolation and the cDNA synthesis. So far, the effort to increase the processivity of RT has met limited success.
The 5′-end of eukaryotic mRNA has a special structure called ‘cap’ [2]. The cap is a 7-methylated GTP attached to the first nucleotide of the mRNA through two pyrophosphates. Now there are several methods to use this cap structure as the target for selecting the full-length cDNA. For example, Edery et al. devised the “Cap Retention Procedure” and used it to make full length-enriched and 5′-end enriched cDNA libraries [3]. Carninci et al. also developed the ‘CAP Trapper’ method and made full length-enriched cDNA libraries [4]. We also developed the so called ‘oligo-capping’ method [5] and used it to make a full length-enriched and a 5′-end enriched cDNA libraries [6]. Although the fraction of full-length clones within a cDNA library varies from library to library, it is 10–20% in most of the cases using conventional methods. Using various cap-based selection methods, this ratio goes up, at least, to 40–50% range and in some cases up to 90%.
These full-length cDNA clones not only serve as the resource for the functional analysis but also give precious information such as transcriptional start sites. We recently set up a special database called DataBase of Transcriptional Start Sites (DBTSS; http://dbtss.hgc.jp) [7]. Using this database, one can identify the core promoter regions and representative 5′ untranslated regions (UTRs). Here, we describe our analysis on the representative 5′ UTR of 4870 genes.
2 Materials and methods
2.1 Oligo-capping method
The oligo-capping method was described in detail [5,6]. This method consists of three steps, (i) removing 5′ phosphates of non-capped RNAs with alkaline phosphatase, (ii) removing the cap with tobacco acid pyrophosphatase (TAP) and (iii) ligating oligoribonucleotides (r-oligos) to decapped mRNAs with T4 RNA ligase. The capped ends of the mRNAs are specifically labeled with a synthetic r-oligo by this method. Such 5′ labeled mRNAs are used for construction of full-length cDNA libraries and 5′ end enriched cDNA libraries as well as the identification of the transcriptional start point of individual genes. We made 132 such libraries so far.
2.2 Full-length cDNA dataset
The detail of the data processing was described [7]. In brief, 5′-end sequences (about 200,000) were compared with human reference sequences (RefSeq) using the BLAST program [8]. Matched sequences were mapped onto the human genome working draft sequence (Golden Path: http://genome.ucsc.edu/) database using the sim4 program [9]. The sequence data were clustered using this mapped data and their 5′-end were compared with that of original RefSeq data. 4870 RefSeq sequences could be extended towards the 5′-end. Of those, the most frequent was selected as the representative for each gene. The full-length cDNA data with the added 5′-end sequences were used in this study. This full-length cDNA dataset can be downloaded from DBTSS (http://dbtss.hgc.jp).
3 Results and discussion
Using the latest version of DBTSS data, we could extend the 5′-end of 4870 RefSeq entries and obtain a representative full-length cDNA dataset for the corresponding 4870 genes. This dataset was used to analyze 5′ UTR and the small open reading frames (ORFs) within the region. Since the data of experimentally validated ‘initiator’ ATG codonswere not available for all the 4870 RefSeq entries, we decided to use the ATG that starts the longest ORF as the ‘initiator’ ATG for this dataset. The length distribution of the 4870 full-length cDNAs and their longest ORFs are similar to that of the original RefSeq entries (data not shown), because the average length difference between this dataset and the original RefSeq entries is only about 40 bases at their 5′-end.
We tested whether there is any ATG upstream of the ‘initiator’ ATG using this 4870 full-length cDNA data set. In a previous report, we analyzed the 5′ UTR region of 1010 genes and found that about 30% of them had at least one ATG in the 5′ UTR upstream to the ‘initiator’ ATG [10]. To our surprise, we found about half (2386 out of 4870) of our dataset had at least one ATG upstream of the ‘initiator’ ATG. In Fig. 1, we show the length distribution of 5′ UTRs measured from the ‘initiator’ ATG (Fig. 1A), from the ‘initiator’ ATG that is also the first ATG (Fig. 1B) and from the ‘initiator’ ATG that is not the first ATG (Fig. 1C). The distribution shows a sharp peak for the 5′ UTRs measured from first and ‘initiator’ ATG with the average of 120 bases. In contrast, 5′ UTRs measured from the ‘initiator’ ATG that is not the first ATG show a broad peak with an average of 330 bases. Thus, there seems to be two types of mRNAs, one with a long 5′ UTR and the other with a short UTR. The long ones tend to have an ATG upstream the ‘initiator’ ATG.
