

1 Introduction
The lac operon of E. coli has served as a paradigm for transcription regulation since it was first described by Jacob and Monod in their seminal work in 1961 [1]. The lac operon, which encodes structural genes for the three enzymes involved in lactose metabolism (β-galactosidase, galactoside permease, and thiogalactoside acetyltransferase), is subject to both negative and positive regulation during transcription, depending on the availability of lactose in the medium [2]. Although regulation of lac operon has been the subject of intense genetic, biochemical, biophysical, and structural studies, the structural information regarding the central enzyme of the system, E. coli RNAP, has been lacking until recently. In the past 5 years, however, spectacular advances have been made in RNAP structural studies, including the solving of crystal structures of bacterial and yeast RNAPs, RNAP complexes with nucleic acids, and domains of RNAP subunits with DNA and transcription factors [3–11]. In this review, we present structural information that is currently available for bacterial RNAPs, with special emphasis on their functional implications for the regulation of lac operon, and attempt to integrate them into preexisting body of biochemical and genetic data.
2 RNAP structure and function
2.1 General overview
The DNA-dependent, multisubunit RNAP of E. coli is an evolutionarily-conserved protein which shares functional and structural relatedness with RNAPs of eubacteria, archaebacteria, yeast, and mammals [12–14]. The catalytically competent core has a conserved subunit composition of with a molecular mass of , and is capable of catalyzing DNA-dependent RNA synthesis, RNA hydrolysis and pyrophosphorolysis. Binding of the bacterial-specific initiation factor σ converts core to holoenzyme, which is capable of specific promoter recognition and efficient transcription initiation [15,16]. All prokaryotic organisms express one or more σ-like factors. Use of alternative σ's allows RNAP to recognize different classes of promoters, thus affording organisms the specificity and selectivity in transcription process required for optimal cell growth [16,17]. In E. coli, which expresses seven σ factors, the -associated holoenzyme (E) transcribes the bulk of its housekeeping genes, including those of the lac operon.
A transcription cycle carried out by RNAP proceeds through three stages: initiation, elongation, and termination, all of which are targets of regulation. During initiation, RNAP holoenzyme binds specifically to two conserved hexamers in the promoter at nucleotide (nt) positions −35 and −10 relative to the transcription start site to form a closed promoter complex (RPc). In a process involving several intermediates, RPc converts to a stable open promoter complex (RPo) in which DNA duplex becomes unwound around −10 region (from −12 to +3). In the presence of rNTPs, RPo begins to synthesize and release short (2–12 nts) RNA products (‘abortive initiation’) without leaving the promoter [18]. After several rounds of abortive initiation, the initiation complex (RPi) enters elongation stage. This transition (‘promoter escape’) is marked by a significant conformational change [18–20], leading simultaneously to loss of RNAP-promoter contacts, possible σ-dissociation [21–23], and formation of a highly processive ternary elongation complex (TC) [18,20]. Elongation by TC continues until it encounters a termination signal encoded within the DNA sequences resulting in irreversible dissociation of core, DNA and RNA.
E recognizes two types of promoters, the so-called −35 and extended −10 promoters [24]. Many promoters, including , belong to the former, having both the −10 and −35 hexamers. The consensus sequence and positions of −35 and −10 hexamers are −35TTGACA−30 and −12TATAAT−7 [24–26]; the hexamers are separate by a 16–18 base-pair-long spacer region of nonspecific sequence [27]. The extended −10 promoters do not have any discernable −35 element, a defect that is functionally compensated by the presence of a dinucleotide -TG- (‘extended −10 element’) at positions −15 and −14 [28]. If present, the extended −10 element can also improve the initiation efficiency at −35 promoters. The −35, −10 and extended −10 elements are involved in direct and sequence-specific interactions with E during RPc formation, and are therefore major determining factors in establishing the equilibrium binding constant for RNAP-promoter interaction and the rate of RPc formation [29]. Some E. coli promoters possess additional cis-element located immediately upstream to the −35 element (nt positions −40 to −60) called the ‘UP-element’ [30,31]. The UP-element, which can be recognized by the presence of an AT-rich sequence, facilitates RNAP binding through its interaction with αCTD of RNAP, and stimulates the intrinsic transcription by up to two orders of magnitude [30,31]. Additional DNA sequences located in and around the promoter can compensate for weak −10 and −35 elements and affect the overall promoter strength. These auxiliary promoter elements, which include the −15 enhancer, discriminator region (DSR), and initial transcribed sequences (ITS), were shown to affect the rate of RPc formation and the efficiency of DNA melting, abortive initiation, and promoter escape [29,32–35].
