

1 Introduction
The group Acanthomorpha comprises all teleosts with true spines in dorsal and anal fins [1,2]. With more than 15 300 species and 314 families, they represent nearly 60% of extant fish diversity. Despite their numerical importance, the phylogenetic relationships within the group were poorly known until recently, leading to its dubbing as “the bush at the top of the teleostean tree” [3]. However, decisive steps have been made during the last 15 years, stemming from comparative anatomy and molecular systematics. In 1990, a number of ichthyologists decided to pool their efforts to improve our understanding of relationships among percomorphs, which represent most acanthomorph diversity [2]. This led to significant advances in the placement of some subgroups, however many of the nodes of the global acanthomorph tree have remained unresolved or poorly defined. The recent increase in efficiency of molecular sequencing techniques has allowed major breakthroughs on the general phylogeny of Acanthomorpha [4–11]. While the trees obtained with these datasets partially agree, many parts of the tree are still subject to disagreement, and additional datasets for new markers with wide taxonomic samplings are still needed.
This study presents more complete datasets based on the work of Chen et al. [6] (partial 12S–16S mitochondrial sequences, 28S nuclear ribosomal sequences and rhodopsin gene sequences) and adds detailed analyses of partial sequences for a new gene, Mixed Lineage Leukaemia-Like (MLL). Partial results for this promising gene, with a smaller dataset, had been presented in [9,12]. One problem remains, though: no matter how many markers are used, the inferred clades must be assessed for reliability, and robustness does not equate with reliability [6,9,12–14]. One solution to this is to infer a tree based on all the available data to maximize the character congruence, while assessing the reliability of the clades by studying their repeatability across separate phylogenetic inferences from each independent dataset, without consideration for their bootstrap support in each [6]. This methodological framework combining separate analyses (taxonomic congruence without consensus trees) and simultaneous analyses [13–15] is summarized in Fig. 1. The MLL gene is a teleostean orthologue of a gene that, in humans, encodes a protein of 4498 amino acids involved in leukaemogenesis [16,17]. Partial sequences for introns 25 and 26 were available in GenBank for some acanthomorphs, but only the presence/absence of one of the spliceosomal introns had been recorded in the original publication [18]. Corresponding partial sequences, as well as sequences for an additional fragment of intron 26, are used here.
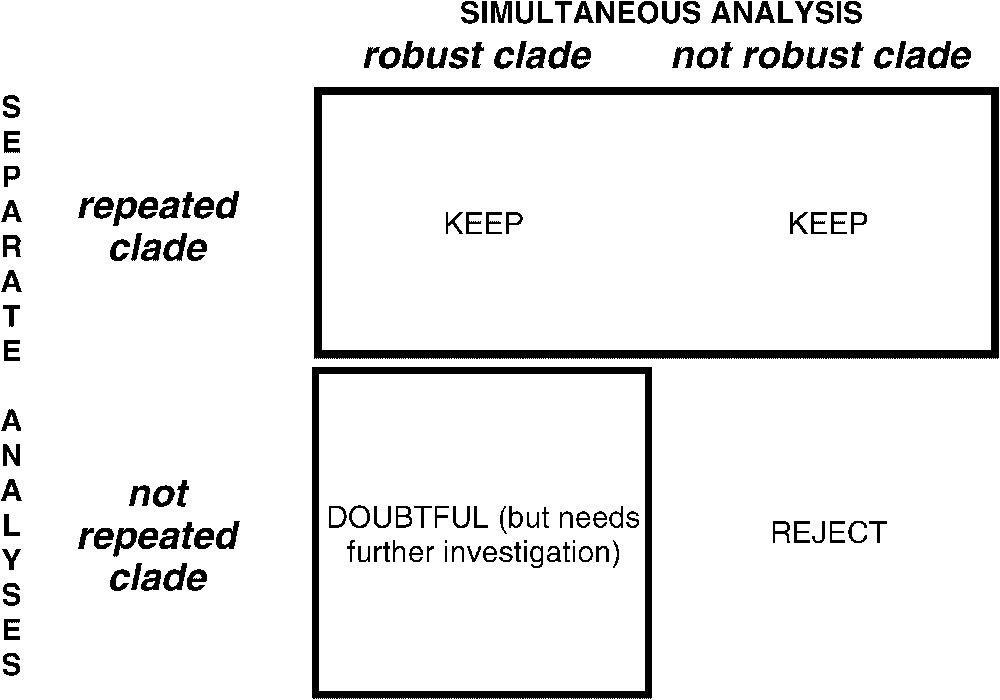
Simultaneous vs. separate analyses. The tree from the simultaneous analysis is kept as the major tree. However, reliability of clades in that tree is taken from their repeatability through separate analyses.
