

1 Introduction
« …κυαμων απɛχɛσθɛ… » – tenez-vous loin des fèves – recommandait Pythagore au VIe siècle av. J.-C. Ces instructions cachaient-elles l'observation que ce végétal était un poison pour certains, ou plutôt correspondaient-elles à des interdits alimentaires religieux de la Grèce antique, liés en particulier à l'aspect de la graine évoquant les portes de l'Enfer ou faisant penser à un testicule ? Il était interdit aux prêtres d'en manger et même de prononcer ce nom. En revanche, Hippocrate ne mentionne pas les fèves dans son traité de diététique et les Latins en faisaient une large consommation [1].
Il faut attendre le milieu du XIXe siècle pour qu'une revue de médecine portugaise publie l'observation d'un malade faisant des poussées ictériques chaque fois qu'il mangeait des fèves [2]. À partir de la fin du XIXe siècle, des cas de plus en plus nombreux d'accidents liés à leur ingestion, voire à l'inhalation de leurs pollens, sont rapportés dans la littérature [1]. Ils concernaient des Italiens, surtout dans le Sud, en Sardaigne et en Sicile, dans ces régions où les fèves sont largement cultivées. Le terme de favisme est alors créé pour décrire cette curieuse susceptibilité.
Une seconde étape dans l'histoire de cette anomalie remonte aux années 1940, où l'on reconnaît son origine génétique : les cas de favismes observés concernaient habituellement des sujets méditerranéens et jamais des sujets originaires de l'Europe du Nord. Par ailleurs, au cours de la seconde guerre mondiale, la primaquine, donnée à titre préventif contre le paludisme dans les troupes américaines du Sud-Est Asiatique, conduisait à des accidents hémolytiques similaires chez des militaires noirs ou méditerranéens.
La troisième étape dans la compréhension du favisme a été la démonstration d'un déficit en glucose 6-phosphate déshydrogénase (G6PD) érythrocytaire chez tous les sujets qui en étaient atteints [3]. Ce déficit, initialement décrit en Méditerranée, est retrouvé dans de très nombreuses populations d'Afrique, du Moyen-Orient et d'Asie. Sa distribution se superpose à celle du paludisme, car, comme nous le verrons plus loin, le déficit en G6PD assure un certain degré de protection contre cette parasitose [4].
On estime aujourd'hui qu'à travers le monde plus de 400 millions de personnes portent un déficit en G6PD [5]. Le gène de cette enzyme est localisé sur le chromosome X, ce qui explique le mode de transmission du déficit, lié au sexe, avec des sujets mâles hémizygotes pour l'anomalie et des femmes, transmettant l'anomalie mais, habituellement, cliniquement indemnes. Toutefois de nombreuses femmes sont homozygotes dans les populations où la fréquence génique est élevée, par exemple elles sont près de 3 % en Côte-d'Ivoire [6] et de 5 % en république du Congo.
Sur la base de mesures d'activités enzymatiques et de paramètres électrophorétiques, on a décrit jusqu'à 400 variants différents, avant qu'ils n'aient été caractérisés de façon plus précise par les techniques de la biologie moléculaire. Le séquençage des gènes a montré que le nombre de variants se situe en réalité aux environs de 150 [7]. La connaissance de la structure spatiale de l'enzyme permet maintenant d'ébaucher des hypothèses expliquant les anomalies en termes de relation structure–fonction [8].
2 La glucose 6-phosphate déshydrogénase
La glucose 6-phosphate déshydrogénase catalyse la première étape de la voie des pentoses : elle transforme le glucose 6-phosphate en 6-phosphogluconolactone, qui s'hydrolyse classiquement en 6-phosphogluconate. Lors de cette réaction, une molécule de NADP+ est réduite en NADPH+ H+. La seconde réaction de cette voie, transformation du 6-phosphogluconolactone en ribulose-6-phosphate, produit également du NADPH, mais, chez les sujets déficitaires en G6PD, elle est totalement perturbée par le ralentissement de la première étape. Le NADPH joue un rôle essentiel dans la réduction des agents oxydants, en permettant, en particulier, dans le globule rouge, de maintenir le pool de glutathion réduit à un niveau normal (Fig. 1). Normalement, le glutathion réduit est en très large excès par rapport au glutathion oxydé [9]. Des travaux récents montrent qu'un rôle protecteur du NADPH sur la catalase érythrocytaire serait tout aussi important dans la lutte contre les stress oxydants [10]. De plus, des études in vitro indiquent que, dans l'endothélium vasculaire et les muscles lisses, la G6PD module la réponse à un stress oxydant en formant du NADPH, qui intervient, d'une part, par la réduction du glutathion et, d'autre part, comme cofacteur de la NO-synthase [11].

