

1 Introduction
Several microbial pathogens have developed different – and however convergent – evolutionary strategies to invade and replicate in the intracellular milieu of eukaryotic cells, escaping from specific host immune-system defenses such as antibodies, complement or NOD molecules [1,2]. Members of the Mycobacterium, Salmonella, Legionella, Chlamydia and Brucella species use diverse approaches to enter into target cells, such as receptor-mediated phagocytosis, macropinocytosis or coiling phagocytosis. Once inside host cells, these microorganisms modify their primary phagosomal vacuoles, generating compartments in which pathogen multiplication can take place [3–7].
The Shigella, Rickettsia, and Listeria species also invade cells through different entry pathways, but have developed an original strategy to handle the damaging mechanisms of phagocytosis: they lyse their internalisation compartments and escape directly to the host cell cytoplasm where they polymerise the cellular actin and spread from cell to cell [8–10].
In this review we will focus on exploitation of the host cell machinery by Listeria monocytogenes that permits the invasion of different types of target cells by manipulating and diverting several cellular receptors and engaging the actin cytoskeleton in interactions that lead to infection.
2 The infection by Listeria monocytogenes
Listeria monocytogenes is a food-borne pathogen that affects essentially immuno-compromised individuals and is associated with several pathologies, such as meningitis, abortions and severe gastroenteritis [11]. Such diverse clinical symptoms are related to one critical feature of this microorganism: Listeria is able, as stated above, to induce its own internalisation into different types of cells including professional phagocytes (macrophages) as well as non-professional phagocytes (enterocytes, hepatocytes, etc.) [12]. This attribute endows Listeria with the capacity to traverse three human barriers – the intestinal, the blood-brain and the feto-placental barriers – explaining the pleiotropy of patho-physiological outcomes associated with listeriosis.
3 Main Listeria effectors required for the intracellular life cycle
The use of several in vitro approaches has permitted the identification of the main bacterial factors required for the intracellular life cycle of Listeria (Fig. 1). Invasion of host cells occurs through the association of at least two bacterial surface proteins belonging to the family of internalins, InlA and InlB, with their specific cellular receptors E-cadherin and c-Met (see below for details). This interaction leads to a receptor-mediated phagocytosis of Listeria in a tight phagocytic vacuole. A pore-forming toxin, listeriolysin O (LLO), lyses then the membrane-bound compartment, allowing the pathogen to reach the host-cell cytoplasm. In this new environment, Listeria is able to proliferate and also polymerises the cellular actin through the activity of the protein ActA: the bacteria now move randomly inside infected host cells and can even reach neighbouring cells by formation of protrusions. These membranous extensions (formed initially by a the plasma membranes of both the primary- and secondary-infected cells) are lysed by two additional bacterial toxins, PlcA and PlcB. The bacteria are found in the cytoplasm of the secondary-infected cell and the intracellular cycle can start again [12].
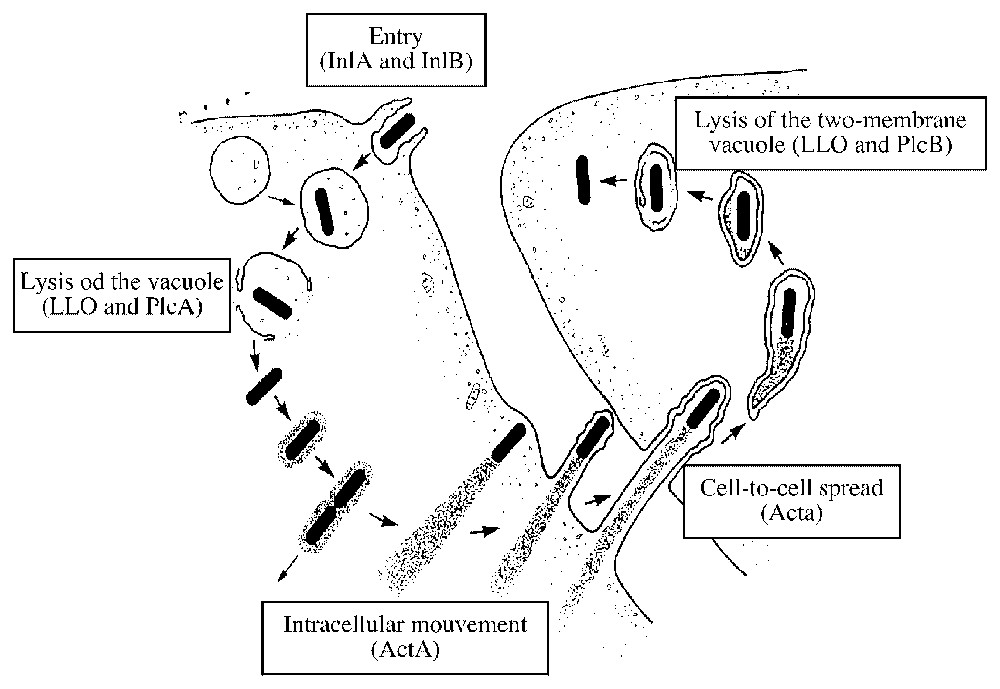
Intracellular infectious cycle of Listeria monocytogenes in mammalian cells. Bacterial factors required for each step are indicated (see text for details).