2 Materials and methods
2.1 Sampling
All sequences from Chen et al. [6] were used, and key-taxa were added to improve the taxonomic overlap between datasets and cut some long branches detected in previous studies. Representatives of groups missing in their study were added, as well as taxa improving the representation of already present groups. The MLL sampling of previous studies [12,18] was extended from 28 to 63 species (Table 1); this extended dataset is used here for the first time but the corresponding fragment of the gene (hereafter called MLL1) was difficult to amplify because of the presence of a spliceosomal intron (intron 25) with a size varying from around 50 base pairs (bp) in most acanthomorph species to almost 700 bp in Hippocampus. This intron has a very high sequence variability, and has repeated stretches of (t) monomers which tend to complicate sequencing and yield sequences of poor reliability that cannot be confidently aligned, except for very closely related taxa. Some sequencing problems with that part of the gene (here referred to as MLL1 [18]) encouraged the use of a different fragment. Starting with the Takifugu rubripes and Tetraodon nigroviridis sequences that were available, efficient primers were designed for a 550-base-pair fragment of the exon 26 (hereafter referred to as MLL2). MLL2 contains no intron and had not been previously used for phylogeny, except for a partial description given in a previous, methodology-focused, publication [9].
(a) Taxonomic sampling; (b) accession numbers
| (a) The classification follows Nelson (1994) except concerning Caproidei [55]. Species sampled only for one of the datasets (generally 12S–16S rDNA) are marked with a *, those with incomplete 28S rDNA sequences with a ?, and the specimens for which a voucher specimen is known to exist with a ©. |
| Osmeriformes: Bathylagidae: Bathylagus euryops; Stomiiformes: Gonostomatidae: Gonostoma atlanticum/bathyphilum; Aplepisauroidei: Synodontidae: Harpadon sp.*; Chlorophthalmoidei: Ipnopidae: Bathypterois dubius; Aulopoidei: Aulopididae: Aulopus purpurissatus*; Myctophiformes: Myctophidae: Electrona antarctica; Hygophum hygomii*; Acanthomorpha: Lampridiformes: Lampridae: Lampris immaculatus/sp., Regalecidae: Regalecus glesne°, Veliferidae: Metavelifer multiradiatus*; Polymixiiformes: Polymixiidae: Polymixia* japonica/ nobilis©; Paracanthopterygii: Ophidiiformes: Carapidae: Carapus boraborensis©/ bermudensis, Ophidiidae: Bassozetus zenkevitchi*, Lamprogrammus niger*, Sirembo imberbis*, Bythitidae: Cataetyx rubrirostris*, Diplacanthopoma brachysoma*; Batrachoidiformes: Batrachoidae: Halobatrachus didactylus©°; Gadiformes: Gadidae: Gadus morhua, Merlangius merlangus, Macrouridae: Trachyrincus murrayi° Coryphaenoides rupestris°, Moridae: Mora moro; Percopsiformes: Percopsidae: Percopsis transmontana*, Aphredoridae: Aphredoderus sayanus©°; Lophiiformes: Ceratiidae: Ceratias holboelli, Lophidae: Lophius piscatorius©°/ americanus/ sp., Antennariidae: Antennarius striatus©°; Zeiformes: Zeioidei: Zeidae: Zeus faber, Zenopsis conchifer©°, Macrurocyttidae: Zenion japonicum*, Parazenidae: Parazen pacificus*, Oreosomatidae: Neocyttus helgae; Beryciformes: Trachichthyoidei: Trachichthyidae: Hoplostethus mediterraneus, Anomalopidae: Photoblepharon palpebratus©*, Anomalops katoptron*; Trachichthyoidei: Diretmidae: Diretmoides veriginae/ sp.©; Berycoidei: Berycidae: Beryx splendens; Holocentroidei: Holocentridae: Myripristis botche/ violacea, Sargocentron rubrum/ microstoma, Ostichthys japonicus*; Stephanoberyciformes: Barbourisiidae: Barbourisia rufa©°, Rondeletiidae: Rondeletia loricata/ sp.°©; Cetomimidae: Cetostoma regani/ sp.