

1 Introduction
DNA integrity is of major importance for transmission of genome information during replication. Accurate transcription, the first step toward gene expression, also requires intact DNA in order to yield functional RNA copies. Lesions may occur within DNA after exposure to endogenous or exogenous oxidative agents including ionizing and ultra-violet radiations [1]. Different transcription features may be altered by the presence of a lesion on a transcribed DNA strand: the fidelity of transcription by the RNA polymerase, the lesion bypass and the transcription rate. In that case, the level of transcription of a gene can be either reduced or increased, depending on the sequence context of the lesion [2].
The T7 RNAP is an enzyme that carries out all the fundamental steps of transcription including initiation, elongation and termination, without the help of activator proteins.
Few studies focused on effect of DNA base modifications on transcription elongation by different RNA polymerases are available. Data from the literature focused either on non distortive DNA lesions such as a basic site, 8-oxo-7,8-dihydroguanine [3,4], 3-methyl-adenine [5], 5,6-dihydrouracil [6] and recently thymine glycol [7] or on bulky damage such as aminofluorene [8], psoralen adducts [9] and cyclobutane pyrimidine dimers [2,10,11]. It was found that these damage blocked transcription elongation with various efficiency. The presence of damage on the non-transcribed strand would result in little if any effect on the transcription [2,10–12]. No evident relationship has been established between lesion structure and putative effect on RNA transcription in terms of arrest, ribonucleotide misincorporation or transcription rate.
To gain further insights into these parameters, we were interested in a family of oxidative damage to thymidine. More precisely, we focused on methyl-modified derivatives of thymidine with three different levels of oxidation, namely: 5-(hydroxymethyl)--deoxyuridine (HmdU), 5-formyl--deoxyuridine (FordU) and 5-carboxy--deoxyuridine (CdU) (Fig. 1). 5-FordU and 5-HmdU derive from the spontaneous decomposition of 5-(hydroperoxymethyl)uracil, formed upon exposure to either ionizing radiation or a type-I photosensitizer. Another pathway of formation of HmdU in DNA is oxidation and deamination of 5-methylcytosine [13].
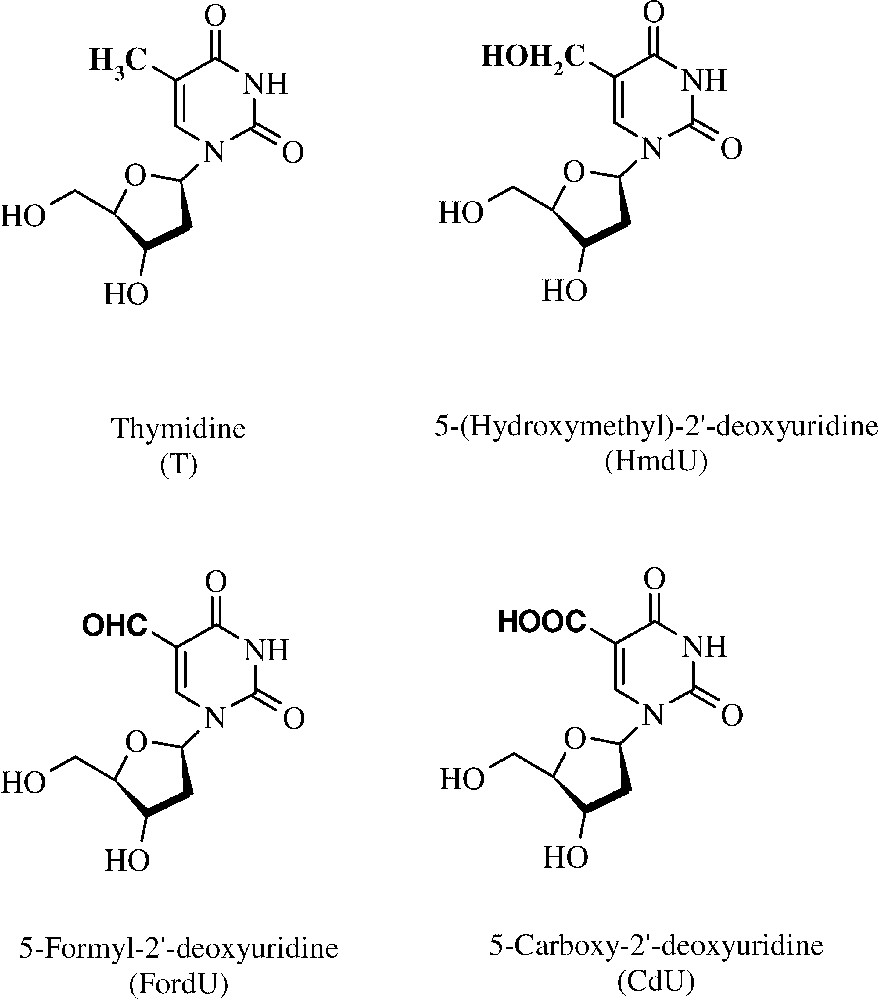
Structure of thymidine and of the three damages resulting from the oxidation of the methyl group of thymidine.
