

1 Introduction
Gene therapy involves the transfer of genetic material, usually in the form of DNA, with the purpose of altering the genetic repertoire of cells for therapeutic purposes [1]. As conventionally practiced, the expression of the newly transferred DNA is in parallel with expression of genes of the endogenous genome, utilizing the endogenous transcription and translation apparatus of the target cells. This review focuses on a new paradigm of gene therapy, the use of trans-splicing to modify the genetic repertoire at the pre-mRNA level to treat genetic and acquired disorders.
Gene expression is a complex multistep process that includes the synthesis of pre-mRNA (transcription), maturation of the and ends of the pre-mRNA transcript (capping and polyadenylation, respectively), the removal of introns from the pre-mRNA, and the export of the mature mRNA to the cytoplasm of the cell, where it is translated [2]. While the sequence of the genome dictates the form of the protein to be produced, the pre-mRNA product of transcription is not always terminally committed to the generation of a single mRNA, but is plastic, subject to processes that lead to the production of one or more different mRNAs from the original primary transcript.
Among the mechanisms that impact on the fate of a pre-mRNA, the most important is alternative exon selection [3,4]. Normally, exon selection occurs in a cis fashion, where the exon sequences of the newly forming pre-mRNA are selected sequentially to , with exon skipping used to expand the genetic repertoire by producing different mRNAs that translate into proteins that are similar, but with subtly different functions [4]. In contrast, in pre-mRNA trans-splicing, the sequence of the gene being expressed is altered at the pre-mRNA level by joining the pre-mRNA to another, independent pre-mRNA [5]. In therapeutic trans-splicing, the genetic information for the independent pre-mRNA is delivered exogenously via a gene transfer vector [6]. As will be discussed in this review, therapeutic trans-splicing can be conceptualized in two categories: ribozyme-mediated and spliceosome-mediated. Therapeutic trans-splicing can be used to alter coding domains, to create novel fusion proteins, to direct gene products to different cellular compartments, and to overcome some limitations to vector-derived gene transfer technology, including gene therapy with large genes or with genes coding for toxic proteins. To demonstrate the potential of therapeutic trans-splicing, we will first review eukaryotic cis-splicing and trans-splicing in nature, and then provide several strategies of ribozyme- and spliceosome-mediated pre-mRNA trans-splicing directed by exogenous gene transfer.
2 Cis-splicing, editing and trans-splicing in nature
In multicellular organisms, pre-mRNA modifications occur via cis-splicing, editing and trans-splicing. As a background to how trans-splicing can be used for therapeutic purposes, we will review the natural processes of pre-mRNA modification.
3 Pre-mRNA cis-splicing
In multicellular organisms, most protein-encoding genes contain introns. Pre-mRNA splicing occurs in the nucleoplasm, and the removal of introns is required for subsequent nucleo-cytoplasmic transport the mature mRNAs [7,8]. Precise and efficient excision of introns is required for faithful transmission of genetic information to the protein synthesizing machinery in the cytoplasm. Errors in splicing secondary to mutations in intron sequences critical for splicing are responsible for several inherited disorders in humans [9,10].
Pre-mRNAs are the primary transcripts from genes transcribed by RNA polymerase II and are direct copies of these genes [7]. The majority of eukaryotic pre-mRNAs contain multiple intervening sequences (introns) that are removed by pre-mRNA splicing [5]. Sequence elements that survive the splicing process are defined as exons. The splicing process, coordinated with other RNA processing events, results in the appearance of mature mRNA. Conserved sequence elements in pre-mRNAs define introns and exons and mark their boundaries [11,12]. In mammalian cells, two families of introns can be distinguished based on these elements and the small nuclear RNAs they interact with: the major class (U2-introns), and the minor class (ATAC, or U12-introns) [11,12]. The process of the splicing of the U2-introns is the focus of the strategies for spliceosome-mediated therapeutic trans-splicing.
The average exon is quite small. Most exons are roughly 150 nucleotides in length with 99% of known exons shorter than 400 nucleotides, although exons over 5000 nucleotides long have been identified. Introns, on the other hand vary greatly in length, the average being about 1500 nucleotides, although some are several hundred thousand nucleotides in length. This dichotomy has lead to the ‘exon definition hypothesis’, in which splicing factors bound to a splice site define the exon by interacting with splicing factors at the next downstream splice site, then switching to interact with factors at the upstream splice site, across the intron, to allow splicing of the introns [11]. In addition, many exons contain internal sequences that either enhance or silence splicing, and other types of splicing enhancers and silencers are found in introns. Exonic splicing enhancers facilitate exon definition by assisting in the recruitment of splicing factors to the adjacent intron, promoting the recognition of both exon/intron junctions during exon definition [13]. Sequences that suppress splicing events at particular sites have also been identified [14].
The upstream exon–intron junction, the splice site of U2-introns, is defined by the consensus sequence AG/GTRAGT (Fig. 1). Downstream from this sequence, introns contain two linked cis elements, the branchpoint and the polypyrimidine tract, followed by the splice site. These elements can be tens or thousands of nucleotides downstream from the splice site. The branchpoint consensus is comprised by the sequence UACUAAC in which the central A is the branchpoint adenosine, at which a ‘lariat’ forms in the splicing reaction [2]. The polypyrimidine tract, Y(11)NYAG/G, immediately precedes the splice site. The distance between the ‘A’ branchpoint and the splice site is usually 18 to 37 nucleotides [15].