°; Percomorpha: Mugiloidei: Mugilidae: Liza sp., Mugil cephalus*; Atherinomorpha: Atherinoidei: Atherinidae: Atherina boyeri*; Bedotioidei: Bedotiidae: Bedotia geayi; Belonoidei: Belonidae: Belone belone, Adrianichthyidae: Oryzias latipes©, Hemirhamphidae, Hemirhamphus sp.; Cyprinodontoidei: Poeciliidae: Poecilia reticulata/latipinna, Gambusia affinis*; Gasterosteriformes: Gasterosteoidei: Gasterosteidae: Spinachia spinachia, Gasterosteus aculeatus*, Syngnathoidei: Aulostomidae: Aulostomus chinensis, Fistulariidae: Fistularia petimba°, Macroramphosidae: Macroramphosus scolopax, Syngnathidae: Syngnathus typhle, Nerophis ophiodon, Hippocampus ramulosus©/sp.; Synbranchiformes: Synbranchoidei: Synbranchidae: Monopterus albus, Mastacembeloidei: Mastacembelidae: Mastacembelus erythrotaenia/ sp.; Dactylopteriformes: Dactylopteridae: Dactylopterus volitans, Scorpaeniformes: Scorpaenoidei: Scorpaenidae: Scorpaena onaria, Dendrochirus zebra*, Helicolenus hilgendorfi*, Triglidae: Chelidonichthys lucerna, Satyrichthys amiscus*; Cottoidei: Cottidae: Taurulus bubalis, Abyssocottidae: Abyssocottus korotneffi*, Cyclopteridae: Cyclopterus lumpus©°, Liparidae: Liparis fabricii©°/ sp., Comephoridae: Comephorus dybowskii*, Psychrolutidae: Cottunculus gobio°, Tetraodontiformes: Tetraodontoidei: Tetraodontidae: Lagocephalus laevigatus, Tetraodon nigroviridis, Takifugu rubripes, Balistidae: Balistes sp., Ostraciidae: Ostracion sp.©°, Molidae: Mola mola, Triacanthodidae: Triacanthodes sp.©°; Pleuronectiformes: Psettodoidei: Psettodidae: Psettodes sp./ belcheri°; Pleuronectoidei: Bothidae: Arnoglossus imperialis, Bothus podas°, Paralichthyidae: Paralichthys olivaceus*, Citharidae: Citharus linguatula, Soleidae: Microchirus variegatus, Solea vulgaris/ solea°, Pleuronectidae: Hippoglossus hippoglossus*, Syacium micrurum; Elassomatoidei: Elassomatidae: Elassoma zonatus©°; Perciformes: Caproidei: Caproidae: Capros aper, Antigonia capros*; Percoidei: Serranidae: Serranus accraensis, Holanthias chrysostictus, Epinephelus aeneus/ coioides, Pogonoperca punctata, Rypticus saponaceus°, Centropomidae: Lates calcarifer (2), Moronidae: Lateolabrax japonicus, Dicentrarchus labrax, Morone chrysops*, Percidae: Perca fluviatilis, Gymnocephalus cernuus, Chaetodontidae: Chaetodon striatus/ semilarvatus, Drepanidae: Drepane punctata/ africana, Pomacanthidae: Holacanthus ciliaris, Haemulidae: Pomadasys perotaei*, Sparidae: Sparus aurata°, Mullidae: Mullus surmuletus*, Menidae: Mene maculata, Polynemidae: Pentanemus quinquarius, Pomatomidae: Pomatomus saltatrix*; Carangoidei: Carangidae: Chloroscombrus chrysurus, Caranx latus*, Trachinotus ovatus, Coryphaenidae: Coryphaena hippurus*, Echeneidae: Echeneis naucrates; Acanthuroidei: Acanthuridae: Ctenochaetus striatus, Acanthurus xanthopterus/ sp., Zebrasoma scopas*, Naso lituratus*, Prionurus maculatus*, Ephippidae: Platax orbicularis*, Luvaridae: Luvarus imperialis*, Scatophagidae: Scatophagus argus*, Siganidae: Siganus canaliculatus/ sp./vulpinus©, Zanclidae: Zanclus cornutus*, Labroidei (sensu Kaufman et Liem 1982): Labridae: Labrus bergylta, Scaridae: Scarus hoefleri, Cichlidae: Haplochromis nubilus/ ismaeli/ sp. brownae©, Astronotus occellatus*; Zoarcoidei: Zoarcidae: Austrolycus depressiceps, Pholidae: Pholis gunnellus, Notothenioidei: Bovichtidae: Bovichtus variegatus, Cottoperca gobio, Pseudaphritis urvillii, Nototheniidae: Notothenia coriiceps, Dissostichus mawsoni*, Channichthyidae: Chionodraco hamatus*, Neopagetopsis ionah; Trachinoidei: Trachinidae: Trachinus draco, Uranoscopidae: Uranoscopus albesca, Ammodytidae: Ammodytes tobianus, Pinguipedidae: Parapercis clathrata*, Cheimarrichthyidae: Cheimarrichthys fosteri, Chiasmodontidae: Kali macrura; Blennioidei: Blenniidae: Parablennius gattorugine, Lipophrys trigloides*, Salaria pavo, Tripterygiidae: Forsterygion lapillum; Gobiesocoidei: Gobiesocidae: Lepadogaster lepadogaster, Apletodon dentatus; Callionymoidei: Callionymidae: Callionymus lyra; Gobioidei: Gobiidae: Pomatoschistus sp./ minutus; Scombroidei: Sphyraenidae: Sphyraena sphyraena, Scombridae: Scomber japonicus, Thunnus sp.