The actin-based motility of Listeria has been reviewed extensively elsewhere [13]. We will discuss here in detail the molecular players and the known signalling events involved in the invasion of host cells by Listeria.
4 The internalin family
The recent sequencing of the Listeria genome [14] revealed the presence of 24 proteins belonging to the family of internalins, which are proteins characterised by the presence of different numbers of leucine-rich repeats (LRR) at their amino (N)-terminal region, followed by an inter-repeat (IR) region and diverse carboxyl (C)-terminal motifs. InlA, the first molecule of this family revealed to mediate invasion into host cells [15], presents at its C-terminus the Leu–Pro–X–Thr–Gly (LPXTG) motif that permits the covalent linkage of the protein to the bacterial peptidoglycan. Listeria encodes 41 LPXTG proteins (only 19 of these molecules belong to the internalin family), which are potential surface-associated molecules that could be involved in interactions with target host cells [16]. An enzyme named sortaseA achieves the anchoring of LPXTG proteins to the proteoglycan [17]. A Listeria mutant presenting a deletion of the sortase A gene shows a diminished survival in the guinea pig compared to an InlA− mutant, suggesting that indeed other LPXTG molecules (belonging or not to the internalin family) could be involved in cellular invasion. This hypothesis is also highlighted by the fact that an inlAB Listeria mutant presents a residual level of internalisation, suggesting again that other surface-associated proteins are necessary for entry.
InlB, the second member of the internalin family showed to be required for invasion of a broad range of cellular targets [18], is attached loosely to the lipoteichoic acids of the bacterial cell wall through a C-terminal domain that contains repeats of 80 amino-acids carrying the Gly–Trp (GW) dipeptide motif at their starting sequence [19]. This protein is consequently easily released in the supernatant of most Listeria strains [20]. Interestingly, some Listeria strains present an InlA molecule carrying a mutation at the LPXTG motif that prevents its covalent linkage to the proteoglycan and also promoting its release to the external milieu [21]. However, only strains expressing the full-length InlA are detected in clinical isolates from human listeriosis [22].
Initially, the study of transposon-induced Listeria mutants that were non-invasive in known target cells permitted the identification of the internalins as bacterial-encoded factors involved in invasion [15]. Subsequently, the coating of latex beads with either InlA or InlB, or their expression in the non-invasive species Listeria innocua demonstrated that these molecules (and in particular, their LLR motifs) are sufficient to promote internalisation [23–25]. These properties enabled later independent studies of the InlA- and InlB-internalisation pathways.