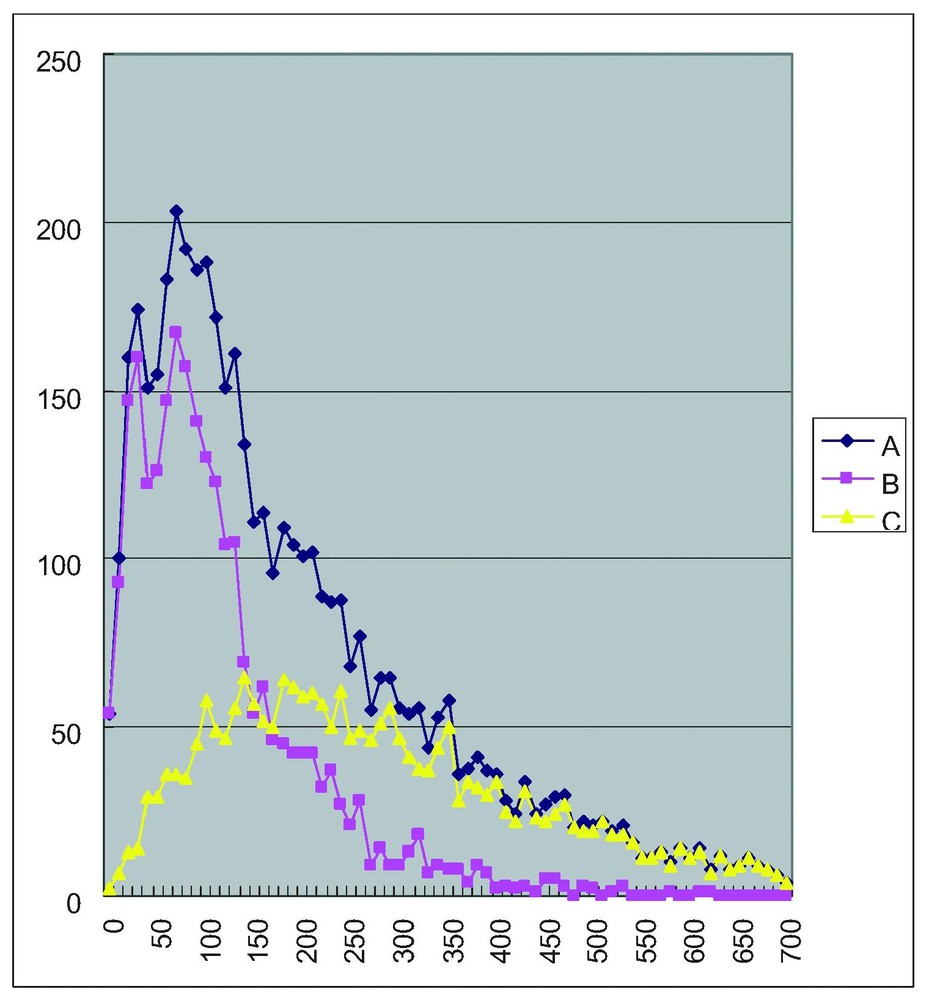
Length distribution of 5′ UTRs. A: Length measured from the ‘initiator’ ATG. B: Length measured from the ‘initiator’ ATG that is also the first ATG. C: Length measured from the ‘initiator’ ATG that is not the first ATG. Vertical axis: number of genes. Horizontal axis: length of 5′ UTR in bases.
The first ATG starts a small ORF different than the longest ORF, if it is not the ‘initiator’ ATG. The average size of such small ORFs is 31 amino acids. Their size distribution is shown in Fig. 2A. These small ORFs can have the topology of either overlapping the longest ORF or not (Fig. 2B). Out of 2386 small ORFs, 469 (ca. 20%) overlapped with the longest ORF. The presence of small ORFs, especially the overlapping ones, could potentially hinder the translation of the longest ORFs. According to the typical translation initiation model, a ribosome does not make a direct entry to the ‘initiator’ ATG. A small (40S) ribosomal subunit is first recruited to mRNA near the cap structure. Then, it linearly ‘scans’ the 5′ UTR for ATG. When it encounters the ‘first’ ATG, it pauses until a large (60S) subunit joins and a complete form of a ribosome becomes ready to initiate translation [11]. Up to 50% of the 4870 genes studied are associated with a potential difficulty to translate the longest ORF, if this classical model is true for all genes. There are several mechanisms already know to complement the classical model such as translation initiation leaks, ‘ribosome re-entry’ and internal ribosome entry sites (for review see [12,13]). It is possible that such mechanisms may play a more pivotal role in the translation in higher eukaryotes than previously anticipated.