*; Stromateoidei: Stromateidae: Pampus argenteus, Stromateus sp.*, Centrolophidae: Psenopsis anomala; Channoidei: Channidae: Channa striata/ sp.; Anabantoidei: Anabantidae: Ctenopoma sp., Belontiidae: Colisa lalia* |
| (b) Sequences obtained for this study are indicated in bold. X01–X04 stands for: from sequence X01 to sequence X04, while X01/X04 stands for: sequence X01 and sequence X04. When the beginning of the accession number is the same, only the last numbers are indicated. |
| 28S rDNA: AJ270039–40/46, AY141465–756, AY372697–730, AY372737–53, DQ021382–98. |
| 12S and 16S rDNA: AY157325, AB028664, AF042475, AF048997, AF049722, AF049724–25, AF049730/32, AF049734–35/40, AF055589–93/95, AF055597–98, AF055600–04/06, AF055609–14/16, AF055618–19, AF055621–25, AF055627/30, AF137213, AF215462, AF221881, AF227680, AF302287/392, AF355009, AF421956, AF488442, AF542204, AF542220–21, AJ421455, AP002928, AP002937, AP002943–44, AP002947, AP004403–08/10, AP004413, AP004421–23/26/28, AP004431–34/41, AY09828/77, AY141325–40, AY141342–410/12–64, AY157326, AY161233, AY368277–82, AY368284–311, D84033/49, Z32702/04/12/21/23/31. |
| Rhodopsin: AB001606, AB084933, AF137212–14, AF148143–44, AF156265, AJ293018, AY141255–324, AY368312–34, U57539/42, U97272/74–75, X62405, Y14484, Y18664/66, Y18672–74/76, Siganus et Elassoma (com. Pers. Chen), DQ021401–04. |
| MLL1: AF036382, AF137230–36, AF137238–44, AF137246–47/49–50, AF137253–62, AY362204, AY363629–67, SCAF15123. |
| MLL2: AF036382, AY362201–03, AY362205–20, AY362222–89, SCAF15123, DQ021399–400. |
2.2 DNA sequencing
Samples were kept in 70% ethanol until extraction following a classical protocol [19]. Sequence-specific amplifications were performed by PCR in a final 50-μl volume containing 5% DMSO, 300 μM of each dNTP, 0.3 μM of Taq DNA polymerase (Quiagen), 5 μl of 10× buffer (Quiagen) and 0.25 μM of each of the two primers (see Table 2 for a list of the MLL1 and 2 primers; the other primers were taken from Chen et al. [6]); 0.1–1 μg of DNA were added depending on species. After denaturation for 2 min, the PCR was run for 40 cycles of (30 s, 94 °C; 30 s, 52 °C; 1 min, 72 °C). The result was visualized on ethidium bromide-stained agarose gels, and purified with the Minelute PCR Purification kit (Quiagen). Sequencing was performed on a CEQ2000 Beckman sequencer, version 4.3.9, with the manufacturer's kit according to instructions. Each sequence was obtained at least twice and checked against its chromatograms in Bioedit [20]. Potential contaminations and mix-ups were eliminated by pairwise sequence comparison and using Blast [21] on GenBank [22] through NCBI (http://www.ncbi.nlm.nih.gov/), and, for dubious cases, another sequencing was performed on a new extraction. All sequences are deposited in GenBank (accession numbers listed in Table 1). Two MLL sequences [18] from GenBank were not used, because they were identical to sequences from distant species: the sequences from Channa sp. and Zeus faber were identical, as were those of Dissostichus mawsoni and Mullus sp. Those genera or related ones were sequenced again, and the contaminations (‘Zeus faber’ AF137241 and ‘Mullus sp.’ AF137248) were detected and removed from the dataset. Also, all sequences of Phycis blennioides used by Dettai and Lecointre [9] have been removed, since careful examination has shown a sample mix-up.