5 The InlA-internalisation pathway: the InlA/E-cadherin interaction allows crossing of the intestinal barrier
The first cellular receptor ever identified for any bacterial invasion protein was the integrin α5β1, the receptor for the invasin of Yersinia enterocolitica: an affinity chromatography approach was used to perform this identification by running cellular extracts of target cells over an invasin column [26]. A similar method was used successfully to isolate the receptor for InlA, E-cadherin, a member of the broad cadherin family of adhesion molecules [27]. E-cadherin is a transmembrane molecule that is involved in the formation of adherens junctions in polarized epithelial cells through homophilic interactions of E-cadherin dimers located both on the basolateral sides of adjacent cells. Genetic and biochemical studies have revealed that the LRR domain of InlA interacts with the outmost extracellular domain of E-cadherin [28]. A proline at position 16 of the E-cadherin is critical for its interaction with InlA, as revealed by the fact that species that express a different residue at this position (for example, glutamic acid in the case of the mouse species) are not permissive to infection by translocation across the intestinal barrier, as opposed to humans and guinea pigs [10,29,30]. The recent crystallography of the InlA/E-cadherin complex demonstrated that the proline at position 16 coincides with the only incomplete leucine-rich repeat on InlA, confirming the exquisite specificity of this interaction [31].
The cytoplasmic domain of the E-cadherin is critical for invasion as demonstrated by a series of experiments showing that an E-cadherin truncated at its transmembrane level and expressing only the extracellular domain is unable to support entry of Listeria in target cells [32]. Several molecules are known to interact with the cytoplasmic tail of E-cadherin contributing to formation of adherens junctions, in particular members of the catenin family. The cytoplasmic part of E-cadherin binding β-catenin is essential for entry. Moreover, since β-catenin is known to interact with α-catenin (which provides a direct structural link to the actin cytoskeleton), an E-cadherin chimera was constructed expressing its ectodomain fused with the C-terminus of α-catenin: this molecule was able to support full invasion of target cells through the InlA-dependent pathway, demonstrating that the E-cadherin/β-catenin/α-catenin link is sufficient to permit Listeria entry [32].
Most probably, other molecules normally associated to the adherens junctions, and in particular to the cytoplasmic tail of E-cadherin, are also involved in the process of cell invasion by Listeria, but their identity remains to be established. In particular, the proteins and lipid effectors that could regulate the dynamic assembly of the entry complex (see below to compare with the actin polymerization/depolymerization rearrangements necessary to the InlB-mediated entry) still have to be identified. Rac-1 and Cdc42, members of the Rho family of small GTPases, as well as the actin nucleator complex Arp2/3 (that have a known role in the formation of junctions), could be positive regulators of the reorganization mechanisms presumed to occur; their roles in Listeria entry are being currently investigated. Recent data suggest that the unconventional myosin VIIa and its ligand vezatin [33] are involved in the entry process, providing a molecular motor for bacterial internalization [34] (Fig. 2).

Model for InlA-dependent entry of Listeria in epithelial cells. Proteins known to play a role in invasion are depicted. The model highlights how mysion VIIa could move along the actin filaments to induce membrane rearrangements during the entry process.
At the organism level, the identified species specificity of InlA concerning its receptor E-cadherin led to the idea of generating a transgenic mice model expressing the human E-cadherin at the intestinal level to evaluate the role of InlA in the crossing of the intestinal barrier. In these animals, Listeria directly targets enterocytes by interacting with the human E-cadherin and this interaction allows the bacterial translocation across the intestinal barrier, followed by its multiplication in the small intestinte lamina propria and dissemination to mesenteric lymph nodes, liver and spleen [10]. This transgenic model not only provides the molecular explanation for the innocuity of Listeria in mice following oral infection but also explains the enteropathogenicity of Listeria in guinea pigs and most probably in humans.
6 The InlB-internalisation pathway
Three cellular receptors have been currently identified for InlB, suggesting that this molecule could play different complementary roles during the invasion process [35] (Fig. 3).
