

1 Introduction
Genome sequencing is producing huge amounts of data and modern experimental techniques are enabling large scale gene expression analysis (see, for example, [1–5]).
Mathematical modelling of gene expression can provide interpretation of experimental results, help to focus new experiments on critical missing data, and predictions can be made about results that are difficult to obtain in vivo. Mathematical modelling can still be used to explore and predict properties of a given system, testing the behaviour of different network structures and parameter values. On the other hand, the inferential power of modelling can be used to deal with ambiguous or unknown information in experiments.
Several theoretical strategies are being used to approach the modelling of gene regulation networks [6–9]. For example, several authors have developed simple representations of real genetic networks and have explored its basic properties [10,11]. Some models are built starting from simplified views of known regulatory networks and then the model parameters are adjusted in order to find results close to the patterns of gene expression observed in vivo [12,13]. Another approach is to infer unknown regulatory mechanisms from known experimental data [14–18]. A different strategy is the design of simple in vivo artificial genetic networks that have specific properties to be tested under several conditions [19–25].
From the mathematical point of view, there are several methodologies to describe genetic regulatory networks. Some authors use discrete models, building, for example, boolean or bayesian networks [26–32]. Others, use several types of continuous models, as differential equations, difference equations and reaction-diffusion equations [33–38]. All these models aim to describe the real system with different levels of detail.
Here we propose a simple mathematical framework in order to build models of gene regulatory interactions using differential equations. Our approach is based on the operon model of Jacob and Monod [39], as a paradigm of the mechanisms of gene regulation in bacteria. The description of these mechanisms starts by establishing a basic model for the quantitative description of protein production. The action of the activators and repressors regulating the expression of each gene are the building blocks of these mathematical models.
We consider that genes are templates for protein production, and regulation occurs due to the interconversion between the activated and the repressed states of the genes. We also assume that both transcriptional and translational mechanisms are described by one overall rate constant. As a consequence, we are able to describe genetic regulatory systems where the control of the protein production is associated with ‘on’ and ‘off’ states of the gene expression. The resulting gene expression patterns predicted by these mathematical models can be compared with the real experimental system in order to be validated and calibrated. A similar strategy can also be used to infer some unknown gene interactions from known expression patterns.
One of our concerns has been to maintain this mathematical framework as simple as possible, enabling the derivation of the qualitative properties of the models, as well as the possibility of constructing simple computer programs for the derivation and integration of the equations for the temporal evolution of protein concentrations. From the methodological point of view, this construction is based on the bi-molecular mass action law and the basic mechanisms do not introduce ad hoc regulatory functions, as is in general used in the mathematical modelling of gene expression [7,13,40–42]. As a consequence, the modelling strategy proposed here is close to the basic principles of chemical kinetics, and do not introduces neither ad hoc threshold effects, nor Michaelis-Menten type functional forms. However, threshold effects will be found as a consequence of the conservation laws associated to the mass action law.
In Section 2, we introduce the basic simplifying assumptions for the construction of the differential equations describing genetic regulatory networks, and we analyze the basic mechanism of protein production resulting from gene expression, without any regulatory mechanism. From Sections 3 to 6, we analyze the simplest cases of positive and negative regulation. It is shown that the proposed models have all the qualitative properties of the regulatory mechanisms analyzed in experiments. Therefore, if a biological mechanism in prokaryotes is described by a known genetic network, in principle, the formalism developed here is a good candidate to describe the most important qualitative features of the biological process under analysis.
In Section 7, as an illustrative example, we apply this framework to model of the negative self-regulation of the trp operon. In Section 8, we analyze the genetic switch involved in the regulation of the lac operon, responsible for the control of the lactose metabolism in Escherichia coli. Finally, in Section 9, we discuss the main conclusions of this paper.
2 The basic model for protein production from a DNA template
The simplest model of protein production from a DNA template without any regulation mechanism can be described by the kinetic diagram:
| (1) |
In chemical kinetics, diagrams of type (1) represent the collision between molecules and subsequent chemical transformation. In a chemical process of type (1), after the production of one molecule of protein P, the gene template, the mRNA and the catalysts become free, and new transcription and translation processes can be initiated. If the collisional interactions that initiates these processes occur randomly in the media, the time evolution of the several components is described by the mass action law, [43,44]. To be more specific, we consider several substances,
| (2) |
| (3) |
Applying the mass action law to the kinetic diagram (1), the time evolution of the concentrations of G, M and P is described by the differential equations:
| (4) |
To simplify the model diagram (1) and Eq. (4), we assume that the concentrations of C1 and C2 are not limiting factors of the reactions in (1), that is, C1 and C2 do not degrade. We also assume that during transcription the mRNA (M) concentration is constant, implying that, by (4),
| (5) |
| (6) |
The general solution of (6) is:
| (7) |
In this simple model for protein production, the use of the mass action law relies on the hypothesis that DNA templates, mRNA molecules and proteins are well distributed inside the cell and that the collisional interaction mechanisms are essentially random. The rate constants k and d are effective velocity constants multiplied by catalysts, nucleotides and aminoacids concentrations. In order to simplify the models, we will always avoid the explicit introduction of other species than gene templates (DNA) and proteins, being the gene concentration always a conserved quantity.