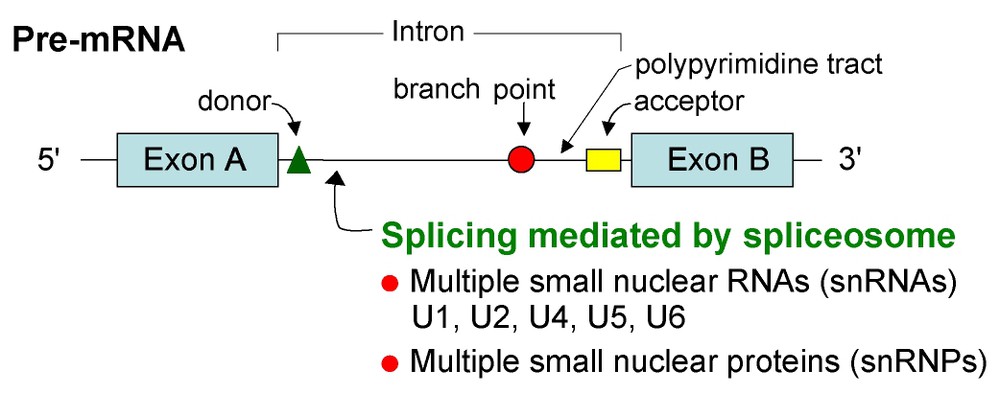
Pre-mRNA cis-splicing. The process of removing introns is catalyzed by a spliceosome formed from the assembly of multiple small nuclear RNAs (snRNAs; U1, U2, U4, U5 and U6) and small nuclear ribonuclear proteins (snRNPs). These splicing reactions occur in the nucleus and generate mRNA molecules from primary RNA transcripts (pre-mRNA). After assembly of the spliceosome, the end of the first exon sequence is ligated to the end of the second exon sequence. In cis-splicing this occurs sequentially, resulting in mature mRNA. The critical recognition sequences in every intron include donor, branchpoint, polypyrimidine tract and acceptor sequences.
In the splicing process, two trans-esterification reactions are involved in the removal of each intron and ligation of the surrounding exons. A complex referred to as the ‘spliceosome’ catalyzes splicing reactions [2,7]. The spliceosome, a complex nearly as large as a ribosome, is composed of four ribonucleoprotein (RNP) particles and additional proteins. Each small RNP particle contains at least one or two small nuclear RNAs (snRNAs) containing an abundance of uracil residues, named U1, U2, U5 or U4/U6 snRNP, respectively. The U1 snRNP recognizes the splice site consensus sequence involving an RNA–RNA interaction between the pre-mRNA and the U1 snRNA and the U2 snRNP recognizes the branchpoint sequence of the major class introns (hence the term ‘U2-introns’) [7,12].
RNA splicing is catalyzed by a spliceosome formed from the assembly of U1, U2, U5, and U4/U6 snRNPs plus protein components. After assembly of the spliceosome, the reaction occurs in two steps. First, the branchpoint A nucleotide in the intron, located close to the splice site, attacks the splice site and cleaves it; the cut end of the intron then becomes covalently linked to this A nucleotide, forming a -- branched nucleotide. In the second step, the -OH end of the exon sequence, created previously, ligates to the exon, thereby cleaving the RNA at the splice site. The two exon sequences are thus joined to each other and the intron sequence is released as a ‘lariat’ structure. These splicing reactions occur in the nucleus and generate mRNA molecules from primary RNA transcripts [2,16].
Other cis elements modulate specific splicing reactions, leading to alternative splicing. It has been estimated that approximately 60% of human pre-mRNAs can be alternatively spliced, resulting in the synthesis of multiple mRNAs encoding different proteins from a single gene [17–19]. From among several possible splice events, various splicing factors determine which splice sites are chosen [4]. Two major groups of splicing factors involved in the selection of splice sites include the heterogeneous nuclear ribonucleoprotein (hnRNP) proteins and the serine/arginine-rich (SR) splicing factors, both of which contain RNA binding domains [11,20]. The choice of splice site in any given cell type depends on the ratio of splicing factors [3,7,17,21–27].
4 Pre-mRNA editing
RNA molecules can adopt a wide variety of conformations and tertiary structures, and are able to engage in a broad range of cellular processes, including the ability to fold to form catalytic centers, a functional entity referred to as a ‘ribozyme’ [28–31]. Naturally occurring ribozymes are used by multicellular organisms to edit pre-mRNAs. This is done by cleaving the target pre-mRNA in two sites, and then joining the resulting and fragments of pre-mRNA into an ‘edited’ new pre-mRNA. In the context that editing involves the joining together of fragments of the same pre-mRNA, it is not trans-splicing per se – for further details of pre-mRNA editing, recent reviews are available [31,32]. However, as will be described later, gene transfer of sequences coding for ribozymes together with ‘extended guides’ (sequences designed to hybridize to a targeted pre-mRNA) can join with endogenous cellular pre-mRNAs in strategies designed for therapeutic ribozyme-mediated trans-splicing.
5 Pre-mRNA trans-splicing
In addition to the dominant cis-splicing process, the eukaryotic cellular spliceosome apparatus appears to be both willing and able to select exons on different pre-mRNA molecules for ligation into a single mRNA via pre-mRNA trans-splicing. Naturally occurring trans-splicing was first observed in lower eukaryotes including trypanosomes and nematodes, and in algal chloroplasts and plant mitochondria, as a process in which pre-mRNAs are processed to produce a mature transcript that contains exons from two different precursor molecules [5,33–42].
In the spliced leader form of naturally occurring trans-splicing, first observed in trypanosomes, a common 39 bp spliced leader (SL) RNA sequence at the end of a small, non-polyadenylated RNA is transferred to pre-mRNAs to form the -terminal exon of mature mRNAs [33–35,41,42]. Uridylic acid-rich small nuclear RNAs are also involved in this form of pre-mRNA processing. The SL RNA provides a cap structure to the end of a family of mRNAs by trans-splicing into a single pre-mRNA at different points, ultimately leading to several protein products from a single transcriptional event [43]. The process occurs in addition to cis-splicing of internal introns in trypanosomal pre-mRNAs, leading to increased diversity of gene expression and economy of cellular resources [41]. Interestingly, some splice acceptor domains in human pre-mRNAs can function as spliced leader addition sites in trypansomes [38]. Similar RNA processing has also been demonstrated in flatworms and plant mitochondria [39,43].