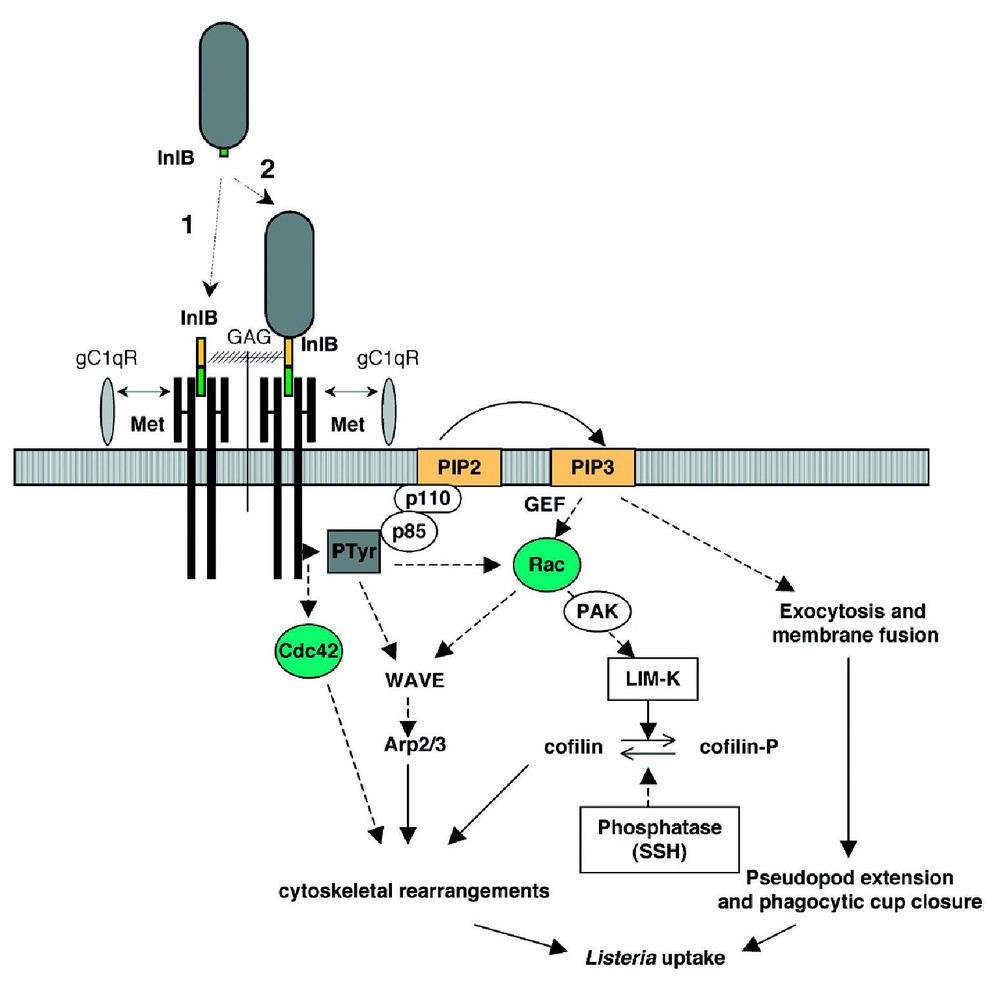
Model for InlB-dependent entry of Listeria in mammalian cells. InlB can dissociate from the bacterial surface by interacting with GAGs (1) and its soluble form can access gC1q-R and Met on the host cell, inducing signals either as a prelude or independently from entry. The surface-bound InlB triggers the activation of Met and the recruitment of adaptor proteins (some of which become tyrosine-phosphorylated) and of the p85-p110 PI 3-kinase. Downstream events involving activation of Rho GTPases and cytoskeletal regulatory proteins lead to membrane rearrangements and bacterial uptake (dashed arrows indicate hypothetical steps).
6.1 gC1q-R
Using also an affinity chromatography approach (see above for E-cadherin identification as the InlA ligand), the receptor for the globular head of the first component of the complement cascade C1q (also known as gC1q-R) was isolated as the first InlB cellular partner [36]. Besides binding the C1q molecule and InlB, gC1q-R has been shown to interact with several viral proteins such as HIV-1 Rev and Tat [37], adenovirus protein V [38], Epstein-Barr virus nuclear antigen-1 [39], hepatitis C virus core protein [40] and protein A from Staphylococcus aureus [41] but the actual biological function of gC1q-R remains elusive. Furthermore, its cellular distribution is controversial, since it has been reported to be present at the mitochondria, the nucleus and the plasma membrane [42,43]. Its mechanism of attachment to membranes is unknown, since it does not possess any trans-membrane domain and is not glycosylphosphatidyl inositol-anchored. In any case, expression of gC1q-R in cells that normally do not express this molecule permits invasion through an InlB-dependent pathway [36]. Indeed, anti-gC1q-R antibodies and the molecule C1q itself compete with InlB for binding to gC1q-R and inhibit the InlB-dependent entry of Listeria in target cells. The absence of obvious cytoplasmic (and potentially signalling) domains in the gC1q-R molecule suggests that it could play a role during invasion as a co-factor for another cellular receptor.