Rôle biochimique de la G6PD à l'entrée de la voie des pentoses phosphates. La production de NADPH maintient à un haut niveau le pool de glutathion réduit et stabilise la structure de la catalase. Ceci permet à la cellule de se défendre contre tout stress oxydant.
La G6PD est une enzyme ubiquitaire présente dans toutes les cellules : Notaro et al. ont relevé en 2000 dans les banques de données plus d'une cinquantaine de séquences homologues à celle de l'enzyme humaine [12]. Cette enzyme est retrouvée dans la plupart des espèces, des micro-organismes à l'homme ; les quelques rares exceptions concernent des micro-organismes vivant dans des milieux pauvres en oxygène ou chez des parasites pouvant profiter de l'activité enzymatique de l'hôte. L'homologie de séquence entre les diverses enzymes est très forte : 94 % entre mammifères et 20 % entre mammifères et micro-organismes [12]. La G6PD humaine comporte 515 résidus, sa structure est identique à celle d'autres primates (chimpanzé ou gorille). Chez l'homme, cette protéine est codée par un gène constitué de 13 exons, situé sur le locus q28 du chromosome X (Fig. 2).

Le gène de la G6PD est localisé dans la région q28 du chromosome X. Il comporte 13 exons et s'étend sur environ 20 kb. Le 1er exon n'est pas traduit et l'intron situé entre les exons 2 et 3 représente à lui seul près des deux tiers de la taille de gène.
La G6PD de Leuconostoc mesenteroides a été la première qui fut cristallisée et étudiée par diffraction de rayons X : sa forme active est un homo-dimère [13]. L'établissement de cette structure a permis d'expliquer, sur le plan des relations entre structure et fonction, le comportement anormal d'un certain nombre de mutants de la G6PD humaine [14]. Plus récemment, un mutant de l'enzyme humaine, la G6PD Canton, où l'arginine 459 est remplacée par une leucine, a pu être cristallisée [15]. La forme active est également un homo-dimère, qui s'associe en tétramère en fonction du pH et de la force ionique (Fig. 3). On observe, dans chaque sous-unité, la présence d'une molécule de NADP+ faisant partie intégrale de la structure de l'enzyme et localisée à distance du site catalytique (Fig. 4a). Comme on le voit sur la Fig. 4b, plusieurs domaines impliqués dans la structure et la fonction de la molécule peuvent être individualisés. Comme on l'a représenté dans le Tableau 1, on peut distinguer 12 régions dans la molécule de G6PD [12]. Notons tout spécialement l'aire de contact entre les deux monomères, le site de fixation du coenzyme et la zone catalytique.

(a) Structure tridimensionnelle d'un tétramère de G6PD. Ce tétramère résulte de l'association de deux dimères fonctionnels (A/B et C/D). Chaque monomère contient une molécule de NADP+ qui lui est étroitement liée. (b) Vue stéréoscopique de ce tétramère ; les molécules de NADP+ sont représentées en orange. Schéma réalisé à l'aide des logiciels SwissProt et Rasmol à partir des coordonnées cristallographiques du fichier 1QKI de la PDB.