Primers used for the amplification and sequencing of MLL1 and MLL2
| Primer name | 5′–3′ sequences | Source | Fragment | ||||||||
| MLL U31 | CCC | TTY | TAY | GGV | GTY | CGC | TC | This study | MLL1 | ||
| MLL U32 | CTT | TCT | ATG | GGG | TTC | GCT | C | This study | |||
| MLL L737 | CGT | CGC | TGT | TGT | TGT | TGT | C | This study | |||
| VenkMLL L | ATR | TTN | CCR | CAR | TCR | TCR | CTR | TT | Venkatesh et al. (1999) | ||
| VenkMLL U | GCN | CGN | TCN | AAY | ATG | TTY | TTY | GG | |||
| MLL U1477 | AGY | CCA | GCR | GTC | ATC | AAA | CC | This study | MLL2 | ||
| MLL U1499 | GTC | AAT | CAG | CAG | TTC | CAG | C | This study | |||
| MLL U1506 | CAG | CAG | TTC | CAG | CCY | CTS | TA | This study | |||
| MLL L2127 | CWG | NTT | TTG | GTC | TYT | TGA | TNA | TAT | T | This study | |
| MLL L2132 | ACC | YGA | TTK | YGG | TCT | YTT | GAT | This study | |||
| MLL L2158 | ARA | GTA | GTG | GGA | TCY | AGR | TAG | AT | This study |
Alignment was mainly performed by hand under BioEdit [20]. The alignments of ribosomal sequence data from Dettai and Lecointre [9] were ameliorated, while still based on secondary structure [6]. The alignment of the loop regions in these datasets was based on several runs of Clustal X [23] with default gap penalties, and was then adjusted manually to avoid discontinuity of individual gaps. Loops were conserved for the analysis, but when the insertion length varied, the gap regions were deleted. The rhodopsin sequences contain no gap, and alignment of MLL coding sequences was performed using the proteic alignment as guideline. The intron 25 exhibited a large variability in size and sequence among acanthomorphs and could not be aligned reliably, so it was removed from the phylogenetic analysis. The alignments are available upon request. A combined dataset was created by concatenation of the sequences for each species. As some datasets contained more sequences than others (12S–16S for example), only taxa that had no more than one missing sequence (excluding MLL1) were included in the combination (Table 3). Although the two MLL datasets cannot be considered to have evolved independently (and therefore, cannot be used as independent corroboration), the sequences were not assembled and analysed together because the two datasets are far from overlapping. For the combined dataset, analyses were performed with and without the incomplete MLL1 dataset. As the taxonomic sampling was different for this dataset, concatenations of sequences were performed when the used species belonged to the same genus, or were non-controversially related according to [24]: Liza sp.–Mugil sp., Ctenopoma sp.–Colisa lalia and Myripristis botche–Sargocentron sp.