6.2 c-Met
A second receptor for InlB was then identified, c-Met (the receptor for the hepatocyte growth factor or HGF/scatter factor), a tyrosine kinase receptor that can actually promote recruitment to the plasma membrane of several proteins already known to be activated through the InlB-dependent pathway [44]. c-Met is a disulfide-linked heterodimer formed by a 45-kDa extracellular α- and a 145-kDa transmembrane β-subunit that contains the tyrosine kinase catalytic domain [45]. In vivo, the interaction between HGF and c-Met has been shown to be critical for the development of the liver, placenta and specific muscle groups [46,47]. In vitro, this interaction leads to the scattering of target cells, a phenomenon that is directly related to the invasive phenotype of cancer cells, in which Met expression is deregulated. InlB, as HGF does but with different kinetics, stimulates the phosphorylation of two Tyr residues in the tyrosine kinase catalytic domain of c-Met, leading to activation of its intrinsic kinase activity, promoting auto-phosphorylation of two additional Tyr residues at the C-terminal domain that function as docking sites for Gab1, Cbl and Shc [44,48]. Phosphorylation of these adaptor molecules induces the recruitment of the phosphoinositide 3-kinase (PI 3-K) p85–P110 and generation of phosphatidylinositol 3,4,5-phosphate (PIP3) at the plasma membrane [48,49]. The direct downstream effectors of PI 3-K remain to be identified. However, formation of InlB-induced actin ruffles at the plasma membrane of target cells is dependent on the PI 3-K activity [48], suggesting that the reorganization of the cortical actin cytoskeleton is under the control of the PI 3-K downstream molecules. The actin nucleator complex Arp2/3 has been implicated in the InlB-dependent Listeria entry process as well as the small GTPase Rac1, the Rac1 downstream effector Lin-11/Isl-1/Mec-3 kinase-1 (LIMK-1) and the LIMK-1 substrate actin depolymerising factor(ADF)/cofilin [50]. Indeed, phosphorylation and inactivation of the ADF by the LIMK-1 induce accumulation of actin and inhibits the closure of the phagocytic cup at the site of bacterial entry, while positive activation of ADF by inactivation of the LIMK-1 leads to a loss of actin assembly at the phagocytic cup and also inhibits bacterial internalization [50], highlighting the importance of a tight temporal control of actin dynamics during Listeria invasion of host cells.
6.3 Glycosaminoglycans
Glycosaminoglycans (GAGs), including the heparin-like molecule heparan sulphate (HS), represent a third kind of cellular receptor for InlB [19]. GAGs are highly charged polysaccharides, present in the surface of all eukaryotic cells, which have been implicated in several modulating functions, such as storage of growth factors (including HGF) at the surface of cells, their protection from the activity of extracellular proteases and oligomerization of growth factors that potentiate their signalling capacities [51–53]. Depletion of GAGs reduces by ten fold the invasion of target cells through the InlB-dependent pathway [19]. Moreover, heparin induces the clustering/aggregation of InlB, suggesting that GAGs could also cluster InlB and increase the signalling potential of bacterial-released InlB by promoting its storage at the cell surface and favouring its interaction with c-Met [19].
6.4 InlB domains involved in ligand binding
The resolution of the crystal structure of InlB [54–56] revealed an L-shaped molecule with a slightly curved tube composed of successive β-strand-loop-310-helix-loop motifs at the N-terminal LRR region and the IR-region projecting at almost a right angle from the base of the LRR tube with the Gly–Trp repeats (which have are structurally related to the Src homology domain 3) at the other end of the molecule. The N-terminal LRR tube presents a fold that is predicted to be suitable for interacting with several ligands [54]. Indeed, the LRR region has been shown to be critical for the binding of InlB to the extracellular domain of c-Met [44,57]. However, the interaction of InlB with gC1q-R and GAGs seems to be dependent on the Gly–Trp repeats region [19,56]. These results would mean that InlB possesses two modular domains that are both required for invasion of target cells, but that involve diverse activation mechanisms by interacting with different cellular components.