We have included in our study another oxidative damage of thymidine, the 5-carboxy--deoxyuridine. 5-Carboxy--deoxyuridine has been shown to be formed by menadione-mediated photosensitization either of thymidine or of 5-formyl--deoxyuridine [14]. The presence of this damage has not yet been reported in cellular DNA, but it provided us with an interesting comparative model for the present study.
A few studies were aimed at evaluating the impact of FordU and HmdU lesions on DNA replication and their enzymatic repair by DNA N-glycosylases [15–17]. However, no data is available concerning the effect of these two oxidized nucleosides on DNA transcription. In the present study, emphasis was placed on determination of the influence of these three lesions on RNA transcription elongation when damage was located within the transcribed strand of the template. For this purpose, we have synthesized single-stranded templates that contained either a thymidine residue or a related methyl oxidative derivative including HmdU, FordU and CdU, at a defined site in the template part of an oligonucleotide downstream from the promoter (Table 1). A single-stranded template was chosen as we were concerned only by the elongation step of the transcription. In this model, no helix opening by T7 RNA polymerase was required. We thus focused on accuracy and processivity of RNA elongation by the enzyme.
Oligonucleotides used for the experiments including the four transcription templates (60T, 60Hm, 60Fo and 60Ca)
| Name | Sequence (–) |
| T7 promoter | GCCTAATACGACTCACTATAGGGG |
| 15c1 | TCT GGT CAC TTC GGA |
| 34std2 | GGGTTGACTCCGTACATGAGTACGGAAGTAAGGG |
| D3 5′TCTGGTCACTTCGGAXCGTGACTGATCTTCGAGCCACCCCTATAGTGAGTCGTATTAGGC3′ | |
| 3′ GGGGATATCACTCAGCATAATCCG5′ |
1 Sequence of the oligonucleotide complementary of the transcript used for the primer extension assay.
2 Internal standard used for calibration.
3 D = In vitro transcription model (T7 promoter hybridized with either 60T, 60Hm, 60Fo or 60Ca) with X = T, HmdU, FordU and CdU.
2 Materials and methods
2.1 Enzymes and reagents
T7 RNA Polymerase, DNase I and rNTPs were purchased from Ambion (Huntingdon, UK) (T7 MEGA™ kit). Radiolabelled nucleotides and T4 polynucleotide kinase were from Amersham Biosciences (Orsay, France). RNase inhibitor, M-MuLV reverse transcriptase (Meylan, France). -Deoxyuridine and 5-trifluoromethyl--deoxyuridine were from Pharma Waldhof (Düsseldorf, Germany) and 5-iodo--deoxyuridine (IdU) from Sigma. NAP25 column and microspin G-25 column were obtained from Amersham Biosciences.