Recent reports indicate that higher eukaryotes do trans-splice RNAs naturally, albeit rarely. Trans-splicing of a pre-mRNA for carnitine octanoyltransferase has been demonstrated in rat liver [44]. In addition to alternative cis-splicing, the human estrogen gene is also subject to natural trans-splicing [45], and the four highly homologous human cytochrome P450 3A genes form active chimeric mRNAs by trans-splicing [46]. Cultured rat cells transformed by an SV40 fragment generate a truncated T antigen which can only be created through trans-splicing of two copies of the pre-mRNA, and HeLa cell extracts can also perform the trans-splicing reaction in vitro [47]. That mammalian cells can perform these reactions naturally set the background for the strategies of therapeutic spliceosome-mediated trans-splicing, when there is a deliberate attempt to alter the information content of a pre-mRNA via the delivery of an exogenous invading trans-splicer transcript.
6 Therapeutic pre-mRNA trans-splicing
Despite the fact that higher eukaryotes rarely trans-splice naturally, they clearly have the capacity to perform programmed trans-splicing reactions when presented with an appropriate precursor RNA molecule. Therapeutic trans-splicing, the focus of this review, is different from strategies using therapeutic-based RNA modifications using RNAi or endonucleases and ligases; for a discussion of these approaches, several reviews are available [48,49]. The strategies for therapeutic trans-splicing can be divided into two general categories: ribozyme-mediated and spliceosome-mediated.
6.1 Ribozyme-mediated therapeutic pre-mRNA trans-splicing
Ribozymes, naturally occurring catalytic RNAs, can mediate the cleavage of a phosphodiester bond in a target pre-mRNA to generate -hydroxyl and ,-cyclic phosphate termini. The strategy for ribozyme-mediated therapeutic pre-mRNA trans-splicing is to transfer a gene coding for ( to ): a hybridization domain that is antisense to a sequence in the target pre-mRNA; the ribozyme per se; and the new pre-mRNA to be trans-spliced into the target pre-mRNA (Fig. 2).
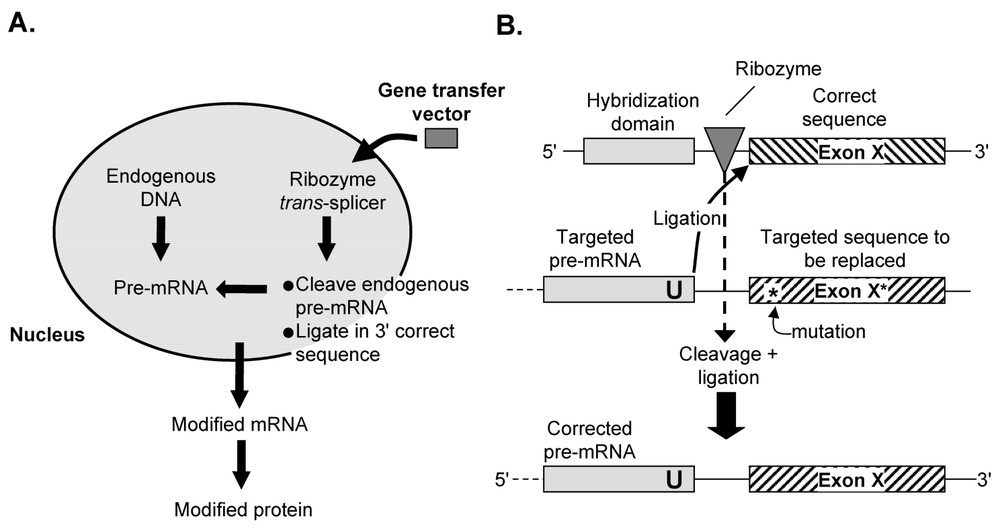
Therapeutic ribozyme-mediated pre-mRNA trans-splicing. (A) Overview. An expression cassette expressing a trans-splicing ribozyme able to perform cleavage and ligation into an endogenous ‘target’ pre-mRNA via a complementary hybridization domain (also referred to as the ‘internal guide sequence’), is delivered to the cell using a gene transfer vector. The modified pre-mRNA is processed, exported and translated to produce a modified protein. (B) Details of ribozyme-mediated trans-splicing, using correction of a mutation as an example. In this model, the endogenous pre-mRNA contains a mutation, indicated by ‘∗’, in a downstream exon (exon X). A corrective trans-splicing ribozyme, introduced by gene transfer, binds to the target based on the hybridization domain, cleaves the endogenous sequence to the target ‘U’, and replaces the defective exon with a corrected sequence. Unlike spliceosome-mediated trans-splicing, any portion of the endogenous pre-mRNA is a valid target and no endogenous sequence elements are required. However, the ribozyme-mediated reaction is reversible, and a steady-state level of the mutant form, to some degree, is unavoidable.
The function of such constructs should not be surprising since spliceosomes are essentially ribozymes, with the catalytic center composed of RNA. Virtually any U residue in a exon can be targeted for splicing by hybridization sequences at the exon-binding site (referred to as the ‘internal guide sequence’) to make it complementary to a target sequence present on the substrate RNA [50–52]. To date, ribozyme-mediated trans-splicing has not proven to be very efficient in vivo. However, if it can be developed to maintain specificity and fidelity, it has the attractive feature of splicing into an endogenous pre-mRNA transcript, thus preserving the natural transcriptional regulatory mechanisms controlling the level of gene expression. Compared to simply delivering a traditional therapeutic genetic payload, a major advantage of this approach is that, by invading a naturally occurring pre-mRNA, regulated expression of the corrected gene product is guaranteed, and the level of expression of the mutant gene product is simultaneously reduced.