In the following, in order to describe the binding of transcription factors that account for activation and repression of proteins, we associate binding sites or operons to genes where transcription factors (transcriptional activators and repressors) can bind in order to promote or repress protein production, Fig. 1. This approach follows the operon model of Jacob and Monod [39].

Jacob and Monod operon model for the regulation of protein production in bacteria. The transcriptional regulators bind to the binding sites of the gene and transcription is activated or repressed. We can consider different binding sites for different transcription factors or, in the competitive case, the same binding site can be occupied by an activator or a repressor.
We will now consider the basic cases of protein production by positive and negative regulation. The interaction between the regulators and inducer or corepressor molecules will not be taken into account in these models. We will only consider the interaction between activator or repressor molecules and the respective DNA binding sites. These interactions obey the mass action law (see, for example, [45]) and this approach will be followed in the next sections.
3 Positive regulation
3.1 The activator total concentration is constant
In the mechanism of protein production by positive regulation, an activator binds to an operator site in the DNA and transcription and consequent protein production (translation) is initiated. Representing the activator concentration in the cell by A, the concentration of template gene by G, and the concentration of binded DNA-activator complex by
| (8) |
We are assuming that genes can be measured in units of concentration (number of molecules or gene templates per unit of volume). This applies to batch cultures where we have several copies of the same gene, and to tissues where we have several copies of identical cells. For cells with only one expressing gene, we can introduce an ergodic argument, and assume that for a time scale larger than the characteristic time of protein production, the protein production is also described by a mass action law mechanism. In fact, this ergodic argument is on the very foundation of the mass action law, in the derivation of Eqs. (3) from molecular mechanisms (2) [46, pp. 89, 291–294]; [43, pp. 166–172].
Applying the mass action law to the above reactions, we obtain the following system of differential equations:
| (9) |
| (10) |
| (11) |
The system of Eqs. (11) has the two fixed points in phase space:
| (12) |
| (13) |
As the first equation in (11) is independent of P, integration by quadratures gives:
| (14) |
In Fig. 2, we show the time solution (14) as a function of the time t, for a particular choice of parameters and initial conditions. For
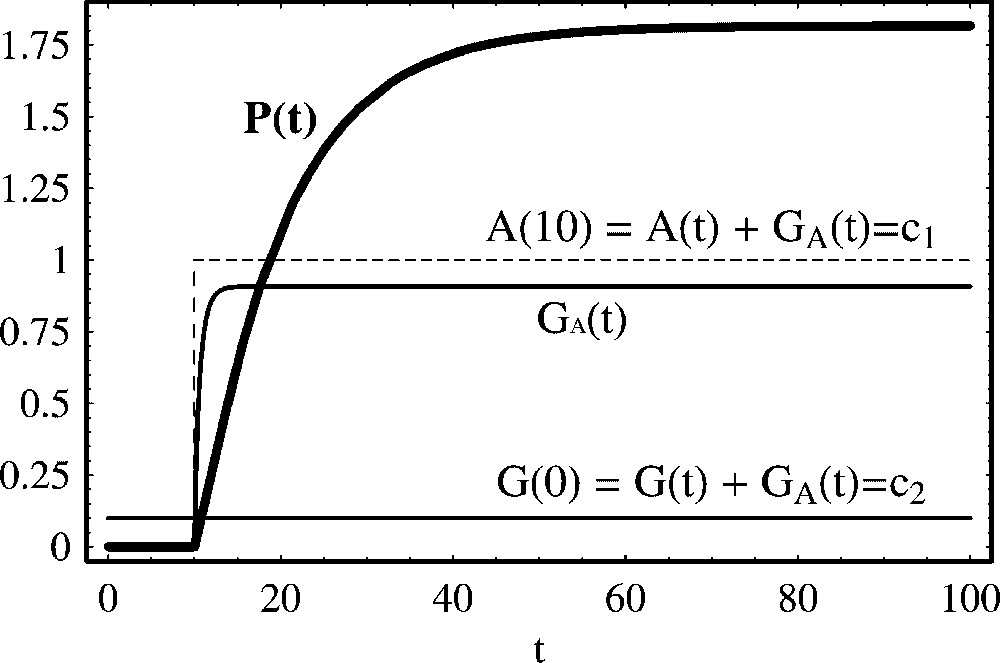
Positive regulation of protein production with constant activator total concentration. We show the temporal evolution of protein concentration calculated from (11). From t=0 to t=10, the activator is absent, A(t)=0.0. In this case, by (10),
Positive regulation of protein production with constant activator total concentration. We show the temporal evolution of protein concentration calculated from (11). From t=0 to t=10, the activator is absent, A(t)=0.0. In this case, by (10), ... Lire la suite