In the absence of external regulatory input, many naturally occurring promoters, including , are relatively weak due to their non-consensus sequence elements and/or suboptimal spacer lengths. However, many prokaryotic promoters are programmed to respond to a variety of regulatory signals that modulate their activities by either increasing or decreasing the rate of productive initiation. In most cases, the signal entails a sequence specific communication between regulatory protein and its cognate binding site located within, near, or at some distance from the target promoter [2,36]. is a prime example of the promoters that respond to both negative and positive regulatory inputs. In the presence of glucose, lac repressor (LR) binds to the operator sites in the lac promoter () region and prevents RNA polymerase (RNAP) from binding to [37]. The repression is removed by lactose, which binds to LR and causes its dissociation from DNA, thus allowing RNAP to bind and initiate transcription from . During glucose starvation, is positively regulated in response to elevated intracellular levels of cAMP by catabolite activator protein, CAP. The CAP-cAMP complex binds to CAP binding sites upstream of , recruits RNAP, and facilitates transcription initiation from [38].
2.2 Structure of RNAP
2.2.1 Escherichia coli
RNAP is the most extensively characterized bacterial RNAP, both genetically and biochemically. However, the structure of this enzyme determined by cryo-electron microscopy (EM) has a relatively low resolution of [39]. The atomic-resolution (high-resolution) crystal structures have been obtained for Thermus aquaticus (Taq) core and Thermus thermophilus (Tth) holoenzyme, at 3.3 Å and 2.6 Å, respectively [3,4]. The subunits of E. coli and Taq/Tth enzymes share substantial sequence homology and are functionally similar [3,4,14,40,41]. Therefore, the structural data obtained for Taq/Tth RNAP can be readily applied to E. coli enzyme. According to available structural data, Taq and Tth RNAPs share the similar crab claw-like shape, of which the top and bottom pincers are made up of the two largest subunits, β and (Fig. 1a and b). The pincers are joined at the back by the N-terminal domains of asymmetrically placed α-subunit dimer (αI- and αII-NTD). The ω subunit is located near the bottom pincer, wrapped around the C-terminus. In all structures, the internal space of the protein between the pincers is intersected by three channels: the main channel with a diameter of 20–27 Å, which accommodates the double-stranded DNA and the DNA/RNA hybrid, and two minor channels that branch off from the major channel to form the upstream-facing ‘RNA exit channel’ and the downstream-facing substrate-accessible ‘secondary channel’. The minor channels are in diameter and serve as exit pathways for the single stranded -terminal RNA and the -terminal backtracked RNA, respectively [3–6,14,42]. The active center of the enzyme with a catalytic triad of Asp residues holding two essential Mg2+ ions is located on the back wall of the primary channel [3–6,42]. The two pincers near the active center are connected by the F-bridge α-helix, which joins the flexible G-loop element to form the wall of the secondary channel (Fig. 1a and b). The RNA exit channel walls are made of the upstream portions of β and pincers (the clamp) including the ‘rudder’, ‘lid’ and the N-terminal ‘Zn-finger’ elements, and the β ‘fork loop’ and flexible ‘flap’.