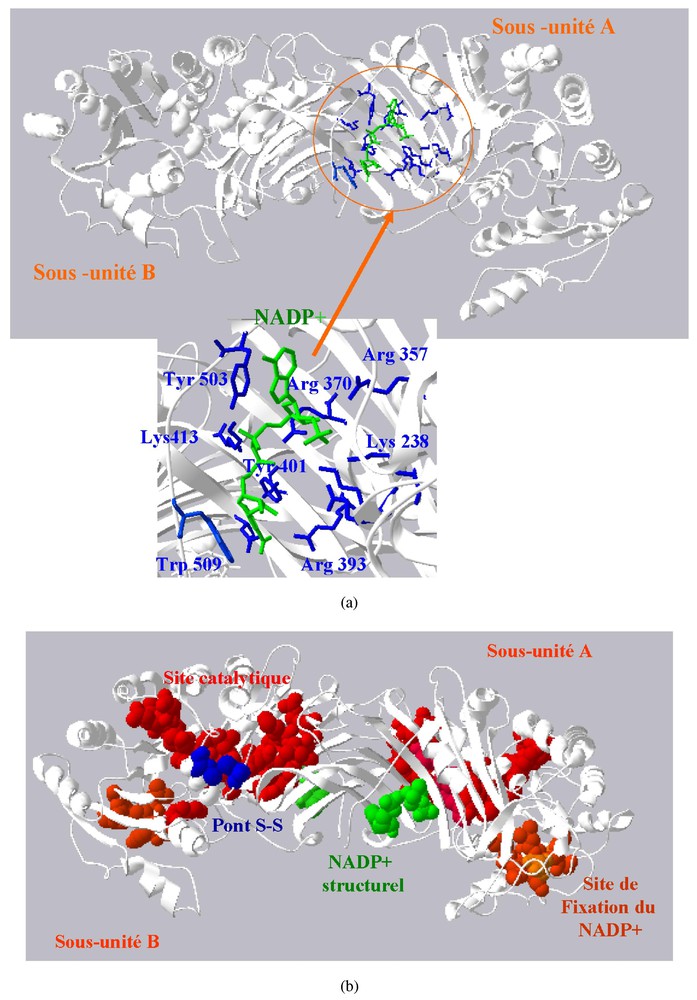
(a) Vue de la région où se fixe la molécule de NADP+ structurelle. Seuls sont indiqués les résidus à son contact dont les modifications conduisent à des enzymes déficientes de type I (cf. Tableau 2). (b) Schéma montrant le site de fixation du coenzyme (région I), le site catalytique (région IV) et la zone de contact entre les sous-unités A et B stabilisées par la molécule de NADP+ (cf. Tableau 2).
Corrélations structure–fonction et nombre de variantes des divers types identifiées dans chaque région
| Région | Résidus | Exon | Fonction | Type I | Type II | Type III |
| Région I | 34–53 | Exons 2–3 | liaison du coenzyme | 3 | 2 | 2 |
| 53–136 | 3 | 5 | 9 | |||
| Région II | 137–148 | Exon 5 | core hydrophobe | 1 | ||
| 149–165 | 1 | 1 | 3 | |||
| Région III | 166–180 | Exon 6 | face au site catalytique | 3 | 2 | 1 |
| 181–192 | 2 | |||||
| Région IV | 193–218 | Exon 6 | site catalytique | 3 | 1 | 1 |
| 219–239 | 2 | 2 | 2 | |||
| Région V | 240–274 | Exons 7–8 | face au site catalytique | 4 | ||
| 275–283 | 3 | 3 | 2 | |||
| Région VI | 284–292 | Exon 9 | hélices α amphipathiques | 1 | 2 | |
| 293–332 | 4 | 3 | 3 | |||
| Région VII | 333–339 | Exon 9 | core hydrophobe | 1 | 1 | |
| 340–346 | 1 | |||||
| Région VIII | 347–362 | Exons 9–10 | core hydrophobe | 2 | 2 | 1 |
| 363–364 | 2 | |||||
| Région IX | 365–376 | Exon 10 | core hydrophobe | |||
| 377–387 | Exon 10 | 7 | 1 | |||
| Région X | 388–404 | Exon 10 | contact entre sous-unités | 7 | 1 | |
| 405–432 | 7 | 2 | ||||
| Région XI | 433–443 | Exon 11 | core hydrophobe | 1 | 1 | |
| 444–450 | 1 | 2 | ||||
| Région XII | 451–464 | Exon 12 | hélices α amphipathiqes | 2 | 4 | |
| 465–515 | 3 | 1 |
Toute cellule nucléée est capable en permanence d'adapter sa teneur en G6PD en réponse à un stress oxydant. Il en est tout autrement dans la lignée érythrocytaire, où la synthèse protéique cesse peu après l'énucléation. Dans un globule rouge, l'enzyme présente est celle qui a été synthétisée dans les précurseurs érythroïdes. Dans le sang périphérique, l'activité enzymatique est à son maximum dans le réticulocyte et le globule rouge jeune. Chez un sujet normal, cette activité initiale est 50 fois supérieure à celle qui est suffisante en l'absence d'un stress oxydant. Avec une demi-vie de 62 jours, le stock d'enzyme disponible diminue régulièrement avec l'âge de la cellule. Chez le sujet normal, cette activité reste largement supérieure aux besoins de l'érythrocyte, ce qui explique la bonne tolérance d'un déficit modéré [16,17].