Information related to each dataset and analysis. For the protein coding genes, in the BMI, each codon position was allowed its own model (1: 1st codon position, 2: 2nd codon position, 3: 3rd codon position). The estimated parameters are not presented for the combined dataset, as they differ for each one of the 11 subsets (five datasets out of which three have different values for each codon position)
| Taxa | Analysed dataset length | Constant sites | Maximum parsimony | Estimates | ||||||
| MP informative positions | Nb. of equipars. trees | Length of most pars. tree | CI and RI values | Used model | Invariable sites proportion | Value of _ parameter | ||||
| 28S | 102 | 876 | 483 | 247 | 127545 | 1831 | CI = 0.28 RI = 0.47 | GTH +I+G | 0.29 | 0.46 |
| 12S and 16S | 146 | 823 | 216 | 509 | 8 | 10063 | CI = 0.114 RI = 0.334 | GTR +I+G | 0.24 | 0.62 |
| Rhodopsin | 122 | 759 | 289 | 384 | 460 | 5278 | CI = 0.151 RI = 0.456 | GTR +I+G | 1:0.34 | 1:0.57 2:0.46 3:1.45 |
| 2:0.52 | ||||||||||
| 3:0.04 | ||||||||||
| MLL1 | 66 | 832 | 197 | 428 | 9 | 3249 | CI = 0.275 RI = 0.395 | GTR +I+G | 1:0.01 | 1:0.29 2:0.29 3:3.24 |
| 2:0.01 | ||||||||||
| 3:0.01 | ||||||||||
| MLL2 | 92 | 554 | 162 | 330 | 24 | 3314 | CI = 0.213 RI = 0.450 | GTR +I+G | 1:0.1 | 1:0.68 2:0.75 3:5.58 |
| 2:0.21 | ||||||||||
| 3:0.01 | ||||||||||
| Combined | 105 | 3021 | 1181 | 1426 | 2 | 18230 | CI = 0.167 RI = 0.355 | GTR +I+G | Parameters estimated separately for all subsets | |
The size of each dataset, number of taxa and number of informative positions for parsimony are given in Table 3.
2.3 Data analyses
Separate and simultaneous analyses have been conducted under maximum parsimony (MP) and Bayesian phylogenetic inference method (BPIM). Under MP criterion, heuristic searches (TBR search, 5000 random addition sequences, gaps coded as missing characters) were conducted with PAUP*4.0b10 [25], as well as 10 000 bootstrap replicates with 10 random addition sequences performed for each. To summarize the repeatability of clades in terms of taxonomic congruence and number of occurrences, supertrees were constructed using PAUP* from maximum parsimony majority-rule consensus trees obtained from each gene separately.