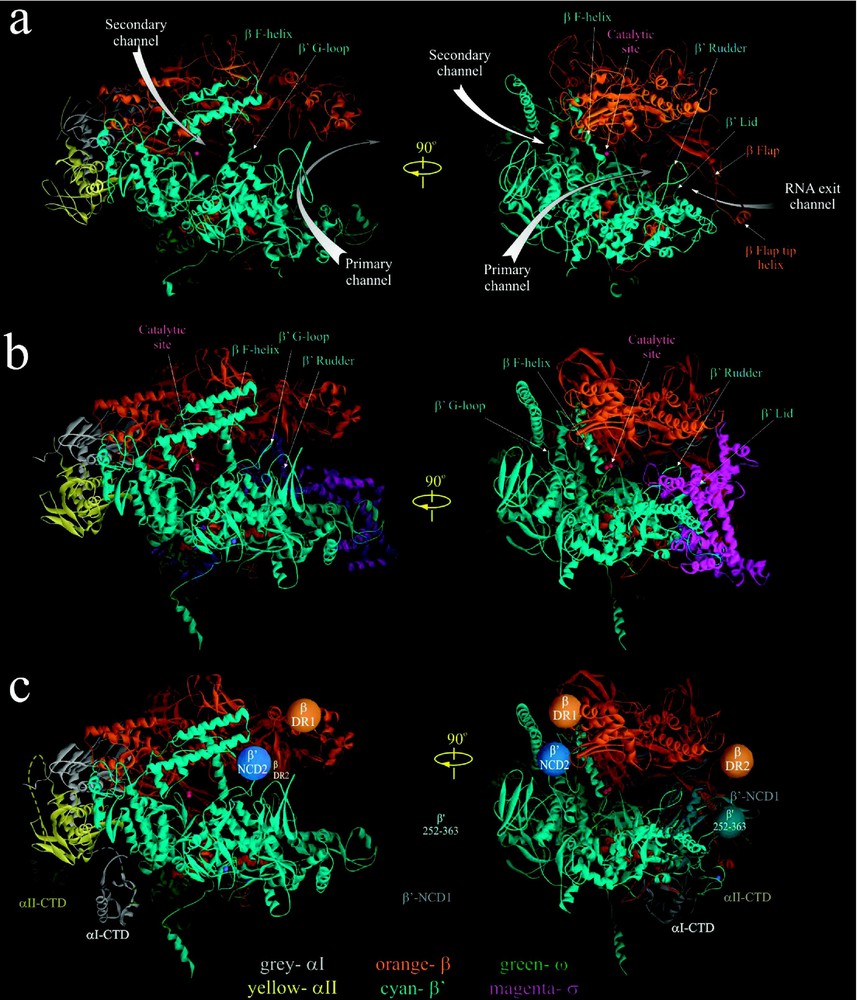
High-resolution crystal structures of Taq RNAP core [3] (a) and Tth RNAP holoenzyme [4] (b). In (c) the approximate locations of αI- and αII-CTD structures [10], as well as positions of nonconserved domains of E. coli β and [39] (orange, blue and cyan balls) are modeled on Tth RNAP structure with σ subunit removed. The structures are shown as ribbons using WebLab ViewerPro program. Subunits are color coded as indicated in the bottom. Small magenta balls indicate the position of catalytic Mg2+ ions. Left panel in a–c is the secondary channel view of RNAP. Right panel showing the main channel view is obtained by rotating the left view 90° clockwise about the vertical axis.
2.2.2 Non-conserved domains
Despite their overall similarity, the Taq/Tth and E. coli RNAPs also have distinct structural dissimilarities. The major differences reside in four large non-conserved domains of β and subunits; E. coli lacks a 283-residue domain present in the Taq/Tth between conserved regions A and B (-NCD1, visible in Tth structure as an extended part of the lower pincer, Fig. 1c), but instead has a 188-residue domain inserted in the conserved G-loop element (E. coli -NCD2), which is absent in Taq/Tth [3,4,39]. -NCD2 is not visible in the cryo-EM map [39]; it is apparently very flexible and disordered, and its location in RNAP is not determined. Two other domains of E. coli β are absent in the Taq and Tth β: a 115-residue element (β-DR1) between conserved regions B and C, and a 99-residue region (β-DR2) between conserved regions G and H. The location of β-DR1 and β-DR2 in E. coli core RNAP was determined by flexible fitting of the high-resolution structure of Taq RNAP into the low-resolution cryo-EM map of E. coli core [39]. The non-conserved domains are dispensable for RNAP assembly and basic function [43,44]; however, they could play an auxiliary or a regulatory role in transcription. For instance, -NCD1 contributes to Tth σ-core binding [4,45]; E. coli -NCD2 interacts with transcript cleavage factors GreA and GreB [46] and may influence RNAP's propensity to backtrack, affecting its pausing and arrest [47]; E. coli β-DR1 is targeted by the bacteriophage T4 termination factor Alc, which selectively induces premature transcription termination on E. coli DNA during infection [43].