Examples of therapeutic ribozyme-mediated trans-splicing include a Tetrahymena ribozyme shown to trans-splice an exon that is attached to its end onto a separate exon RNA in mammalian cells [53]. Cytotoxicity was demonstrated in yeast using a trans-splicing ribozyme to splice diphtheria toxin coding sequences onto a viral target RNA [54]. Similar targeted ribozymes have been used to revise mutant transcripts that are associated with several human genetic and malignant diseases, and fused an in-frame diphtheria toxin A chain onto the hepatitis C virus mRNA leading to the destruction of infected cells by apoptosis [31,50,53–59]. Trans-splicing ribozymes have been used to repair mutant p53 RNA in human cancer cells [57] and to repair a mutant canine skeletal muscle chloride channel [53]. In the canine muscle study, repair efficiency was low (1.2%) by quantitative RT-PCR, but patch-clamp analysis of individual cells yielded a wide range of repair efficiency, with 18% of cells showing some functional restoration and some cells showing full wild-type function, suggesting that repair efficiency varies from cell to cell. Wild type globin sequences have been trans-spliced into sickle globin mRNA to ‘repair’ the mutant transcripts in vivo via a ribozyme-mediated trans-splicing mechanism in erythrocyte precursors isolated from patients with sickle cell disease [55]. Ribozyme-mediated trans-splicing has also been used to correct aberrant transcripts in myotonic dystrophy, by shortening the trinucleotide repeat expansion found at the end of the human myotonic dystrophy protein kinase mRNA in vitro [60].
6.2 Spliceosome-mediated therapeutic pre-mRNA trans-splicing
There are two basic approaches to therapeutic spliceosome-mediated trans-splicing, including: (1) gene transfer-directed pre-mRNA trans-splicing into an endogenous pre-mRNA; and (2) segmental trans-splicing, in which fragments of genes are delivered by separate gene transfer vectors, and the pre-mRNAs joined by trans-splicing to code for an intact protein. Although circumstances exist in which transient expression of a trans-splicer is desirable, such as the ablation of tumor cells or tissue remodeling, most applications require long-lasting or permanent expression of the therapeutic trans-splicer pre-mRNA. Gene transfer vectors capable of providing such extended expression include retrovirus, lentivirus and adenovirus-associated vectors. Although these vectors are all capable of providing the proper structures for trans-splicing, the degree to which the splicing signals incorporated into the transgene interfere with vector production and levels of intracellular expression need to be evaluated on a case by case basis.
7 Gene Transfer-directed pre-mRNA trans-splicing into an endogenous pre-mRNA
The most common strategy of therapeutic trans-splicing is directed toward modification of an endogenous pre-mRNA, where a therapeutic pre-mRNA derived from an exogenously constructed and delivered expression cassette is used to transcribe a new partial pre-mRNA that, via trans-splicing, repairs clinically relevant mutant transcripts (Fig. 2). Like ribozyme-mediated therapeutic trans-splicing, spliceosome-mediated therapeutic trans-splicing into an endogenous pre-mRNA preserves the natural transcriptional regulatory processes controlling pre-mRNA levels. This property makes these strategies of pre-mRNA trans-splicing attractive approaches to the treatment of genetic disorders associated with mutant genes that are highly regulated or that encode potentially deleterious mutant proteins.
The ability of higher eukaryotes to perform trans-splicing suggested that trans-splicing might be used as a treatment of genetic diseases [6]. Strategies that operate through the repair of mRNA are especially attractive for this application, since they can theoretically correct both dominant and recessive disorders [61,62]. Directed trans-splicing, targeting an intron upstream of the defective exons, should enable the replacement of the defective portion of an mRNA molecule, reducing the number of mutant transcripts and replacing them with equal numbers of the repaired transcript. In this scenario, the trans-splicing target mRNA remains under the control of its endogenous promoter, assuring proper tissue specificity and regulation of expression.
In the most common strategy, spliceosome-mediated trans-splicing into endogenous pre-mRNA utilizes gene transfer to the cell of pre-mRNAs engineered to contain a set of sequence elements which promote cis-splicing of the targeted intron, including a branchpoint, polypyrimidine tract and a splice acceptor site (Fig. 3). Interaction of the trans-splicer pre-mRNA and the target pre-mRNA occurs through a critically important ‘hybridization domain’ by virtue of its complementary sequence which is inverted with respect to the remainder of the trans-splicer to preserve the antiparallel configuration of the hybrid. The hybridization domain anchors the trans-splicing pre-mRNA to the specific target intron. In the early developments of this form of trans-splicing, the hybridization domain was constructed such that it spanned the cis-elements in the target required for efficient splicing, i.e., the branchpoint and polypyrimidine tract. However, recent studies have demonstrated optimal trans-splicing with a hybridization domain targeted considerably more within the intron, and not impinging at all on the aforementioned sequence elements [63]. Trial and error have led to the general principal that targeting the size and location within the intron of the hybridization domain for most efficient trans-splicing is a gene-specific function that must be optimized on a case by case basis. Although the example given illustrates the strategy for replacement of exon sequences to the hybridization domain (Fig. 3), similar strategies have been developed for exons, as well as for the replacement of internal exons in a process involving two separate splicing events [64].