3 Physiopathologie des déficits en G6PD
La G6PD B est la forme sauvage, normale. Les études électrophorétiques ont mis en évidence un polymorphisme fréquent, d'activité normale, où l'asparagine en position 126 est remplacée par un aspartate. Cette variante est appelée G6PD A. On connaît plusieurs cas où une seconde mutation a été trouvée sur ce même allèle, entraînant une diminution modérée d'activité, ces variants sont appelés G6PD A–. La fréquence de la G6PD A– est voisine de 20 % dans les populations noires africaines [6]. On en connaît trois types, que l'on distingue par l'anomalie qui s'ajoute à la substitution Asn → Asp présente en position 126 [6]. Il s'agit des substitutions 68 Val → Met, 227 Arg → Leu, ou encore 323 Leu → Pro.
Dans le cas des G6PD A–, l'activité initiale de l'enzyme, mesurée dans les réticulocytes d'un sujet hémizygote, est très proche de la normale (8,8 U contre 9,7 U). Le phénomène majeur est une instabilité modérée de la protéine, qui réduit sa demi-vie à 13 jours (normale = 62 jours). Ainsi, au bout de 21 jours, l'activité résiduelle n'est plus que de 30 % de l'activité initiale et, au bout de 63 jours, de 3 %, ce qui est encore suffisant pour assurer les besoins de la cellule. Au bout de 90 jours, cette activité ne représente plus que 0,8 % de celle d'un globule jeune et la survie cellulaire est alors menacée par tout stress oxydant. Chez les porteurs hémizygotes d'une G6PD A–, l'activité enzymatique globale de la population érythrocytaire se situe entre 5 et 15 % de la normale [16,17]. Ce déficit peut être compensé par une légère stimulation de l'activité médullaire, ce qui se traduit par une réticulocytose environ double de la normale (entre 1 et 2,5 % versus 0,8 %), sans manifestation anémique. Une classification internationalement admise, établie en fonction de l'activité enzymatique et des effets cliniques, situe la G6PD A– dans le type III (Tableau 2) [18].
Classification des variants de la G6PD en fonction de leur activité
| Type* | Critères |
| Classe I | Déficit enzymatique avec anémie hémolytique non sphérocytaire chronique |
| Classe II | Déficit enzymatique sévère avec activité enzymatique inférieure à 10% de la normale |
| Classe III | Déficit enzymatique discret ou modéré avec activité enzymatique comprise entre 10% et 60% de la normale |
| Classe IV | Activité comprise entre 60 et 150% de la normale |
| Classe V | Activité accrue, supérieure à 150% de la normale |
* On doit ajouter à cette classification les variantes dont l'activité enzymatique est totalement abolie et qui, de ce fait, sont létales.