Small ORFs in 5′ UTR. A: Length distribution of small ORFs in 5′ UTRs. Vertical axis: number of genes. Horizontal axis: length of small ORFs in amino acid residues. B: Topology of the small ORF in relation to the longest ORF. Hutched box: small ORF, Closed box: the longest ORF, Horizontal line: mRNA, Bar with diamond: first ATG, Bar with closed circle: ATG for the longest ORF, Bar with triangle: stop codon of ORFs. The upper number shows the number of genes whose small ORF overlaps with the longest ORF. The lower number shows number of genes that does not.
At present, there is no evidence that these small ORFs are translated or have functional roles. It might be very interesting to test the presence of the corresponding small peptides within cells. We also did preliminary categorization of the 4870 RefSeq entries according to functional groups and asked whether they contain small ORFs or not (Table 1). All functional categories contained both genes with small ORFs and without them. Also, the ratio between the two is not extremely different from 50–50. However, there are some differences according to the functional categories. For example, the ‘matrix’ and ‘mitochondria’ as well as ‘cell cycle’ and ‘cytoskeleton’ categories show a relatively low ratio of small ORFs, whereas the ‘transcription’ and ‘channel’ categories have more genes with small ORFs. Although there are several examples of the small ORFs that play some regulatory roles in translation [12,13], further study are needed to clarify the full picture of the roles that the small ORFs play within the cells.
The 4870 RefSeq entries were first linked to Locus Link [14] entries and all the fields in the Locus Link were searched for the presence of keywords. If the keywords were found, the corresponding cDNA sequence data were searched for the presence (Yes) or absence (No) of a small ORF in the 5′ UTR
| Keywords | Small ORFs in 5′ UTR | ||||
| Number | % | ||||
| Yes | No | Total | Yes | No | |
| Adhesion | 59 | 88 | 147 | 40 | 60 |
| Apoptosis | 43 | 55 | 98 | 44 | 56 |
| Cell cycle | 54 | 76 | 130 | 42 | 58 |
| Channel | 48 | 38 | 86 | 56 | 44 |
| Cytokine | 21 | 22 | 43 | 49 | 51 |
| Cytoskeleton | 59 | 82 | 141 | 42 | 58 |
| Disease | 31 | 43 | 74 | 42 | 58 |
| DNA repair | 28 | 23 | 51 | 55 | 45 |
| DNA replication | 20 | 24 | 44 | 45 | 55 |
| Endoplasmic reticulum | 38 | 51 | 89 | 43 | 57 |
| Golgi apparatus | 26 | 14 | 40 | 65 | 35 |
| Immune | 53 | 55 | 108 | 49 | 51 |
| Matrix | 42 | 75 | 117 | 36 | 64 |
| Mitochondria | 40 | 79 | 119 | 34 | 66 |
| Nuclear | 87 | 86 | 173 | 50 | 50 |
| Oncogene | 62 | 58 | 120 | 52 | 48 |
| Organelle | 8 | 10 | 18 | 44 | 56 |
| Receptor | 226 | 224 | 450 | 50 | 50 |
| Signal transduction | 178 | 220 | 398 | 45 | 55 |
| Syndrome | 30 | 27 | 57 | 53 | 47 |
| Transcription | 234 | 169 | 403 | 58 | 42 |
| Translation | 39 | 51 | 90 | 43 | 57 |
| Transporter | 123 | 116 | 239 | 51 | 49 |
| Total | 1549 | 1686 | 3235 | 48 | 52 |
Acknowledgements
We thank H. Hata and every member of the HGC-IMSUT sequencing team for their excellent sequencing work. We are also thankful to T. Hasui and J.M. Sugano for helpful discussions and to Y. Makita for technical support in database construction.