3.2 The activator degrades
In the simple model (8), the activator does not degrade in the process. If the activator A is itself a protein, we must consider an extra mechanism accounting for the activator degradation:
| (15) |
| (16) |
| (17) |
Eq. (17) has one stable fixed point with coordinates
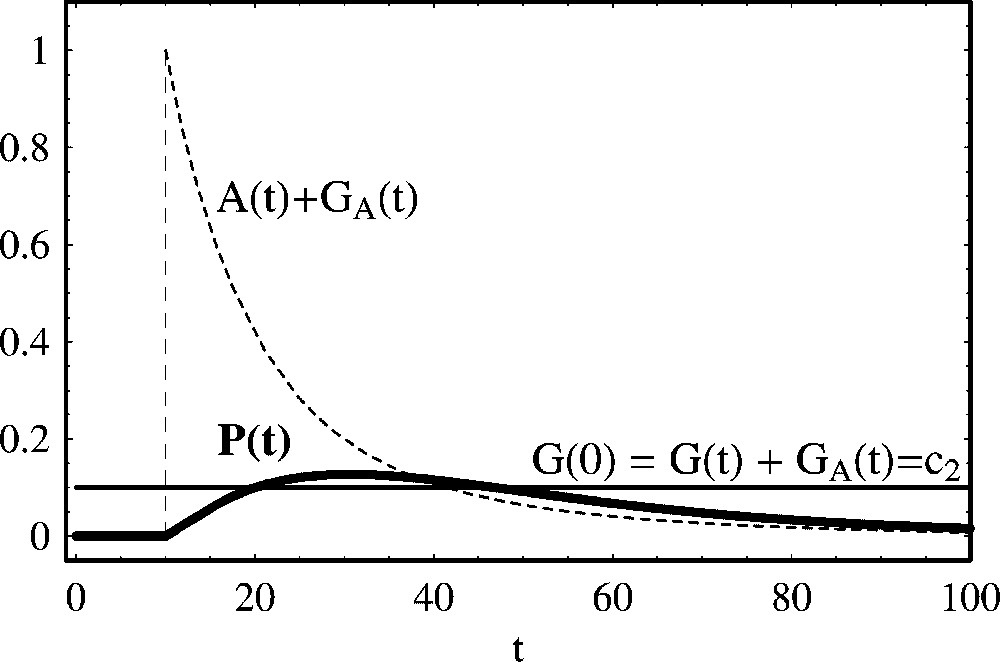
Positive regulation of protein production with a degrading activator. We show the temporal evolution of protein concentration calculated from (17), for the same parameter values and initial concentrations of Fig. 1. Initially, the activator is absent and there is no protein production. At t=10, we have introduced the activator with the initial value A(10)=1.0, and the degradation rate of the activator protein has been set to
Positive regulation of protein production with a degrading activator. We show the temporal evolution of protein concentration calculated from (17), for the same parameter values and initial concentrations of Fig. 1. Initially, the activator is absent and there ... Lire la suite
3.3 Self-activation
An important case occurs when the produced protein activates its own production. For this situation, the activation mechanism is:
| (18) |
| (19) |
| (20) |
In Fig. 4, we show the temporal evolution of the concentration of a self-activating protein, for two different initial protein concentrations.
Self-activation of protein production. We show the temporal evolution of protein concentration calculated from (20), for protein initial concentrations P(0)=1.0 and P(0)=0.05. At t=0, the protein concentration is larger than zero, and the gene has no additional activating mechanisms. If the initial concentration of gene templates available for protein production is above some definite threshold value defined by
Self-activation of protein production. We show the temporal evolution of protein concentration calculated from (20), for protein initial concentrations P(0)=1.0 and P(0)=0.05. At t=0, the protein concentration is larger than zero, and the gene has no additional activating ... Lire la suite
If

Threshold effect for positive regulation with a self-activator, for the parameter values
Threshold effect for positive regulation with a self-activator, for the parameter values
4 Negative regulation
4.1 The repressor total concentration is constant
In negative regulation of protein production, an inhibitor binds to an operator site and mRNA transcription is not initiated. Representing by R the concentration of the repressor, the template gene by G, and the binded complex by
| (21) |
| (22) |
| (23) |
| (24) |
As in the case of activation, the system of Eqs. (24) has two fixed points with coordinates:
| (25) |
| (26) |
The solution of the system of Eqs. (24) is:
| (27) |
In Fig. 6, we plot the time evolution of protein production as a function of time. In order to compare the effect of activation and repression agents, we have taken the same parameter values as in the case of Fig. 1. In this case, there exists protein production in the absence of repressor, but when we connect the repression mechanisms, the production process stops and asymptotically in time, the steady state of the protein reaches a low value but away from zero.