La variante « méditerranéenne » (188 Ser → Phe) est plus sévère que la variante A–. Son activité dans les réticulocytes se situe à 20 % de la normale et, dans le sang périphérique, à moins de 5 %. Sa demi-vie de 8 jours témoigne d'une instabilité nettement plus marquée que celle de la G6PD A– et entraîne sa classification dans le type II. Elle est trouvée dans le pourtour méditerranéen, le Proche- et le Moyen-Orient où, selon les régions, elle intéresse 10 à 25 % de la population, voire plus, localement. Cette variante, bien tolérée en l'absence de stress oxydant, peut conduire à des accidents hémolytiques graves sous l'effet de multiples facteurs déclenchants. La diminution d'activité G6PD de globules rouges normaux et anormaux en fonction de leur âge est représentée sur la Fig. 5.
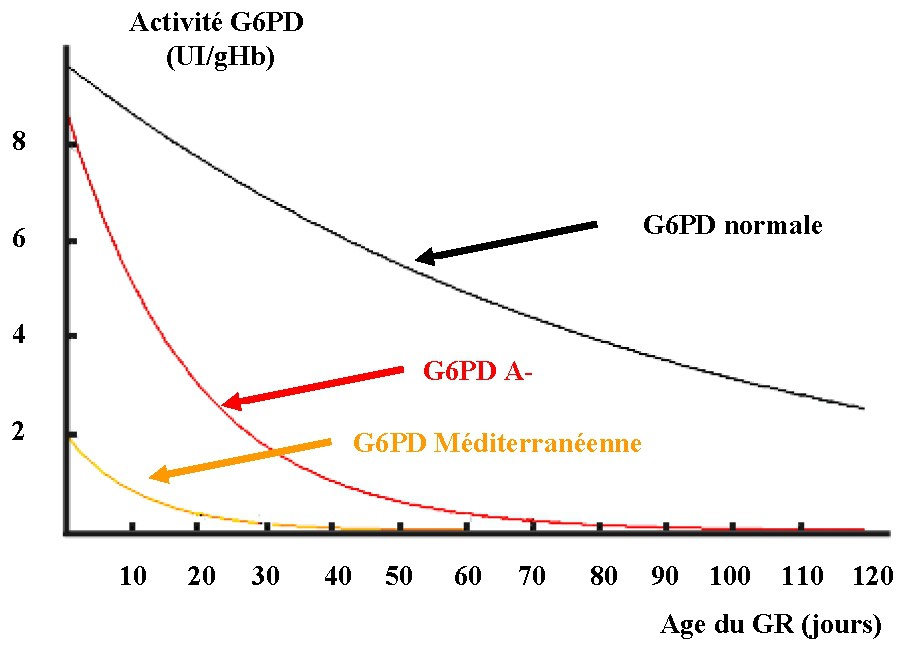
Décroissance d'activité de la G6PD en fonction de l'âge de l'érythrocyte chez un sujet normal ou porteur d'un déficit enzymatique (G6PD A– et G6PD méditerranéenne).
Lors d'un stress oxydant, chez les sujets porteurs d'un déficit en G6PD, la transformation de l'hémoglobine en méthémoglobine est accélérée par la présence d'un excès de radicaux libres. D'autres dérivés oxydés d'hémoglobine, tels que les hémichromes, se forment et précipitent en corps de Heinz, qui se fixent à la membrane érythrocytaire. Ces derniers sont à leur tour à l'origine de radicaux libres oxygénés, particulièrement toxiques chez le déficitaire en G6PD, puisque les mécanismes de réduction sont défaillants. Les globules rouges fragilisés sont éliminés par les macrophages lors de leur passage dans le filtre splénique. Dans les accidents les plus graves, on observe une hémolyse intra-vasculaire parfois importante, avec alors des effets sévères sur la fonction rénale. Ces agressions oxydantes peuvent être provoquées par des aliments, des médicaments, ou divers produits chimiques, dont la liste, connue dans ses grandes lignes, s'accroît régulièrement [19]. Elles peuvent entraîner des accidents hémolytiques gravissimes chez les porteurs d'un déficit en G6PD. La prévention de ces accidents pourrait toutefois être facilement organisée par un diagnostic généralisé du risque, et une bonne information permet aux déficitaires en G6PD de mener une vie tout à fait normale, exempte de tout accident hémolytique. La question est toutefois de savoir si le principe de précaution doit exclure les porteurs d'un déficit en G6PD du bénéfice thérapeutique que pourraient leur apporter certaines molécules, sous le prétexte d'un accident exceptionnel rapporté dans la littérature.