2.2 Oligonucleotide synthesis and characterization
Oligonucleotides were synthesized using the phosphoramidite chemistry on solid support. The phosphoramidite building block for each DNA base lesion was chemically synthesized and then incorporated at a defined site into a 60-nucleotide-long oligonucleotide using an automated synthesizer (392 DNA/RNA Synthesizer, Applied Biosystems). Four oligonucleotides were synthesized (Table 1): the control sequence containing a thymidine (T) at position 44 from terminus and three other modified oligonucleotides in which either HmdU, FordU or CdU were inserted at the same position. The oligonucleotides were deprotected using appropriated conditions and were further purified by preparative electrophoresis on a 15% denaturing polyacrylamide gel in TBE. The composition analysis of the oligonucleotides was performed by HPLC–MS/MS analysis after enzymatic hydrolysis [18]. All the oligonucleotides gave the expected composition (data not shown).
5-(Hydroxymethyl)--deoxyuridine phosphoramidite derivative was synthesized in four steps from -deoxyuridine following the method reported by Sowers and Beardsley [19].
5-Formyl--deoxyuridine containing oligonucleotide was obtained by mild oxidation of an oligonucleotide containing 5-(1,2-dihydroxyethyl)--deoxyuridine. The phosphoramidite building block of 5-(1,2-dihydroxyethyl)--deoxyuridine was prepared from IdU in seven steps, as already described [20]. Before use, 1 nmol of 5-(1,2-dihydroxyethyl)--deoxyuridine containing oligonucleotide was oxidized in water at room temperature by 50 equiv of NaIO4 during 1 min (98% yield). This led to 5-formyl--deoxyuridine-containing oligonucleotide. It was subsequently desalted on a microspin G-25 column.
The 5-carboxy--deoxyuridine phosphoramidite building block was synthesized from 5-trifluoromethyl--deoxyuridine in four steps [21].
2.3 Oligonucleotide radioactive labelling
The 15-mer oligonucleotide (50 pmol) was incubated at 37 °C for 30 min in a 25-μl mixture that contained 50 mM Tris-HCl, pH 7.6, 10 mM MgCl2, 10 mM 2-mercaptoethanol, [γ-32P]-ATP (2.5 μl, 10 mCi ml−1) and 15 units of T4 polynucleotide kinase. The residual [γ-32P]-ATP was eliminated by purification on a microspin G-25 column.
2.4 Oligonucleotide transcription
Typically, 12 pmol of the templates were annealed with 10 pmol of T7 promoter in 10 μl of TES buffer (10 mM Tris-HCl, 1 mM EDTA, 100 mM NaCl, pH 8.0). The mixture was heated at 95 °C for 1 min and then slowly cooled down at 4 °C.
Transcription kinetics were performed in 50 μl with 0.5 μl of T7 enzyme Mix 10 U RNase inhibitor and 1.5 mM ATP, 1.5 mM CTP, 1.5 mM GTP, 0.75 mM UTP, 0.5 μl [α-32P]-UTP (10 mCi ml−1) in 1X transcription buffer (40 mM Tris-HCl, pH 8, 20 mM MgCl2, 10 mM NaCl, 0.25 mM EDTA, 1 mM DTT, 0.05 mg ml−1 BSA).
Transcriptions for the transcript identification were conducted in 20 μl with 0.2 μl of T7 enzyme Mix (Ambion), 2 U RNAse inhibitor and 3.75 mM of each rNTPs. After completion of the incubation, 1 μl of DNase I was added and the reaction mixture was further incubated at 37 °C for 15 min. The volume was adjusted to 25 μl with RNase free water and resulting RNA strands were purified on microspin G-25 column.