7 Other bacterial molecules involved in adhesion and invasion
Other bacterial surface proteins of the internalin family (different from InlA or InlB) or LPXTG-anchored proteins could be potentially involved in the interaction between Listeria and the plasma membrane of host cells. An unexpected molecule that seems to play a role in recognition of target host cells is the surface protein ActA, previously known to be responsible for the intracellular actin-based motility of Listeria [58]. The N-terminal region of ActA presents three stretches of 40 to 54 amino acids containing a unusual high content of clustered positively charged residues in close proximity to hydrophobic residues, a pattern that strongly resembles the domains involved in HS recognition in HS-binding proteins. An ActA− mutant is indeed less efficient in attachment and invasion of target cells than wild-type Listeria [59]. Furthermore, while binding of wild type bacteria is affected in cells deficient in HS, the ActA− mutant exhibits identical diminished binding to cells, regardless of their expression of HS, again suggesting that the impaired attachment is probably due to absence of HS recognition and highlighting a role of ActA in target cell binding [59].
Another molecule that could be involved in facilitating Listeria invasion is the pore-forming toxin LLO. Recently, it has been shown that wild type bacteria (but not a LLO− mutant) induce calcium fluxes in target cells [60]. Interestingly, addition of plasma membrane calcium channel antagonists or chelation of extracellular calcium (but not blockade of calcium release from intracellular stores or intracellular calcium chelation) reduced Listeria entry in host cells, suggesting that mobilization of extracellular calcium by LLO (and activation of downstream calcium-dependent signalling) is required for efficient cell invasion [60].
8 Concluding remarks
The expression of two different invasion proteins by Listeria monocytogenes raises the interesting possibility that synergy might exist between the two internalisation pathways used by this pathogen. Several arguments favour this hypothesis: firstly, the inlA and inlB genes are part of the same operon and thus are co-expressed and co-regulated; secondly, E-cadherin and c-Met co-localise when they are co-expressed in cells (Fig. 4). An attractive hypothesis is that InlB released from bacteria can induce cell scattering, junction opening and other signalling events that lead to the interaction of InlA with its receptor, explaining how Listeria reaches the normally non-accessible E-cadherin during the infectious process. Other events might be also involved, such as the retargeting of E-cadherin from adherens junctions to the apical pole of intestinal epithelial cells, as has been observed after activation of c-Met by HGF [61]. These hypotheses are under current investigation.
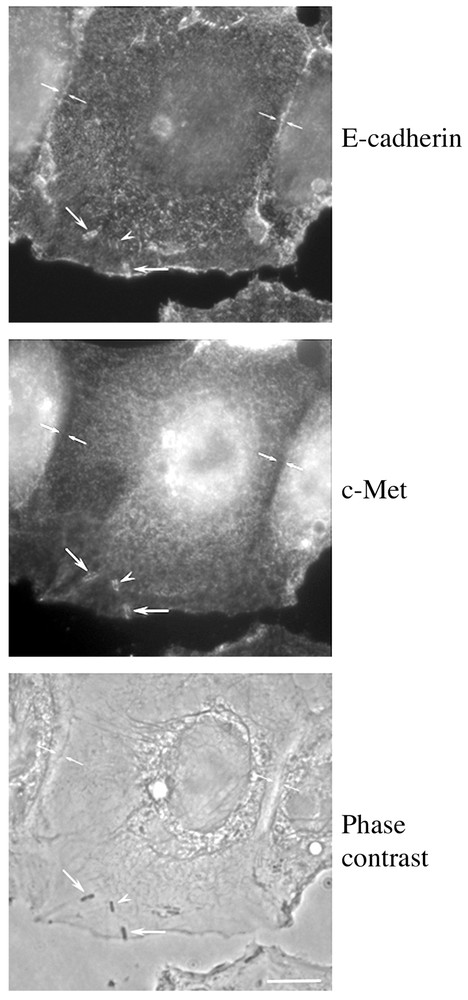
Recruitment of E-cadherin and c-Met during Listeria entry in LoVo cells. The receptors for InlA and InlB co-localise at the entry site of several bacteria suggesting that a synergy between these two internalins could foster the invasion process (bar: 10 μm).