Certaines variantes, comme les G6PD A–, méditerranéenne ou Canton sont, de par leur fréquence, de véritables polymorphismes, alors que d'autres, retrouvées dans les mêmes populations, sont plus rares. Des mutations privées, trouvées à l'occasion d'accidents hémolytiques, sont régulièrement décrites, affectant alors n'importe quelle population. Il s'agit parfois de néomutations.
La liste des 150 variants de la G6PD montre, dans tous les cas, sauf deux, qu'une anomalie de structure est à l'origine de la diminution d'activité enzymatique. Il s'agit, soit de mutations faux-sens (parfois plusieurs sur un même allèle), soit de courtes délétions, sans décalage du cadre de lecture, dans des régions répétitives. Les exceptions concernent un cas d'épissage anormal et un cas de décalage du cadre de lecture, toutes deux affectant la seule traduction de l'exon C-terminal, et permettant vraisemblablement la synthèse d'une protéine tronquée, encore pourvue d'un reliquat d'activité.
Dans certains cas, la protéine peut être instable, parce que mal repliée, ouvrant sa partie centrale hydrophobe au milieu aqueux environnant ; ailleurs, la substitution modifie les contacts au voisinage de l'interface entre les deux sous-unités, empêchant la réalisation de la structure dimérique active. Plusieurs mutants de type I concernent la région où se fixe la molécule de NADP+, qui fait partie de la structure de l'enzyme et stabilise la géométrie de l'aire de contact entre monomères. C'est dans la région X (Tableau 1) et son voisinage que se localise le plus grand nombre de variants de type I. Elle est codée par les exons 10 et 11, et correspond à la zone de contact entre sous unités.
Des anomalies génétiques aboutissant à un défaut total de synthèse, comme une mutation altérant les séquences consensus d'épissage, ou des délétions avec décalage du cadre de lecture, n'ont jamais été trouvées chez des sujets déficitaires en G6PD. Elles sont probablement létales [8].
Les manifestations hémolytiques des déficits en G6PD sont habituellement observées chez les sujets mâles, hémizygotes pour l'anomalie. En réalité de nombreux cas sont également trouvés chez des femmes, le plus souvent lorsqu'elles sont homozygotes pour un déficit ou hétérozygotes composites, et plus rarement lorsque l'inactivation d'un chromosome X par lyonisation s'effectue essentiellement au détriment du chromosome normal.
Les techniques de biologie moléculaires et en particulier le séquençage direct de l'ADN des sujets déficitaires en G6PD permet de caractériser aujourd'hui facilement un nombre croissant de nouveaux mutants. La sélection des sujets étudiés fait que, le plus souvent, ils se classent dans les types I ou II. Des listes de variants avec leurs principales propriétés sont disponibles sur Internet [19,20].
4 Facteurs déclenchant une crise hémolytique
Les agents susceptibles de provoquer un accident hémolytique ont en commun la propriété d'être des substances ayant des propriétés oxydo-réductrices. C'est le cas des fèves, qui contiennent deux glycosides, la vicine et la convicine, dont l'hydrolyse conduit à la divicine et à l'isouramil, composés dont les propriétés oxydantes peuvent être rapprochées de celles de la quinine [21]. Les fèves sont en effet connues pour avoir un certain effet antipaludéen. Des accidents ont parfois été observés avec des boissons toniques contenant de la quinine.