2.5 Transcript identification
The purified transcript (2 μl) was annealed with 5 pmol of the 15-mer 32P-labelled oligonucleotide in 50 μl of M-MuLV-RT 5X reaction buffer (250 mM Tris-HCl, pH 8.3, 375 mM KCl, 15 mM MgCl2, 50 mM DTT). The mixture was heated at 85 °C and slowly cooled at 4 °C. A reverse transcription on the templates was then performed using the following procedure: 0.1 pmol of radioactive labelled template was incubated in a 10-μl mixture containing 50 mM Tris-HCl, pH 8.3, 75 mM KCl, 3 mM MgCl2, 10 mM DTT, dNTPs (0.1 μM), 2 U M-MuLV reverse transcriptase and RNase free water. The mixture was incubated at 37 °C for 30 min. Gel loading buffer was added to the samples to stop the reactions. Samples were denatured at 95 °C for 3 min and subsequently loaded onto a 20% denaturing polyacrylamide gel. After the electrophoresis, the gels were exposed to phosphorimager screens. The bands corresponding to the transcribed strand and to the 34-mer standard labelled oligonucleotide were quantified using the Biorad Quantity One v4.2.2 software.
3 Results
3.1 Transcription reaction
To obtain reproducible transcription rate of the short template models, we used the T7 MEGA™ kit from Ambion that has been optimized for short synthetic templates. In order to have the possibility to observe low variations in the transcription kinetic of the different oligonucleotide templates, we reduced the recommended amount of T7 enzyme mix for the kinetic experiments. Thus the ratio enzyme/template in each reaction tube was lowered. The minimal amount of enzyme necessary to observe a quantifiable signal, especially for the templates T and HmdU was 0.5 μl in 50 μl of reaction mixture. Rounds of transcription were performed under these enzyme limiting conditions.
3.2 Nature of the transcribed strand
To analyse the behaviour of T7 RNAP when it encounters an oxidative damage of the methyl group of thymine, several transcription experiments were carried out using the synthesized templates.
First, we evaluated the influence of the lesions on transcription achievement. We could observe that each transcription reaction produced the full-length runoff transcript (40 nucleotides long). From kinetic experiments (Fig. 2), the absence of production of stalled transcripts was also noticed, suggesting that the enzyme does not arrest at the lesion sites. In a second time, we wanted to check the fidelity of the T7 RNAP when it encountered a lesion. For this purpose, a primer extension reaction was performed. After the transcription, the DNA template was digested by DNase I. The RNA transcript was then annealed with a complementary 15-mer oligonucleotide and a reverse transcription was performed with each of the four nucleoside triphosphate in independent reaction tubes. It was inferred from these experiments that in each template the oxidized base was recognized as a thymine by the T7 RNAP (Fig. 3).
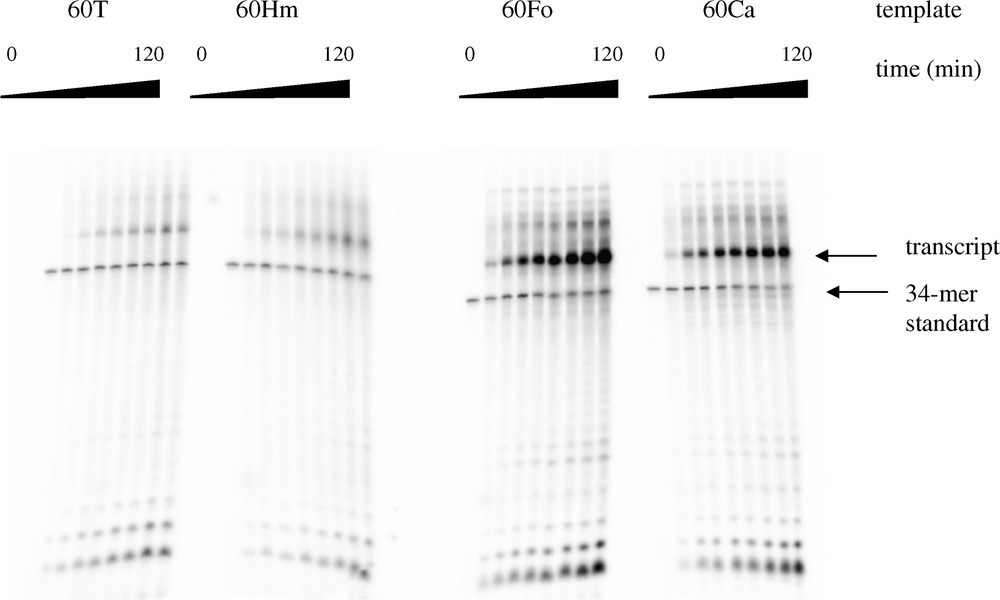
Transcription kinetic of the four templates. The transcription was performed with limiting amounts of enzyme (see § Materials and methods). A 34-mer oligonucleotide was introduced as an internal standard in the reaction mixture. Aliquots of the transcription reaction were withdrawn at various incubation times (0, 10, 20, 30, 40, 50, 60, 90, and 120 min) and the amount of transcript was quantified after electrophoresis on a 20% polyacrylamide gel.