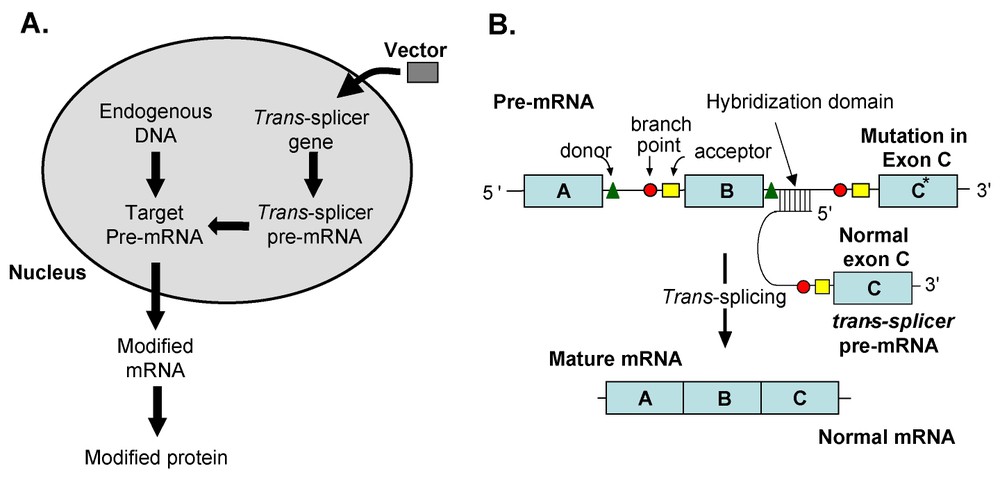
Therapeutic spliceosome-mediated trans-splicing modification of an endogenous pre-mRNA. (A) Overview. An expression cassette expressing a trans-splicing pre-mRNA, directed to an intron in an endogenous ‘target’ pre-mRNA via a complementary hybridization domain, is delivered to the cell using a gene-transfer vector. The trans-splicer pre-mRNA contains sequence elements that attract the spliceosome to generate a corrected mature mRNA from the interaction of the endogenous pre-mRNA and newly introduced pre-mRNA. (B) Details, using spliceosome-mediated trans-splicing correction of a mutation as an example. In this model, the endogenous pre-mRNA has a mutation in the C* exon. With spliceosome-mediated trans-splicing, to the pre-mRNA product, an exogenous corrected exon (C) is introduced by gene transfer, resulting in an mRNA product A–B–C, replacing the mutant exon C* with a wild-type sequence exon C. The critical elements are illustrated, including hybridization domain, branchpoint, splice donor, polypyrimidine tract and splice acceptor.
An early demonstration of the potential of spliceosome-mediated trans-splicing into endogenous pre-mRNA was the demonstration that spliceosome-mediated RNA trans-splicing could be used to alter human chorionic gonadotropin polypeptide mRNAs and to repair mutant lacZ transcripts in cell culture [6,65]. Another demonstration of this approach is the correction of the cystic fibrosis transmembrane conductance regulator (CFTR) F508 mutation in human cystic fibrosis airway-epithelial cells grown in culture and in animal xenografts [66,67]. The repair at the pre-mRNA level resulted in partial restoration of Cl− transport in the CFTR-deficient cells to 12 to 15% of the level observed in wild-type CFTR-containing cells, theoretically sufficient to correct the disease.
Spliceosome-mediated trans-splicing has also been used to reprogram mutant pre-mRNAs in vivo in experimental animal models of hereditary disease. The best two examples of the use of trans-splicing to an endogenous mRNA to correct a phenotype in vivo are the repair of factor VIII hemophilia [68] and X-linked immunodeficiency with hyper IgM (HIGM1) [63].
7.1 Correction of factor VIII hemophilia
Hemophilia A is a sex-linked bleeding disorder caused by deficiency of coagulation Factor VIII [69]. Spliceosome-mediated trans-splicing into endogenous mutant factor VIII has been used to repair the defective mRNA in a Factor VIII knockout murine model [68]. In this study, a Factor-VIII trans-splicer expression cassette carried by either plasmids or adenovirus gene transfer vectors was administered intravenously to Factor VIII knockout mice. Factor-VIII activity measured in the plasma peaked at 3 weeks and could be detected for up to 8 weeks, although the levels declined from peak values. At 3 weeks after administration of the adenovirus Factor VIII trans-splicer, 80% of the mice survived a tail-clip challenge, while no naive or mock-injected animal survived.
7.2 Correction of CD40L mutations in HIGM1
X-linked immunodeficiency with hyper IgM (HIGM1) is a rare, X-linked recessive immunodeficiency disease characterized by failure of immunoglobulin isotype switching from IgM to IgG, IgA, and IgE, caused by mutations of the gene encoding CD40 ligand (CD40L), a cell surface ligand expressed by CD4+ T cells in a regulated fashion [70,71]. Lack of CD40L on the surface of activated CD4+ T cells prevents the interaction with the CD40 glycoprotein on B cells, macrophages and dendritic cells that is essential for immunoglobulin class switching by B cells and macrophage activation [71]. Unable to perform this activation, individuals with HIGM1 suffer from recurrent bacterial infections, especially with opportunistic microorganisms such as Pneumocystis carinii (P. carinii) [72]. HIGM1 is a serious and often fatal disease with only 20 to 40% of patients reaching the third decade of life [73,74]. Currently the only available therapy for HIGM1 is bone marrow transplantation [72,73].
Although gene therapy is an obvious approach to a potential cure for HIGM1, studies by Brown et al. [75], using ex vivo retrovirus-mediated CD40L gene transfer to bone marrow cells in a mouse CD40L knockout (CD40-KO) model, corrected the CD40L deficiency state and stimulated humoral and cellular immune functions, but also induced the development of T lymphoproliferative disease. This observation is a good example of the need for regulated expression of potent genes like CD40L, in that even low-level constitutive expression of CD40L results in abnormal proliferative responses in developing T lymphocytes.