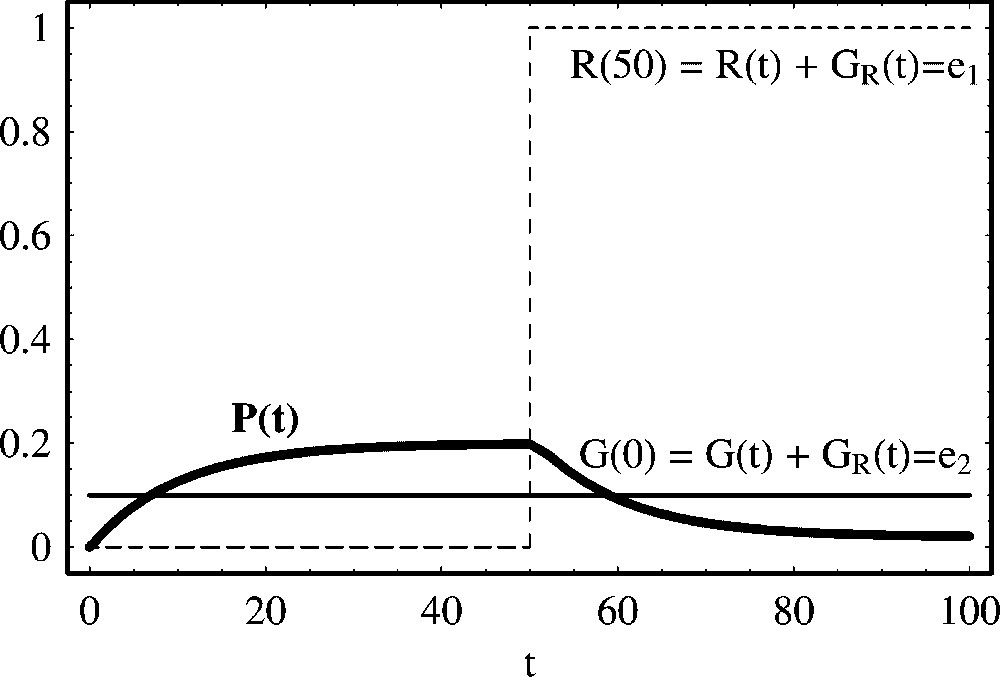
Negative regulation of protein production with constant repressor concentration. We show the temporal evolution of protein concentration calculated from (27). From t=0 to t=50, the repressor is absent (R(0)=0.0) and, therefore, protein production is initiated. At time t=50, we have introduced a repressor R(50)=1.0 and protein decay is initiated. After the introduction of the repressor, asymptotically in time, the protein concentration attains a low equilibrium value,
Negative regulation of protein production with constant repressor concentration. We show the temporal evolution of protein concentration calculated from (27). From t=0 to t=50, the repressor is absent (R(0)=0.0) and, therefore, protein production is initiated. At time ... Lire la suite
4.2 The repressor degrades
In the simple model (21), the repressor does not degrade during the process. If the repressor R is itself a protein, we must consider the extra mechanism accounting for repressor degradation,
| (28) |
| (29) |
| (30) |
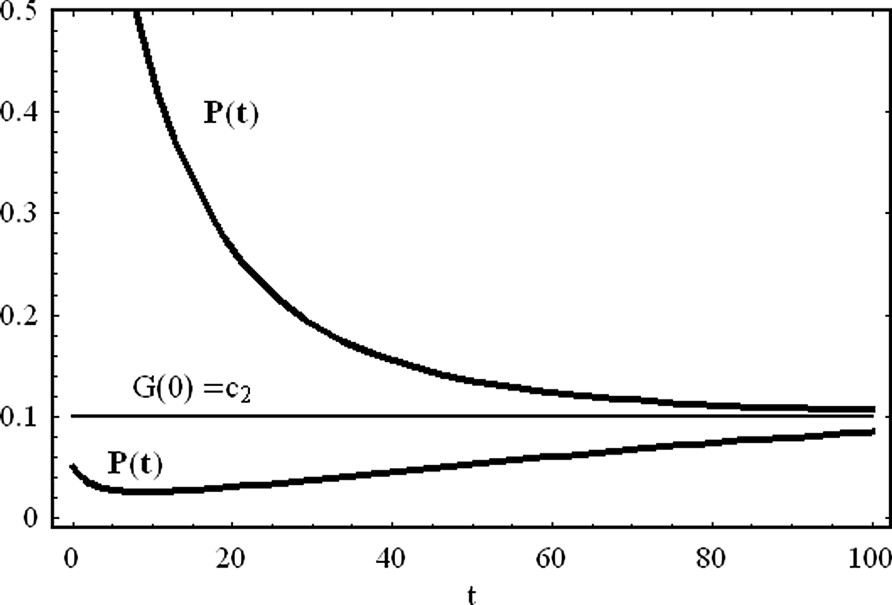
In the case of a repressor agent that degrades during time, the asymptotic protein concentration assumes a positive value,
4.3 Self-repression
An important case is when the produced protein represses its own production. For these situations, the mechanism is:
| (31) |
| (32) |
| (33) |
If
| (34) |
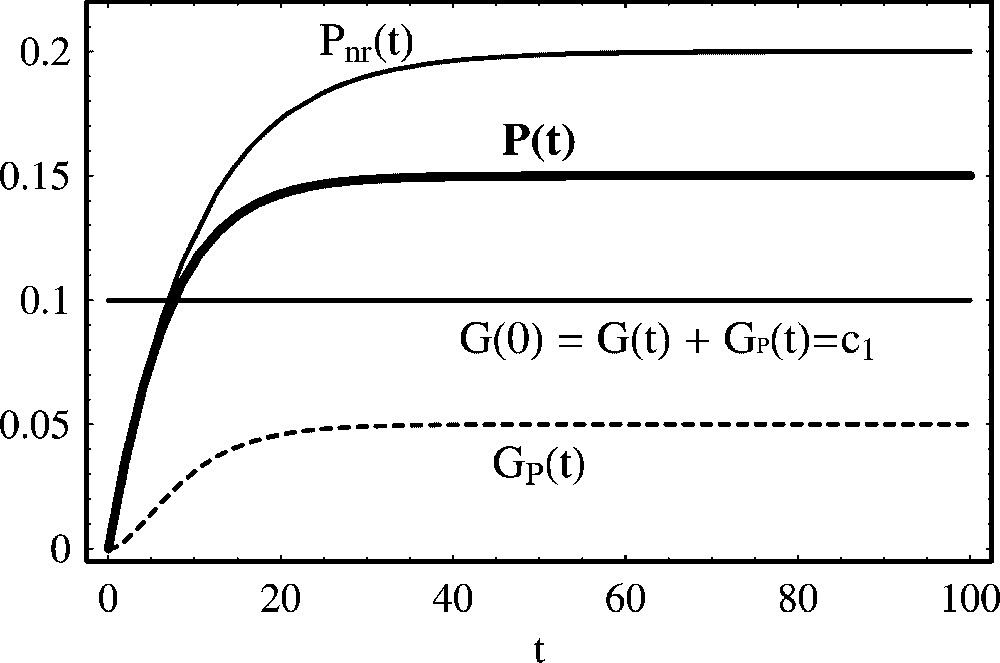
Self-repression of protein production. P(t) represents the temporal evolution of a self-repressing protein as described by mechanism (31).
5 Positive and negative regulation with the regulators binding the same operator