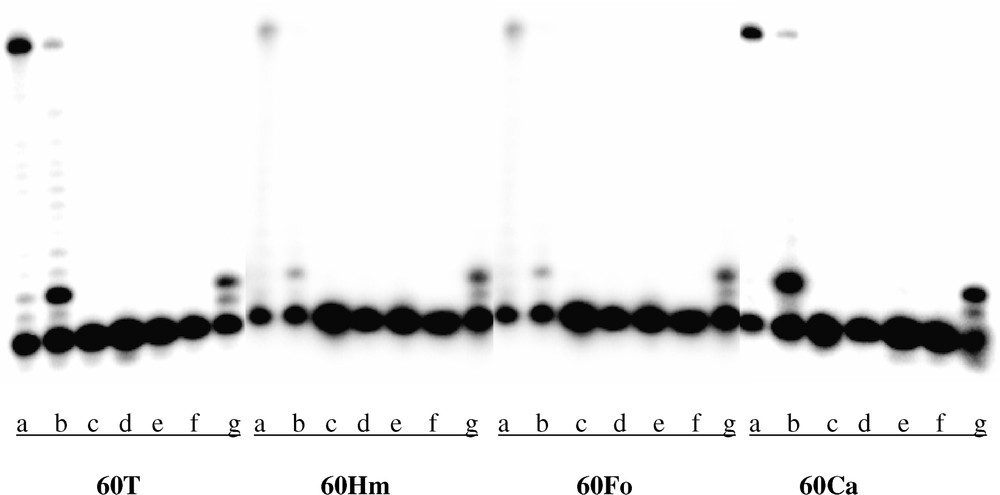
Determination of transcript sequence by a primer extension assay. Transcripts resulting from the transcription of the control (60T), the HmdU (60Hm), the FordU (60Fo) and the CdU (60Ca) containing oligonucleotides were hybridized with a complementary 32P-labelled 15-mer oligonucleotide. The M-MuLV reverse transcriptase conducted the elongation for 1 h at 37 °C with (a) 10 μM dNTPs mix, (b) 0.1 μM dNTPs mix, (c) control without dNTPs, (d) 0.1 μM dATP, (e) 0.1 μM dCTP, (f) 0.1 μM dGTP, (g) 0.1 μM dTTP.
3.3 Transcription kinetic
Influence of the three oxidized bases on transcription kinetic by T7 RNAP was investigated. Templates were annealed with the T7-promoter and transcription was operated in the presence of [α-32P]-UTP, necessary for subsequent quantification of the transcript. Furthermore, we added to each reaction mixture a radioactive labelled 34-mer oligonucleotide used as an internal standard. The amount of the different transcripts could then be normalized with respect to the amount of internal standard, thus limiting effects of pipetting errors (Fig. 2).