The reported RNAP structures still lack several elements. These include a 109-residue long portion of the non-conserved -NCD1 (), and an 80-residue long α-CTD with a 14-residue flexible linker that connects it to the αNTD. The atomic structures of αCTD in complex with CAP and DNA are now available [10] and its approximate position in RNAP can be modeled (Fig. 1c). The αI and αII CTDs recognize and bind the UP promoter element, and serve as targets for many transcriptional activators [30,31].
2.3 Structure of -core interactions
E. coli and -like factors of other bacteria share four regions of sequence homology designated 1 to 4, which are further divided into subregions [16,17] (Fig. 2). All conserved regions have been implicated in either σ-core or σ-DNA interactions ([45,48] and references therein). In Taq and Tth holoenzyme structures, the σ-subunit is visible as a V-shaped structure partially wedged between the upper and lower pincers (i.e. β/ subunits) of core on the upstream face of the enzyme [4,5]. The crystallographically resolved portion of σ comprises four structural domains, , linker domain LD and σ4, connected by short flexible linkers. These four domains contain conserved regions 1.2, 2.1–2.4; 3.0–3.1; 3.2; and 4.1–4.2, respectively. The three α-helical domains and σ4 are located on the enzyme's surface (Figs. 1b and 2), stretched over the upstream opening of the primary channel, while σLD is buried inside the primary channel (Fig. 2). σLD forms a hairpin loop that approaches the catalytic pocket, and emerges underneath the β-flap via RNA exit channel. In the available holoenzyme structures, σ2 and σ4 domains are located apart, which is an appropriate distance for these domains to contact, respectively, the −10 and −35 elements of the promoter DNA in RPc. σ3 is positioned to interact with the extended −10 region of the promoter and the −15 enhancer element.
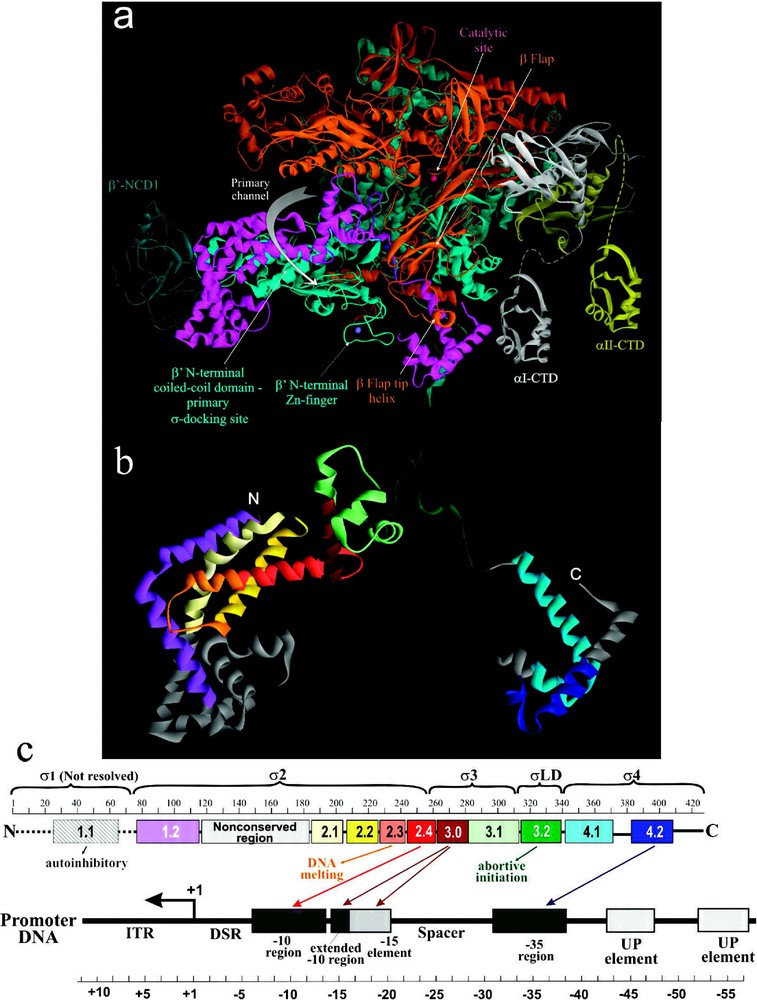
The structural and functional organization of σ. (a) View of Tth RNAP holoenzyme obtained by rotating the left view shown in Fig. 1b 180° about the vertical axis with color coding as above. (b) Ribbon diagram of σ from Tth holoenzyme structure [4]. Colored regions correspond to the evolutionarily conserved domains of σ as shown in (c). (c) Functional map of σ. Top diagram is a linear representation of σ showing structural domains and conserved regions (numbered and color-coded boxes). Bottom diagram shows DNA promoter regions and interactions made by σ DNA binding domains.