We now extend this model to the case where the template DNA has overlapping binding site where an activator or an inhibitor can bind, leading to the activation or repression of protein production. This can be seen as the case of competition by the same binding site.
Adopting the same strategy of the preceeding sections, we now have:
| (35) |
| (36) |
Eqs. (36) have the conservation laws:
| (37) |
| (38) |
The first two equations in (38) are independent of P, and we can determine their fixed points. Equating
| (39) |
In the simulation of Fig. 8, we show that when a repressor is present, even in small concentrations, and competes for the same binding site as the activator, its effect is to decrease the equilibrium value of the produced protein.

Competition of a positive and a negative regulator for the same binding site. We show the temporal evolution of protein concentration calculated from (38). From t=0 to t=10, both the activator and the repressor are absent. From t=10 to t=50, the activator A is present. From t=50 to t=100, both the activator A and the repressor R are present. From t=100 to t=150, only the repressor R is present. Other parameter values of the simulation are:
Competition of a positive and a negative regulator for the same binding site. We show the temporal evolution of protein concentration calculated from (38). From t=0 to t=10, both the activator and the repressor are absent. From ... Lire la suite
6 Positive and negative regulation with the regulators binding different operators
For the case where the template DNA has two operon sites, one for the activator and another for the inhibitor, the reaction mechanisms are:
| (40) |
| (41) |
| (42) |
| (43) |
In the simulation of Fig. 9, we show that when the activator and the repressor have different binding sites, the effect of the repressor is more pronounced when compared with the competitive case of Fig. 8.