For each experiment, the bands corresponding to the transcribed strand (TS) and to the 34-mer oligonucleotide (C) were quantified. The relative amount of transcript (TSrel) was calculated: TSrel = TS/C for each band. The most elevated level of transcript obtained in each series of experiments that included the three oxidized bases and the control was set up to 100. The latter value corresponded to the amount of transcript obtained with the FordU template after 120 min of transcription. Finally, average results from three series of experiments were calculated and curves (percentage of the maximum amount of transcript/min) were established and expressed on the 0–100 scale. Initial reaction kinetics, corresponding to the slope of the linear part of the curve, were calculated using the data obtained between 0 and 30 min (Table 2). Relative global transcription yield was obtained by comparison of the amount of transcript for each lesion after 120 min of transcription with respect to the control (Table 3). As for transcription initial slope, it was observed an increased transcription yield for the oligonucleotides that contained a methyl oxidized derivative of thymidine compared to thymidine. Increase was significant for CdU (3.7 times higher) and FordU (4.9 times higher) containing oligonucleotides (Table 3).
Values of the initial slope of the transcription curves determined between 0 and 30 min reaction for the four templates. Slopes were calculated from transcription curves and are expressed as percentage of the maximum amount of transcript/min
| Oligonucleotide | 60T | 60Hm | 60Fo | 60Ca |
| Initial slope | 0.37 | 0.53 | 1.55 | 1.29 |
Relative transcription yield of the four templates. The amount of transcript obtained from the control template 60T was chosen as the reference and set up to 1
| Oligonucleotide | 60T | 60Hm | 60Fo | 60Ca |
| Relative transcription yield | 1 | 1.6 | 4.9 | 3.7 |
4 Discussion
Data from the literature show that DNA damage induced by ultraviolet radiation, gamma radiation, oxidative stress and chemicals may interfere with the transcription process mainly in two different ways:
- (i) elongation complex may either totally or partially arrest at the site of the lesion resulting in the release of the RNAP. Both prematurely terminated transcript and/or low amount of full-length transcript are then produced. Elongation complex release would be a message sent to repair enzymatic systems to initiate the repair of the transcribed strand. Besides, a correlation between extent of polymerase arrest and extent of transcription-coupled repair has been found for T7 RNAP in many cases [2];
- (ii) enzyme may bypass the lesion either with or without a stalling at the damage site and misincorporate one nucleotide opposite the lesion causing base substitutions in the transcript.
As what is observed with the DNA polymerases [16,17,22], neither HmdU nor FordU blocks the T7 RNAP in the primer extension reaction. The same feature was obtained with CdU. However, it has to be noticed that contrary to what happened with bacterial DNA polymerase for FordU [16,17], the T7 RNAP only obeys the A rule when it bypasses the three lesions studied. Hence none of the HmdU, FordU and CdU lesions affected fidelity of the process. These results may be put together with the results obtained by Liu et al. [6] who observed with the same kind of short models accurate bypass of 5,6-dihydrouracil (DHU) by both SP6 and T7 polymerases. Presence of the DHU lesion only induced a brief polymerase stalling at the lesion site.
The most unexpected result of our study was the marked increase in the initial transcription kinetic (Table 2) and in the transcription yield (Table 3) for lesion-containing template compared to reference template. This increase was in the following relative order: FordU > CdU > HmdU > T.
It has been shown that conformation of the template could affect stability of the initiation complex in relation with the interplay between template, enzyme and transcript [23]. This parameter has been mostly studied through its effect on the production of abortive transcripts. However, the latter results were obtained using different promoter conformations and may not be directly in relation with our observations.