The extreme N-terminal portion of σ polypeptide (), which includes poorly conserved region 1.1, is not resolved in either holoenzyme, or RNAP-DNA binary complex, or free σ structures. σ1.1 possesses an autoinhibitory function: it obscures the DNA binding regions of free σ before it binds core ([49] and references therein). It also facilitates the RPo formation and transcription initiation at some promoters, while inhibiting initiation at others [50]. Additionally, σ1.1 may be involved in the initial σ binding to core by interacting with β flap domain [51].
Recent biochemical and biophysical evidence suggest a multistep and cooperative process of σ-core binding [45,48,49,51,52], which is characterized by a in the range of 10−9 M. Such high binding affinity derives from multiple independent interactions between discrete domains of σ and different parts of the core. However, most of the potential contacts in σ-core interface, including electrostatic (salt bridges), polar (hydrogen bonds) and non-polar (hydrophobic and van der Waals) interactions are relatively weak and distributed over a large area [45]. For the most part, these contacts are limited to the β and -subunits of core. The strongest interaction is observed between σ2 and coiled-coil domain (), which serves as the major σ docking site. Less strong interaction is observed between β-flap and σ4 [53], and between σ3 and β region I [45] (Figs. 1b and 2). In the presence of specific activators, σ4 also interacts with α-CTD [54].
2.3.1 Conformational flexibility
Structural organization of RNAP is described as a fixed core mass surrounded by four mobile modules [39,49]. The fixed core module comprises two αNTDs, ω subunit, and parts of β and surrounding the active site. The mobile modules include: half of the lower pincer (‘clamp module’) comprising the N-terminus of (1–624) and the C-terminus of β (1054–1115), the two β N-terminal modules β1 ( and ) and β2 () that make up the top β pincer, and the β-flap module (). These mobile modules confer considerable conformational flexibility to RNAP structure. The most dramatic demonstration of this flexibility is the swinging motion of the clamp, β1, and β2 modules, inferred from comparing the structures of Taq and E. coli core enzymes, which results in the opening of the claws by [39,49]. The initial opening of the claws is thought to be essential during transcription initiation when the template DNA strand must enter the primary channel and reach the catalytic cleft. The subsequent closing of the clamp may help RNAP to tightly hold RNA-DNA hybrid in position during elongation ([49] and references therein), and may be essential to TC processivity.
The intrinsic flexibility of RNAP is also evident during its conversion from core to holoenzyme, which leads to changes in the positions of all structural domains of core by 2 to 12 Å. The RNA exit channel, which now accommodates σ3, becomes constricted by the β flap domain which is shifted by towards σCD compared to Taq core [3–5,39,49]. Even more pronounced is the altered orientation of the β ‘flap-tip’ helix (), which is shifted by relative to its position in the core. Additional evidence of RNAP flexibility comes from comparing the structures of Taq and Tth holoenzyme; the σ regions 2.4 (R249) and 4.2 (R394) in these structures are separated by a distance of 67 Å and 58 Å, respectively. Moreover, in the structure of Taq RNAP-DNA binary complex, these regions are separated by 63 Å. Such plasticity may explain the ability of RNAP to accommodate promoters with spacers and discriminator regions of quite different lengths.
2.4 RNAP-promoter interactions
Structural information on how RNAP recognizes and binds promoter DNA was gleaned from two crystallographic studies: the 2.4-Å-resolution structure of Taq σ4 in complex with −35 element DNA (from position −26 to −37) [9], and the 6.5-Å-resolution structure of Taq holoenzyme binary complex with fork-junction promoter DNA [8], which partially mimics the RPo. The latter complex contained ds DNA from position −12 to −45, and the ss nt-DNA from −11 to −7. Complemented with vast biochemical, biophysical and genetic data accumulated in the last 20 years, these studies led to construction of structural models of binary RNAP-DNA complexes RPc and RPo [8,49].