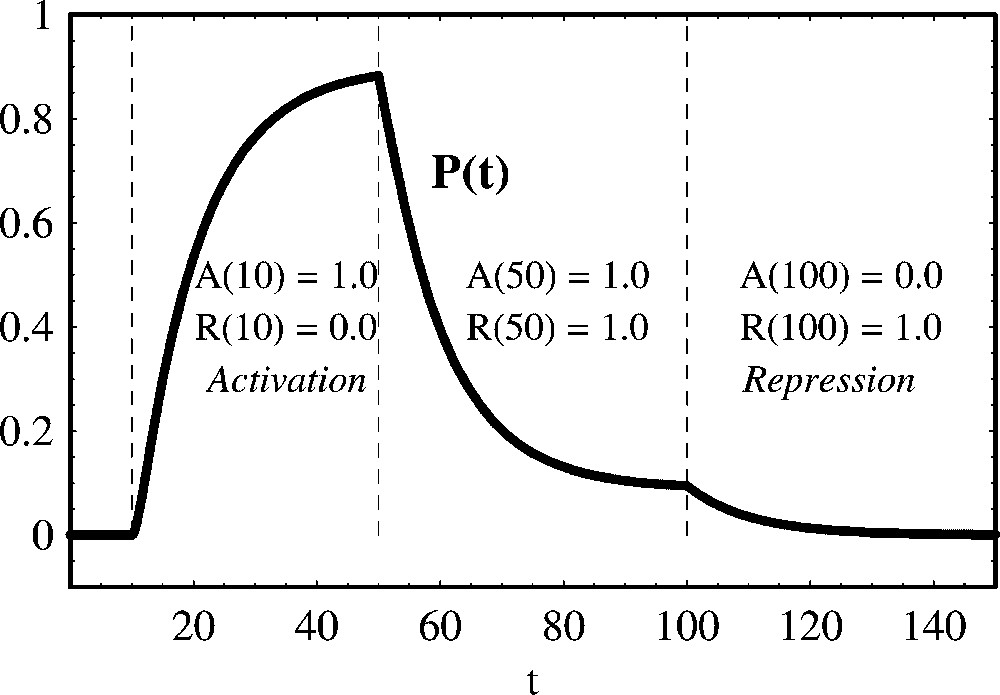
Positive and negative regulation of protein production with the regulators binding different operators. We show the temporal evolution of protein concentration calculated from (43). From t=0 to t=10 both the activator and the repressor are absent. From t=10 to t=50 the activator A is present. From t=50 to t=100 both an activator A and a repressor R are present. From t=100 to t=150 only the repressor R is present. Parameter values are:
Positive and negative regulation of protein production with the regulators binding different operators. We show the temporal evolution of protein concentration calculated from (43). From t=0 to t=10 both the activator and the repressor are absent. From ... Lire la suite
7 The negative regulation of the trp operon
The tryptophan (trp) operon is an example of a repressible system. The mRNA coded by the trp operon is translated into five polypeptides that assemble the enzymes responsible for the biosynthesis of the aminoacid tryptophan. Both the synthesis and the activity of these enzymes are controlled by the level of tryptophan in the cell. The cell also produces a repressor protein (R) that is only active if bounded to a tryptophan molecule (
| (44) |
| (45) |
| (46) |
Solving Eqs. (46) in order to get
| (47) |
The system of Eqs. (47) has two fixed points in phase space. If
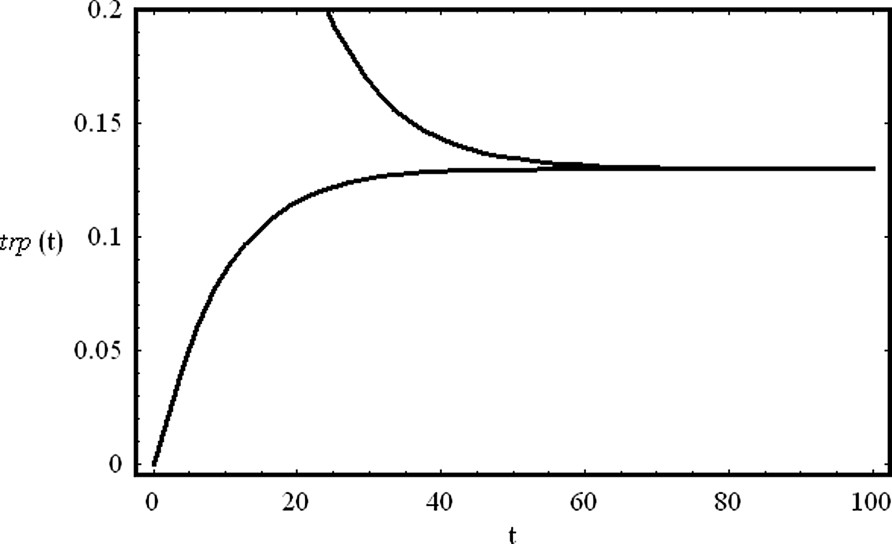
The negative regulation of the trp operon. We show the temporal evolution of the concentrations of the aminoacid trp from two different initial conditions (trp(0)=0.0 and trp(0)=1.0). The production of the aminoacid tryptophan is self-repressed as described by the mechanism (44). Parameter values are:
The negative regulation of the trp operon. We show the temporal evolution of the concentrations of the aminoacid trp from two different initial conditions (trp(0)=0.0 and trp(0)=1.0). The production of the aminoacid tryptophan is self-repressed as ... Lire la suite
8 The genetic switch in the regulation of the lac operon
In Escherichia coli the main source of carbon is glucose. Lactose is only used as an alternative in the absence of glucose. The lac operon controls the expression of a set of genes that code for proteins involved in the metabolism of the disaccharide lactose. In the presence of glucose, the lac operon is off and the genes under its control are not transcribed. The lac operon (G) is controlled by a transcriptional activator, the ‘catabolite activator protein’ (CAP), and a transcriptional repressor, the ‘lac repressor’ (REP), binding different operators. A decrease in glucose concentration inside the cell leads to the rise of CAP induced by cAMP (
In this framework, the regulation of the lac operon is described by the mechanism:
| (48) |
| (49) |
The simulations in Fig. 11 show that the lac operon behaves like an ‘on/off soft switch’: the operon is turned on only when the concentration of