Modified bases could also interfere with genetic signals modulating the process of transcription elongation. Hence, certain sequences are known to induce T7 RNAP as other RNA polymerases to either pause or release at specific sites [24–26]. More precisely, pausing may be induced by sequences that encode RNA hairpins. The latter structures could interfere either with formation of RNA.DNA hybrid or directly with RNAP active site [24,27,28]. Chan et al. [28] have reported using short length templates that pausing of the RNAP depends not only on the nascent RNA hairpin but also on the -proximal RNA or DNA sequences, the bases within the active site and on the downstream DNA duplex. Certain base substitution within one of the latter components can decrease the pause half-life by factors varying from 3 to 10 fold. We thus analysed more precisely the sequence of the DNA template and found that its transcription could lead to the formation of a hairpin characterized as a 5-bp stem and an 8-bp loop structure (Fig. 4a). This corresponds exactly to the characteristic of the hairpin structure, but with a different sequence, as studied by Chan and Landick [29], with the E. coli RNA polymerase (Fig. 4b). The authors hypothesized that the pause hairpin and the DNA downstream sequence could slow down nucleotide addition to the pause complex. Although the enzymes are different, the possibility that the sequence we arbitrary chose plays a critical role in the pausing of the RNAP could be considered. Moreover, presence of a lesion within DNA, at the site of the hairpin loop, could modify the interaction with the enzyme leading to an accelerated incorporation of the nucleotides. This could explain the unexpected results that were obtained. Structural studies have shown that HmU containing DNA is more flexible than T containing DNA [30]. It may not be impossible that either this flexibility or the base structure interferes with the binding of RNAP during the elongation step at this critical site. Structural data are lacking concerning the other lesions. Of course, complementary experiments using the same modified sequences inserted into plasmid templates have to be conducted in order to sustain this hypothesis.
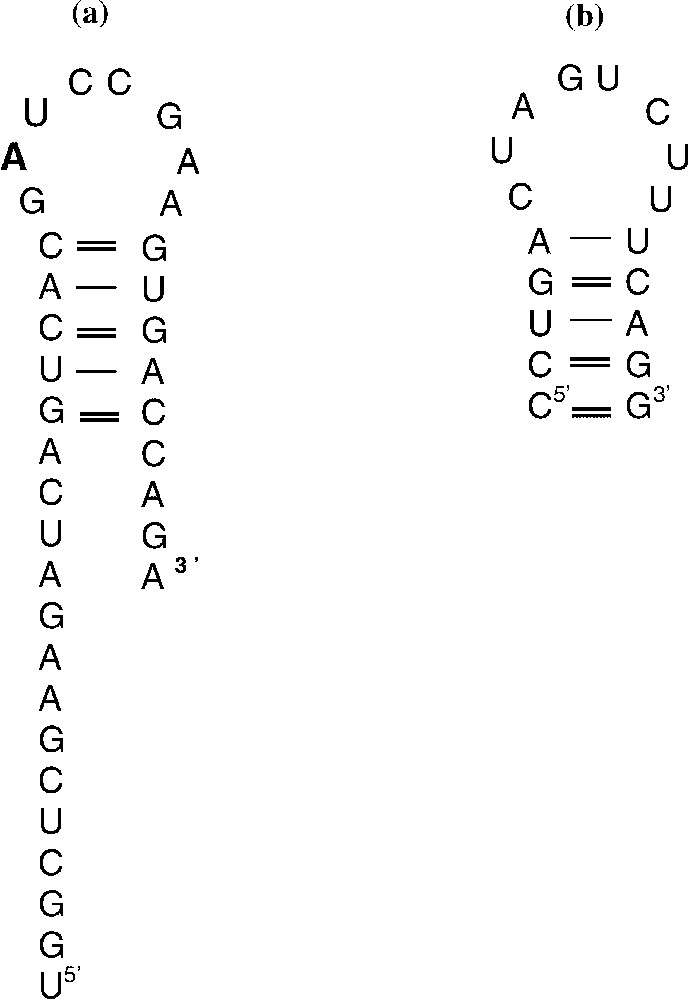
(a) Hairpin structure formed by the RNA resulting from the different template transcriptions. (b) RNA hairpin structure described by Chan et al. [28].
Anyhow, it would be very interesting to gain further insights into the effects of the presence of DNA lesion at specific known pause site for RNAP and to precisely study the effect of normal base substitution by a damaged base on transcription rate and on pausing signals.