Since a major advantage of trans-splicing over conventional gene therapy is that the corrected genes are regulated by their endogenous regulatory machinery, we initiated a study to restore CD40L function in the HIGM1 knockout mouse model by corrective spliceosome-mediated trans-splicing [63]. To evaluate the ability of this strategy to correct a CD40L knockout mouse (KO), lentiviral trans-splicing vectors were constructed with normal exons 2–5 of CD40L cDNA with a hybridization domain targeted to the first intron of the endogenous CD40L pre-mRNA. Bone marrow cells from CD40L-KO mice were modified with these vectors and transplanted into syngenic CD40L-KO mice. RT-PCR and sequencing of CD40L-specific RT-PCR products from spleens of recipient mice 10 weeks after transplantation showed that trans-splicing occurred correctly in recipient mice. CD40L-KO mice immunized with keyhole limpet hemocyanin (KLH) and evaluated for IgM and IgG1 responses against KLH demonstrated that trans-splicer recipients produced both IgM and IgG1 against KLH while CD40L-KO mice were able only to produce antigen specific IgM not IgG1. Regulated expression of CD40L on CD4+ T cells occurred following CD3 stimulation in primary and secondary transplanted mice, and there was marked attenuation of proliferation of P. carinii in the lung following respiratory tract challenge with the organism. Importantly, assessment of the trans-splicer corrected CD40L-KO mice over 1 year demonstrated no evidence of lymphoproliferative disease. These studies demonstrate the feasibility and safety of correcting endogenous CD40L in HIGM1 by trans-splicing, with functional correction of the genetic defect and without the adverse consequences of unregulated CD40L gene expression.
8 Segmental trans-splicing
In a new paradigm of spliceosome-mediated therapeutic trans-splicing, referred to as ‘segmental trans-splicing’ (STS), genes are delivered in separate gene transfer vectors, and the two pre-mRNA fragments are joined by spliceosome-mediated trans-splicing to create an intact pre-mRNA (Fig. 4). The applications of therapeutic STS are still being explored, but it has already been shown to be useful in delivering large genes, as well as toxin genes.
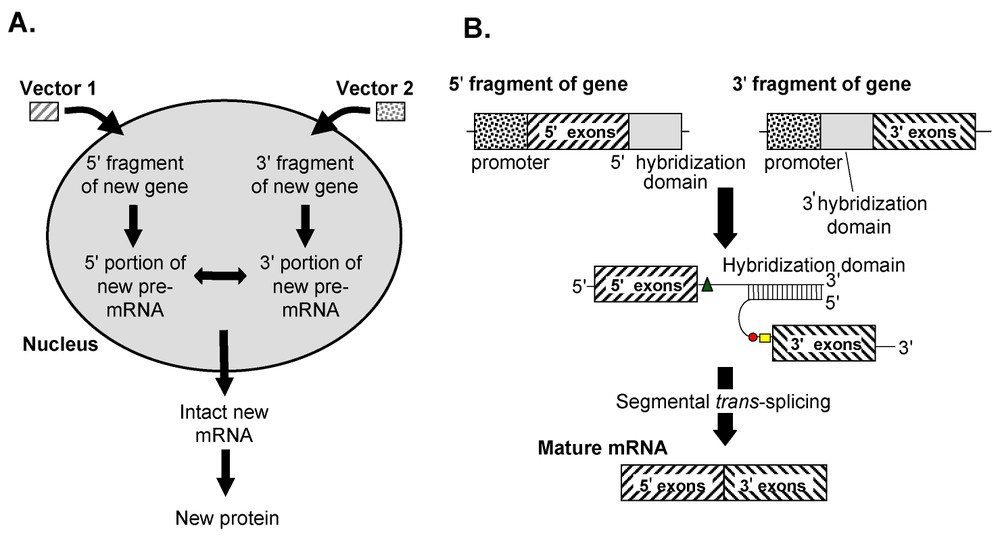
Therapeutic spliceosome-mediated segmental trans-splicing to join two exogenous pre-mRNAs. (A) Overview. and DNA fragments with relevant regulatory and hybridization sequences (also referred to as ‘donor’ and ‘acceptor’ fragments) are introduced to cells using two gene transfer vectors, each coding for a partial pre-mRNA with complementary hybridization domains and splice donor and branchpoint plus splice acceptor sequences on the donor and acceptor expression cassettes, respectively. Following transcription of the donor and acceptor expression cassettes, the two pre-mRNAs interact through the unique complementary hybridization domains. The intranuclear interaction of the pre-mRNA products results in the stoichiometric generation of full-length, functional mRNA mediated by splicesomes. (B) Details. Two engineered individual DNA fragments coding for and fragments of pre-mRNA of the coding sequence an entire gene are delivered using separate gene transfer vectors. These DNA sequences contain a ‘donor’ sequence that includes a promoter and coding sequences of the N-terminal portion of the protein of interest, and a ‘acceptor’ sequence that includes a promoter and coding sequences of the C-terminal portion of the protein. The end of the donor fragment and end of the acceptor fragment contain complementary foreign hybridization sequences that facilitate efficient trans-splicing of the two pre-mRNA fragments. Neither the donor nor acceptor pre-mRNAs alone generates any protein product, but when both are present, the hybridized donor and acceptor pair are recognized by the spliceosome as a functional unit, and the intronic sequences are spliced out to generate a mature functional mRNA.