2.4.1 RPc
In RPc, the ds promoter DNA lies on the surface of holoenzyme, outside the RNAP active-site channel (Fig. 3a). The RNAP-bound ds DNA appears to be bent at three places: at position around −25, where DNA may bend or kink by to accommodate variable spacer length [9], at the −35 element region, where a bending is induced by insertion of σ4 helix-turn-helix motif into the major groove [9], and at further upstream −45 region, where αCTD-DNA interaction may take place [55]. The DNA bending at −35 region may be important for a proper orientation of DNA towards αCTD and for binding upstream transcription activators [10,11].
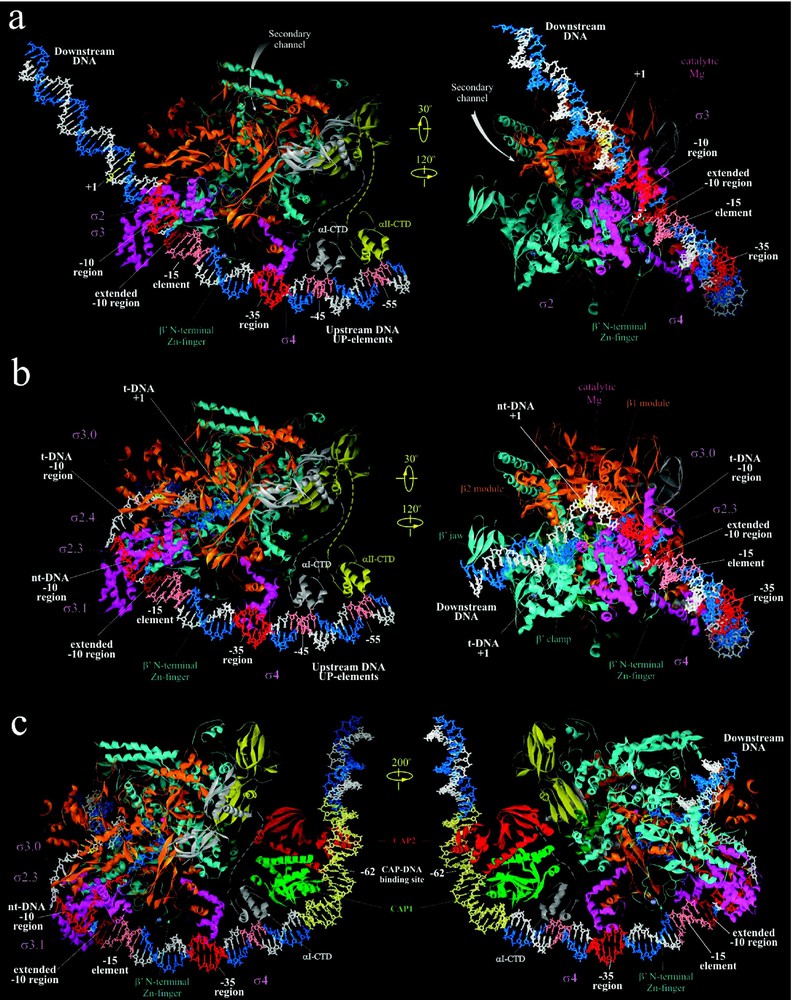
Structural models of RNAP-promoter DNA complexes. (a) Model of Taq RPc based on [8]. (b) Model of Taq RPo adapted from [8,10]. (c) Model of ternary complex of CAP, Taq RNAP and promoter DNA adapted from [10]. Right panel views of (a), (b) and (c) are similar to the view of Tth holoenzyme in Fig. 2a, but tilted forward by 25°. Left panel views are obtained as indicated. Proteins are shown as ribbons and color coded as in Fig. 1. DNA is shown in stick representation with template and non-template strands colored blue and white, respectively, except for various promoter elements colored as indicated.