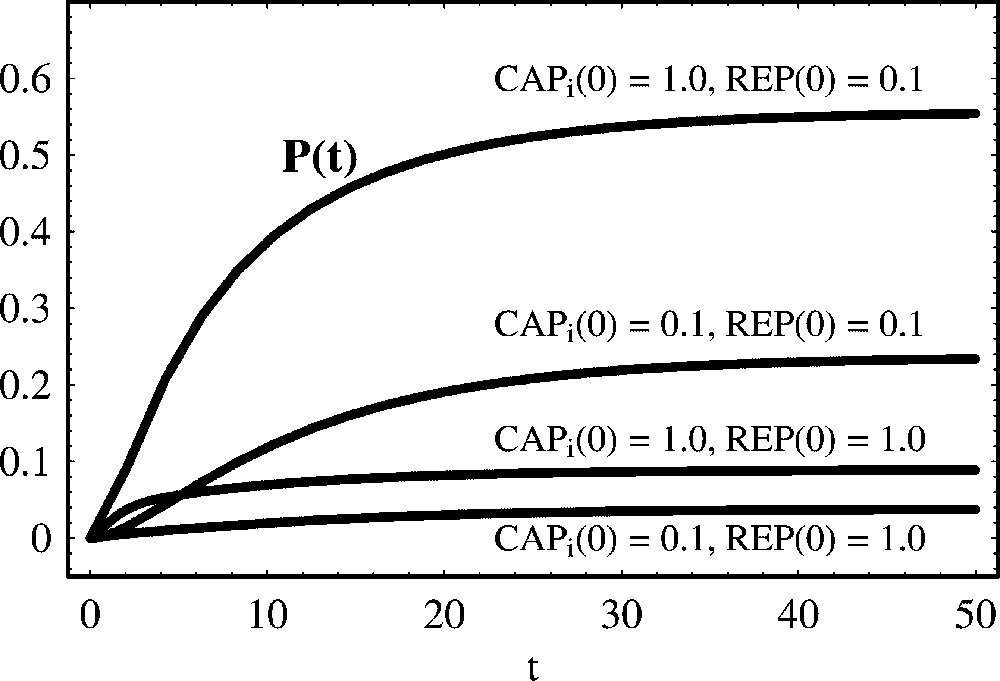
The regulation of the lac operon. This is a case of positive and negative regulation of protein production, with the regulators binding different operators. We show the temporal evolution of protein concentration for different initial concentrations of the activator
The regulation of the lac operon. This is a case of positive and negative regulation of protein production, with the regulators binding different operators. We show the temporal evolution of protein concentration for different initial concentrations of the activator ... Lire la suite
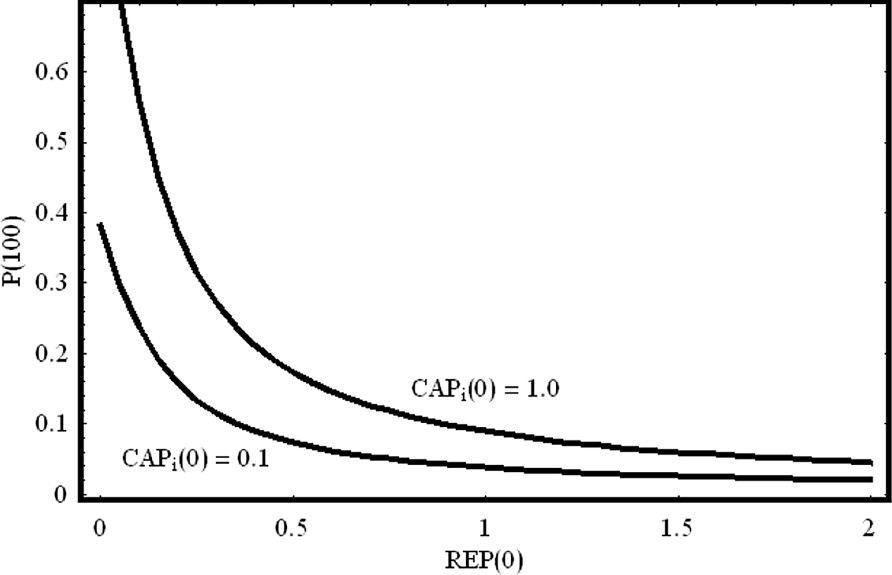
On/off switch of the lac operon. We have plotted the protein concentration for t=100 as a function of the initial concentration of the repressor REP. For fixed activator initial concentration, as the repressor concentration increases linearly, the equilibrium value of the produced protein decreases exponentially. We have chosen two different initial conditions for the activator
9 Discussion and conclusions
We have developed a class of differential equation models describing the basic biological mechanisms of regulation of gene expression. The models are easy to handle, enabling a straightforward comparison with experiments in order to be validated and calibrated. The basic assumptions in the construction of this simple framework are the following:
- – (1) genes are considered as templates for protein production;
- – (2) gene transcription is activated or repressed by transcriptional regulators binding to the regulator sites of the gene;
- – (3) all the mechanisms are bi-molecular and derived from the mass action law;
- – (4) both transcriptional and translational mechanisms are described in time by the same rate constants.
We have explored and explained the quantitative behaviour of basic simple gene regulatory mechanisms. For example, when we compare nonregulated protein production with activated protein production, the equilibrium protein values become dependent of the activator initial concentration, and therefore, are not only limited by the rate constants of the production mechanisms. In the case of nonregulated protein production, the equilibrium values are limited by the rate at which production and degradation mechanisms occur.
If the protein activator concentration degrades during time, the produced protein, after reaching a maximum concentration value, decays to zero. This contrasts with the case of protein self-activation, where the equilibrium values of the produced protein depend on the reaction rates of the processes involved in the production mechanisms and on the concentrations of the gene template. In this last case, if the concentration of the gene template is bellow a certain threshold, protein concentration decays to zero.