As described above, all naturally occurring pre-mRNA trans-splicing, and all previous attempts to utilize trans-splicing using gene transfer, have involved the replacement of a portion of a pre-mRNA with an alternative leader or alternative exon sequence [76]. Since the recognition of these alternative RNAs for the pre-mRNA is based on hybridization, the efficiency of these systems, as represented by the kinetics of bi-molecular reactions, is limited by the dependency of hybridization on the initial concentrations of the molecular participants. Thus, limiting amounts of either the pre-mRNA or the trans-splicing alternative RNA establish the maximal degree to which the reaction can proceed. To address this issue, we recently developed a new strategy of in vivo gene transfer termed ‘segmental trans-splicing’, in which individual ‘donor’ and ‘acceptor’ DNA sequences, delivered in vitro or in vivo, generate pre-mRNAs with and splice signals respectively, and complementary hybridization domains through which the two pre-mRNAs interact, facilitating trans-splicing of the two mRNA fragments. In STS, the kinetic bottleneck limiting the degree to which the reaction can generate an altered mRNA is eliminated by generating two separate RNAs at high concentrations, which contain unique binding sequences to ensure a single bimolecular event. Once this binding has occurred, the splicing reaction that ensues is presumably indistinguishable from traditional cis-splicing. The trans-splicing constructs used in STS are designed for maximal efficiency of trans-splicing, but neither the donor or acceptor constructions produce a viable pre-mRNA without the other [77].
We demonstrated the effectiveness of STS using α-cobratoxin as a model system, based on the robust and easily identifiable phenotype associated with this toxin [78], using the high-volume, hydrodynamic tail vein injection technique that results in the efficient transduction of liver with plasmid expression vectors [77,79]. Two individual vectors containing the donor and acceptor domains of cobratoxin, respectively, were delivered, each of which is transcribed to create a partial pre-mRNA, engineered to bind only to each other. While the Poisson distribution problem of distribution of two different vectors to each cell remains a theoretical challenge, since a cell must get both vectors to produce a product, the robust expression of the α-cobratoxin phenotype in mice in this study indicates that sufficient numbers of cells are transduced by both vectors to generate an easily identifiable, unambiguous result. By comparison to studies with α-cobratoxin protein injected intravenously, it was demonstrated that at least 2 μg of the toxin must have been secreted following administration of 7.5 μg of the donor and acceptor plasmids. It is remarkable that the level of α-cobratoxin produced by trans-splicing was comparable to that produced from similar amounts of intact α-cobratoxin cDNA, a sufficiently robust expression that could be exploited in a variety of gene therapy strategies.
8.1 The large-gene problem
STS can be used to expand the size and complexity of the payload of gene therapy vehicles, which should enable new strategies to emerge for the delivery of therapeutic genes too large to fit into conventional gene transfer vectors. In this regard, the capacity of adeno-associated virus is 4.5 kb, and conventional E1–E3-adenovirus is 7.5 kb, while the capacity of retrovirus and lentivirus vectors is limited to less than 10 kb [80,81]. Creating gene therapy vectors for large cDNAs such as von Willebrand factor gene (von Willebrand disease, 8.6 kb), dystrophin (muscular dystrophy, 11.0 kb); fibrillin 1 (Marfan syndrome, 8.6 kb); neurofibromatosis-1 gene (Von Recklinghausen disease, 8.4 kb), ataxia telangiactasia mutated gene (ataxia telangiectasia, 9.2 kb), adenomatous polyposis colon gene (colon cancer, 8.5 kb); and Huntingtin (Huntington's Disease, 9.4 kb), among others, has been a limitation for most vectors, especially when using their native promoters to drive regulated expression. Assembling these molecules in target cells by segmental trans-splicing should be readily possible.
8.2 The toxic-gene problem
Although there is considerable interest in the use of toxins as potential therapeutic agents for cancer, the delivery of intracellular toxins by gene therapy has been hampered by technical difficulties with preparation of the vectors due to the fact that expression of the toxin kills the packaging cells during generation of the vector. As for the large gene issue, STS provides a solution to this problem by generating the toxic gene product from two non-toxic pieces a process in which two engineered individual DNA fragments coding for and fragments of pre-mRNA of a gene for the toxin are delivered to mammalian cells. These DNA sequences include a ‘donor’ sequence that includes a promoter and coding sequences of the N-terminal portion of the toxin of interest and a ‘acceptor’ sequence that includes a promoter and coding sequences of the C-terminal portion of the toxin.
A recent study in our lab focused on the use of STS to deliver an intracellular toxin to treat cancer. As an example, we used Shigatoxin, a potent intracellular toxin produced by Shigella dysenteriae and some strains of E. coli [82–84]. The toxin is a multi subunit protein made up one molecule of the A subunit responsible for the toxicity of the protein, and five molecules of the B subunit responsible for binding to a specific cell type. Once inside the cell, the A subunit of Shigatoxin, an N-glycosidase that modifies the RNA component of the ribosome to inactivate it, causes the cessation of protein synthesis and death of the cell. Removing the coding sequence for the B subunit ensures that any subunit A produced by a cell will remain intracellular, and have no effect systemically. We demonstrated the utility of STS for the generation of intracellular toxins by assembling Shigatoxin A subunit mRNA from non-toxic pre-mRNA segments delivered to tumor cells via adenovirus vectors in vitro and in vivo. Other potent intracellular cytotoxin or apoptosis inducing genes which could be used in this manner include, diphtheria toxin-A chain, ricin-A peptide, caspases, or Fas-ligand [85]. Although there exist considerable technical obstacles to the delivery of toxins by STS to anything other than foci of localized tumor cells in vivo, the apoptotic presentation of tumor antigens to cells of the immune system may provide an additional level of anti-tumor activity.
Importantly, while intratumoral administration of the and fragments of the Shigatoxin A subunit suppressed tumor growth, it did not cause systemic toxicity. Since, in this strategy, only the cells infected with both the donor and the acceptor STS fragments should be killed, the risk of systemic leakage of the gene transfer vector is minimized. For instance, if the risk of leakage of one virus is 1%, the risk of leakage of both virus vectors should be 0.01% and the chance of both vectors reaching the same cell in distant organs is negligible. These properties suggest there is an extremely low risk of STS side effects by systemic intoxication.