All sequence-specific contacts in RPc with the conserved −10, extended −10, and −35 elements of the promoter are mediated by the σ-DNA recognition elements: regions 2.2–2.4, 3.0, and 4.2, respectively (Fig. 2). Interaction with −10 element occurs through base-specific contacts of σ region 2.4 residues (reviewed in [56]). According to the structure, the interacting residues are most likely Q260 and N263 (numbering according to Taq σ70), which face the major groove of the DNA at position −12 and could interact with either A of the template strand or T of the non-template strand, or both. The essential conserved basic residues in regions 2.2 and 2.3, R237 and K241, are positioned to interact with the phosphate DNA backbone of the non-template strand at positions −15/−14 and −13, respectively. The extended −10 element is recognized by two residues of σ region 3.0, H278 and E281 [57] that are facing the major groove of the extended −10 element. E281 makes base-specific interactions with T at position −13 of the non-template strand, whereas H278 may interact nonspecifically with the negatively charged DNA backbone at positions −17/−18 of the non-template strand. Additionally, residues R274, V277, H278 and E281 of σ region 3.0 may be involved in base-specific and nonspecific interactions in the major groove of the ‘−15 enhancer’ element (−17/−12 segment) [32]. More precise assignment of σ residues is not possible yet due to the lack of a high-resolution structure of RPc.
The atomic structure of the complex of Taq σ4 with −35 LacUV5 promoter element provided more detailed information on σ region 4.2-DNA interactions [9]. These interactions occur through ten conserved residues of the helix-turn-helix motif of σ region 4.2 [9,56]. Among these, four key residues are responsible for base-specific DNA recognition: R409, E410, R411 and Q414. On the template strand, the side chain of R409 interacts with −31G and −30T through hydrogen bonds and van der Waals contacts, respectively, and the side chain of E410 makes hydrogen bond and van der Waals contacts with −33C. R413 may have van der Waals contacts with −32T. On the non-template strand, Q414 and R411 establish hydrogen bond and van der Waals interactions with −35T. Additionally, residues R413, R387, L398, E399 and R379, T408 provide nonspecific but strong ionic, polar and van der Waals interactions with phosphate and ribose backbone at positions −31, −32, −33 of the template or −35 and −36 of the non-template DNA.
Depending on the length of the spacer, the extent of DNA bending and the presence of non-canonical enhancer elements between −35 and −10 regions, such as −15 enhancer, residues of σ region 3.0 (R274, V277, H278, E281) and N-terminal Zn-binding domain (R35, T36, L37, D42, K71) may be involved in base-specific/nonspecific interactions in the major groove of −13/−17 and in the minor groove of −18/−22 segments, respectively [32].
2.4.2
The proposed model structure of RPo was constructed based on the structure of RNAP-fork junction DNA [8]. It includes both strands of DNA from −60 to +25, the trajectory of which was inferred from footprinting data (Fig. 3b) [8,49]. Unlike RPc, where ds DNA downstream of position −5 does not have strong contacts with RNAP, in RPo both strands of DNA up to +20 position are fully enclosed inside the RNAP main channel (Fig. 3b). The location of the upstream portion of ds DNA (from −60 to −17) is similar to that in RPc, however, at −16 the DNA makes a sharp 37° bend toward the RNAP. The two DNA strands separate at position −11, and take drastically different paths downstream for ∼15 nucleotides until they reanneal at position +3, thus creating the ‘transcription bubble’.
The initial melting of DNA is thought to nucleate from the A/T bp at position −11 [58]. Highly conserved aromatic residues of σ region 2.3, F248, Y253, and W256, are exposed on the surface of σ and positioned to interact with the unpaired bases of the non-template strand of the transcription bubble [8,49]. F248 and Y253 are proximal to −8/−9 and −9/−10 bases, respectively. W256 appears to stack on the exposed face of the , forming the upstream edge of the transcription bubble. More significantly, W256 may play a role in capturing the exposed, or ‘flipped’ A base at the crucial non-template strand −11 position. The non-template single strand DNA (from −2 to +4) further continues its path in a groove formed between β1 and β2 modules. The interactions of DNA from −7 to −2 with RNAP (if any) are unclear. A cluster of conserved basic residues of σ regions 2.4 and 3.0 (R259, K285, R288, and R291) pulls the template strand (from −7 to +3), through electrostatic interactions, into the tunnel composed of portions of σ2, σ3, β1, the lid, and the rudder [8,49]. The DNA then moves between the active site wall and σ LD hairpin loop into RNA–DNA hybrid binding channel, juxtaposing DNA +1 position to the catalytic center. The ds DNA downstream of +5 to +12 is held inside yet another protein tunnel, ‘the downstream DNA binding clamp’ formed mostly by jaw domain and portions of β2 and clamp (Fig. 3b) ([49] and references therein).