When the protein is produced by default and exists a negative regulation mechanism, the equilibrium value of the protein concentration is lower in the presence of the repressor and depends on the repressor initial concentration. If in addition the repressor molecules degrade, the resulting protein concentration reaches a higher asymptotic positive value. In the case of self-repression, the produced protein reaches lower equilibrium values when compared with the nonregulated case. Without regulation, protein concentration at equilibrium only depends on the reaction rates of the reactions.
It follows from these simple models that the threshold effect observed in some experimental systems is associated with the existence of conserved quantities derived from the mass action law. In general, the conserved quantities change the position of fixed points in phase space and, in some cases, leads to bifurcation of equilibria. In particular, in the case of protein production by self-activation, depending on the initial concentration of the gene, different asymptotic states of the protein are reached (Fig. 5).
We have also analyzed the two cases where both an activator and a repressor are regulating the production of the same protein. In the simulation of Fig. 8, we can see that when a repressor is present, and competes for the same binding site of the activator, its effect is to decrease the equilibrium value of the produced protein. When the activator and the repressor have different binding sites, Fig. 9, the effect of the repressor is more pronounced, leading to even lower protein concentrations.
The models of positive and negative regulation of protein production are the building blocks for more complex mechanisms of gene regulatory interactions, as we have shown in the cases of the trp and the lac operon. Analyzing the mechanism of self-repression of the tryptophan synthesis, we have shown how this process contributes for the regulation of the aminoacid concentration inside the cell. It is shown that the regulation mechanism of the lac operon behaves like a soft on/off switch, and we have analyzed the dependence on the concentrations of the activator and the repressor.
These models depend on biologically relevant parameters, are robust to the variation of the parameters and have low computational cost. For complex networks of gene regulation, the approach developed here enables the construction of general computer programs for the derivation and integration of the equations describing the temporal (and spatial) evolution of the protein concentrations. The predicted gene expression patterns can then be compared with the real experimental system in order to be validated and calibrated.
Acknowledgments
This work has been partially supported by Fundação para a Ciência e a Tecnologia (Portugal), under the framework of POCTI project C/FIS/13161/1998, and by the grant PRAXIS XXI/BD/18379/98.