Ces propriétés oxydantes sont partagées par une longue liste de médicaments. Les anti-paludéens ont été les premiers décrits comme responsables d'accidents hémolytiques. La chloroquine (4-aminoquinoline), par exemple, est un composé amphipatique légèrement basique qui pénètre dans les vacuoles alimentaires riches en catabolytes de l'hémoglobine produites par le parasite dans les érythrocytes impaludés. En diminuant le taux de glutathion disponible, ce médicament rend plus difficile leur détoxication et accroît l'effet toxique des radicaux oxygénés libérés. La primaquine (8-aminoquinoline) agit par un de ses métabolite hépatique qui, par le biais d'un stress oxydant sur l'erythrocyte, consomme du GSH [22]. L'utilisation de doses supérieures aux doses thérapeutiques entraîne même chez un sujet normal l'effondrement du GSH, phénomène évidemment majoré chez le déficient en G6PD. Sulfamides et chloramphenicol [23] font également baisser le taux de GSH d'une façon dose dépendante. Des accidents ont été décrits aussi bien avec des sulfamides bactéricides qu'hypoglycémiants [7,8,24].
Certains médicaments comme des anesthésiques ou des antibiotiques agiraient en inhibant l'activité de la G6PD [25]. Les accidents les plus graves que nous avons observé au cours de ces dernières années sont survenus lors de chimiothérapie d'induction comportant des générateurs de radicaux libres (F. Galactéros, observations personnelles).
Les médicaments qui sont de puissants oxydants sont dangereux à toutes doses, alors que d'autres ne le sont qu'au-dessus des doses thérapeutiques habituelles [7,8]. Les grandes différences de susceptibilité individuelle sont à rattacher à de multiples facteurs, comme les différences génétiques dans le métabolisme des médicaments. Un déficitaire en G-6PD, acétylateur lent, est sans doute plus sensible à l'effet hémolytique d'un sulfamide que ne l'est un acétylateur rapide [26]. L'effet d'agents oxydants environnementaux vient sans doute se surajouter et conduit à une réponse très variable d'un cas à l'autre [27,28].
5 Déficit en G6PD et protection contre le paludisme
Un aspect particulier au déficit en G6PD est la protection qu'il assure contre le paludisme. Ce déficit, comme nous l'avons déjà mentionné, se superpose à la zone d'infestation par P. falciparum. Plus curieusement il se superpose également aux régions de culture de la fève et peut se rencontrer dans des régions où d'autres facteurs oxydants sont d'usage courant, comme les teintures cutanées par le henné [27–29] ou une alimentation particulièrement épicée [30]. Cette situation témoignerait d'une co-évolution entre peuples, plantes et parasites [28]. Dans le cas de la G6PD A–, la coexistence d'un stimulus oxydant modéré augmenterait les résistances de la cellule au développement du parasite [31].
En réponse à toute infection, des composés oxydants sont libérés dans les cellules et conduisent, dans le globule rouge, à des altérations membranaires avec formation d'épitopes reconnus par le système immunitaire. Ces manifestations sont normalement atténuées par les systèmes réducteurs de l'hématie. En cas de déficit en G6PD, ces mécanismes de défense érythrocytaires sont défaillants et les cellules altérées sont hémolysées. Une telle situation est un avantage pour l'hôte lors de l'infestation palustre, puisque le parasite est particulièrement sensible aux agents oxydants : le globule rouge d'un sujet déficient en G6PD, plus riche en composés oxydants, est un milieu peu favorable à son développement. Les expériences de culture de parasites effectuées sur des globules de sujets déficients pour la variante méditerranéenne le montrent plus nettement que chez les porteurs de la variante A– [32,33]. Cet effet n'est cependant que transitoire, car le parasite s'adapte rapidement mais cette adaptation est toute relative, car il reste particulièrement fragile à tout nouveau stress oxydant [34]. La protection contre le paludisme à P. falciparum concerne aussi bien les femmes hétérozygotes que les hommes hémizygotes, alors que certaines études anciennes semblaient limiter cette protection aux seules femmes hétérozygotes [27].
6 Conclusions
Un nombre considérable d'allèles de G6PD déficiente est observé dans les zones tropicales ou sub-tropicales actuellement ou anciennement impaludées. Cette distribution se superpose à celle des hémoglobinopathies, Hb S, Hb C et Hb E, et surtout à la multitude des allèles thalassémiques. Le déficit en G6PD illustre peut-être un phénomène d'évolution convergente dans l'adaptation à un milieu hostile.