The model structure of RPo does not allow unambiguous identification of the amino acid residues involved in interactions with the ss and ds DNA of the promoter, specifically in the active site channel and in the downstream DNA binding clamp. However, it provides a comprehensive view of RNAP-DNA interactions which lead to promoter melting and formation of RPi. More detailed features of these interactions can be predicted based on the model and tested experimentally.
2.5 lac Operon
Unlike the ideal/consensus promoter DNA used in the structural studies and modeling of RPc and RPo, the Plac of the lac operon in E. coli deviates significantly from canonical promoter [24–26]. These deviations include substitutions of consensus G for T at −34 in the −35 element (TTTACA), and AA for GT at −9/−8 in the −10 element (TATGTT). also has an 18 bp-long spacer, which is one bp longer than the optimal 17 bp-length. Inspection of structural data reveals how these changes might affect the interactions between promoter elements and σ subunit that are essential for transcription initiation. Specifically, the loss of base pair recognition by E410 of σ4 together with suboptimal spacer would cause substantial decrease in initial promoter binding by RNAP and the rate of RPc formation [29]. Additionally, the non-consensus −10 element might destabilize the interactions of NT strand bases with aromatic residues (F248, and Y253) of σ region 2.3 resulting in decreased efficiency of DNA melting and RPo formation. The promoter mutations that increase the activity of include compensatory substitutions in −35 and −10 elements, insertions that alter the spacer length, and mutations in the −15 enhancer region [29,32].
In vivo, transcription initiation from is stimulated under conditions of positive regulation by transcription activator protein CAP. Although activation by CAP principally affects the RNAP-binding step at , [55] it may also exert a stimulatory effect on the rates of RNAP isomerisation, RPo formation, and even promoter escape ([32,35] and references therein).
During transcription activation, CAP-homodimer complexed to its effector, cAMP, specifically binds to its cognate 22 bp binding site centered at position −62 of and bends the DNA 80° (Fig. 3c). CAP then recruits RNAP by interacting with one of its two αCTDs, which can be either αCTDI (CTD of the α subunit that interacts with β) or αCTDII (CTD of the α subunit that interacts with ) ([38] and references within). The CAP-αCTD interaction is mediated by ‘activating region 1’ (residues 156–164, 209 of E. coli CAP) of the downstream subunit of CAP dimer and the ‘287 determinant’ of αCTD (residues 285–290, 315, 317, and 318 of E. coli αCTD). αCTD, in turn, interacts nonspecifically with the ribose-phosphate backbone of the DNA minor groove immediately downstream of CAP-binding site on DNA centered at position −43 [10,55]. These interactions, which are mediated by the ‘265 determinant’ of αCTD (residues 265, 294, 296, 298, 299, and 302), are relatively weak in the native . However, if nonspecific sequences at −43 region are replaced by A/T-rich UP element, a stronger binding by αCTD is observed and more efficient transcription activation is elicited. Lastly, transcription activation by CAP requires specific interactions between αCTD ‘261 determinant’ (residues 257, 258, 259, and 261) and the α-helical segment 593–604 of E. coli σ4, specifically residues K593, R596, K597, H600, P601, R603 and S604 [54]. Thus, recruitment of αCTD by CAP appears to merely tether RNAP at the promoter site, while subsequent specific αCTD-σ4 interaction positions the holoenzyme at the promoter (likely at −35 element) leading to stable RPc formation and efficient initiation.
A structural model of the ternary initiating complex containing CAP, RNAP and DNA (Fig. 3c) was constructed [55] by combining the crystal structures of CAP-αCTD-DNA complex [10], σ4-(−35 element) complex [9], and RNAP-DNA complex [8]. The model provides new insight into the central role played by αCTD in the CAP-mediated transcription activation at . αCTD, by providing three discrete interaction interfaces, serves as a thee-way bridge connecting CAP, DNA, and RNAP. The model also supports the view that simple recruitment through protein–protein adhesion, with minimal number of contacts, is sufficient for transcription activation at intrinsically weak promoters such as .
Acknowledgments
Research in S.B.'s laboratory is funded by a grant from NIH. We are grateful to Richard Ebright for providing coordinates of the modeled RNAP-DNA-CRP-αCTD complex. We apologize to those whose work was not cited because of space limitations.