8.3 True trans-splicing vs concatemerization
The process of segmental trans-splicing combines two (or more) pieces of genetic information at the pre-mRNA level. This is a different process than combining two of pieces of genetic information at the DNA level, either via insertion into the genome or via recombination of the two individual DNA segments in the nucleus. An example of the latter process is the use of two adeno-associated virus (AAV) gene transfer vectors to combine two fragments of a gene that are too large to fit into the small (4.4 to 4.5 kb) genetic cargo capacity of an individual AAV vector [80,81,86]. Unlike spliceosome-mediated segmental trans-splicing that occurs via RNA splicing at the pre-mRNA level, AAV vector-mediated combination of two DNA segments occurs by concatemerization, after which transcription yields a single pre-mRNA that is then spliced by normal cis-splicing [87–93]. While AAV vector DNA concatermization clearly works, these vectors can concatamerize in all possible arrangements (head to tail, head to head, tail to tail), most of which are non-productive in terms of the desired full length RNA product, with the concomitant transcriptional burden generating some unusable products. In this context, segmental trans-splicing theoretically is likely less wasteful of cellular resources. A head-to-head comparison of STS with AAV vector concatermization has not been assessed, and thus it is not possible at this time to estimate which is more efficient and less prone to errors.
9 Future prospects
Ribozyme- and spliceosome-mediated trans-splicing are strategies to replace a defective exon with a corrected version of the same gene segment. This ability to interrupt the processing of a naturally occurring gene by the invasion of an alternative sequence provides the opportunity to perform unique experiments in protein evolution. In addition, gene segments can be deliberately ‘mixed and matched’ at will, at least in theory, and the resultant products assayed for the development of novel activities, transcriptional responses to stimuli, cellular trafficking, and regulation. Such experiments may provide insight into eukaryotic evolution, as well as a new paradigm for drug discovery.
The notion of mixing protein domains may be even simpler using the segmental trans-splicing paradigm. Since there is no limit to the number of donor and acceptor domain vectors that could be generated with the same binding domain, it would be possible to use STS to create diversity within a restricted group. Trans-splicing can also be employed to add ‘tags’ to an mRNA in order to direct it to an alternate cellular compartment. Signal peptides can be added, for example, to direct a portion of the production of a given protein to be secreted. Other signals could direct mutant proteins toward degradative compartments, to avoid the complications of improper protein processing as seen, for example, in the serpinopathies [94].
In theory, there is no limit to the number of segments that could be assembled using the STS paradigm, aside from potential limits imposed by the judicious choice of binding domains. If combined with gene transfer vectors that integrate, this strategy could provide long lasting large gene expression in appropriate cells, irrespective of the sites of integration.
Additional control of gene expression can be conferred in genes transferred by STS by the use of promoters with dual specificities. For example, the segmental assembly of an intracellular toxin or an inducer of apoptosis in a cancer cell could be arranged such that the production of one segment is under the control of a liver specific promoter, such as the albumin promoter, and the second segment is under the control of a cancer specific promoter, such as the survivin promoter [95]. Such an arrangement would ensure that only cancerous liver cells would express both segments and be eliminated. This is relevant to potential cancer therapies in the context that a single active vector containing the cDNAs for intracellular toxins cannot be produced, since they kill the cells in which they are grown and amplified. Thus, STS provides a means of on-site generation of intracellular toxins for therapeutic use, without exposure to any toxic material during manufacture of the precursors.
A critical question requiring further study for repair of mutant RNAs is the precision and specificity of the repair process. Trans-splicing must be highly specific in order to limit the number of illegitimate and possibly toxic or immunogenic products generated. In one published report, spliceosomal trans-splicing seemed to lack specificity, with the trans-spliced element appearing on unintended target RNAs in mammalian cells [96]. Attempts to modify the trans-splicer to suppress nonspecific splicing failed to have the desired effect. Although these observations raise the possibility that nonspecific spliced products may be produced by gene therapy constructs delivering spliceosome-mediated trans-splicing payloads, it has been suggested that increasing the size of the hybridization domain may improve specificity [62]. Segmental trans-splicing should not suffer from this limitation, since no endogenous pre-mRNA is targeted. Ribozyme-mediated repair may also suffer from a lack of specificity [97], but recently described modifications to the ribozyme address this issue [98]. Pilot experiments have been sufficiently promising to warrant additional studies aimed toward development of more specific trans-splicing agents. Likewise, preclinical studies directed toward therapeutic applications of trans-splicing performed to date have been encouraging, demonstrating that trans-splicing can amend mutant transcripts associated with a variety of human diseases.
Significant improvements are also needed in the efficiency of trans-splicing before it can be considered a mature therapeutic modality. Reported efficiencies for spliceosome-mediated pre-mRNA trans-splicing have rarely exceeded ten percent, with most being closer to two percent [62,65–67], although segmental trans-splicing may be more efficient [77]. Evidence suggesting that nascent transcripts assemble with splicing factors and undergo splicing co-transcriptionally raises some theoretical obstacles to improving the efficiency of trans-splicing [99,100]. The linkage of splicing and transcription suggests that the two sets of machinery, both complex and very large, must operate in close proximity to each other. This may impose steric limits to the amount of any trans-splicer that might gain access to the nascent pre-mRNA, and thus to the degree of trans-splicing that occurs [101]. The inclusion of specific exonic enhancer sequences into the trans-splicer pre-mRNA has been shown to improve the efficiency of naturally occurring trans-splicing in vitro [102], but this process has not been investigated in therapeutic trans-splicing strategies. Such strategies may lead to significant improvements in the efficiency, specificity and safety of therapeutic trans-splicing.
Acknowledgments
We thank N. Mohamed and Tienne Virgin-Bryan for help in preparing this manuscript. These studies were supported, in part, by P01 HL51746; the Will Rogers Memorial Fund, Los Angeles, CA; and The Malcolm Hewitt Wiener Foundation, Greenwich, CT.