BPIM was used as implemented in MrBayes 3.0 [26], with the following parameters: 4 chains, 2 million generations, sampling of every 10th tree and discarding of the first 50 000 trees after checking the ‘burnin zone’. No Bayesian search was run on the combined dataset including MLL1, as more than half of the sequences are missing for this dataset, and the parsimony method is the one that deals in the clearest way with the missing data present in the combined datasets.
As the adopted approach involves comparing trees obtained from independent datasets, the trees from [8,10] were used in the comparison.
3 Results
Dataset information is given in Table 3; the majority rule consensus trees inferred by BPIM for MLL1 and MLL2 are presented in Fig. 2a and b. The tree inferred from the combined dataset (minus MLL1) is presented in Fig. 3.
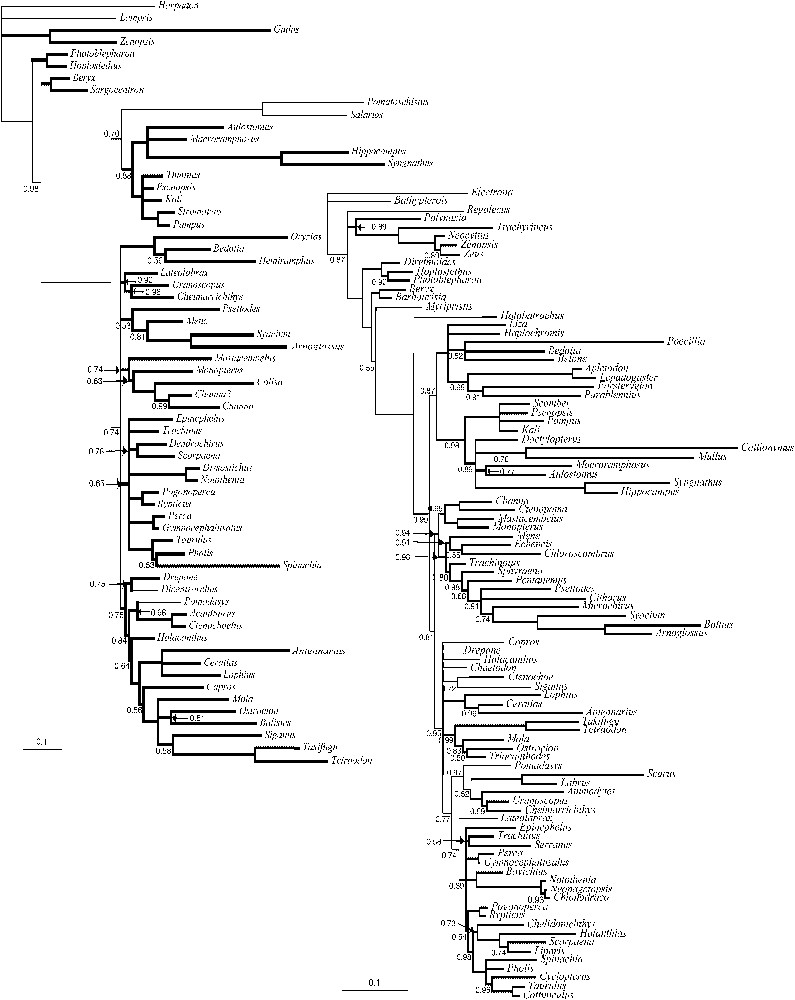
Majority rule consensus of trees inferred by BPIM from the two MLL datasets. Bold branches mark repeated clades. Only the names of the genera are indicated. For the names of the species, see Table 1. The values of the posterior probabilities are indicated under the branches. A (left): Majority rule consensus of the 25 000 trees sampled for the MLL1 dataset (2 million generations, 3 analyses, 25 000 first trees discarded as ‘burnin’). B (right): Majority rule consensus of the 25 000 trees sampled for the MLL2 dataset (2 million generations, 3 analyses, 25 000 first trees discarded as ‘burnin’).
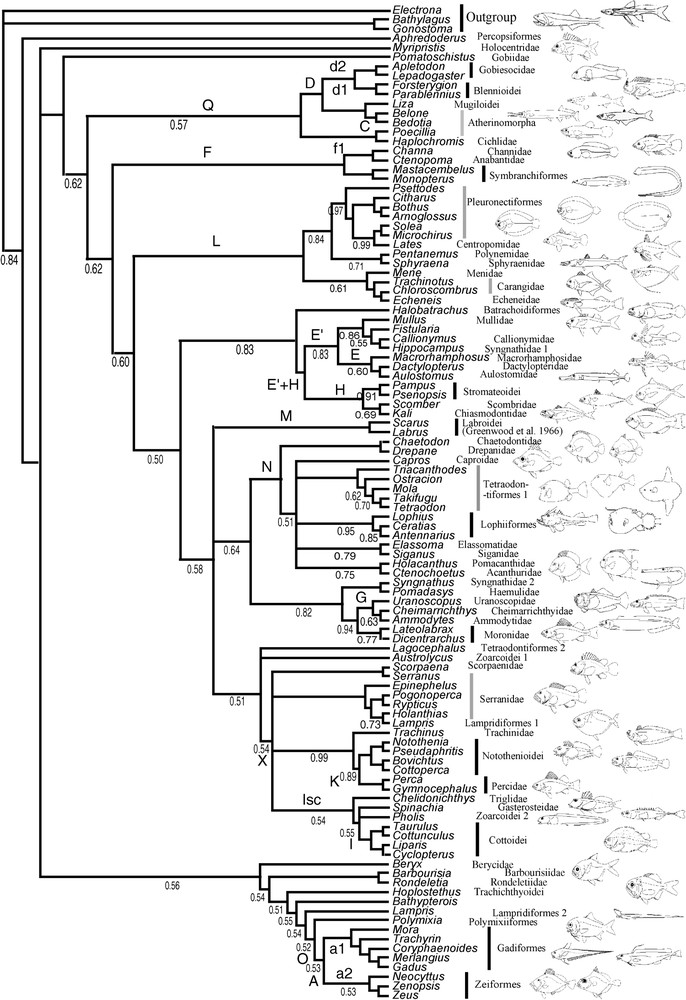
Majority-rule consensus tree of 25 000 trees sampled from the analyses by BPIM of the combined dataset (2 million generations, 3 analyses, 25 000 first trees discarded as ‘burnin’) with a model per gene, including a model per codon position. Names of repeated clades are shown with a letter (see Table 4). The values of the posterior probabilities are indicated under the branches. Note that the tree could have been rooted in Bathypterois. In that case, the most basal group would have been Regalecus + clade O.
A χ-square composition heterogeneity test did not show significant heterogeneity among taxa for MLL1 or for MLL2, unlike the rhodopsin dataset (the only other coding sequence). The differences in amounts of variable positions among first, second and third positions of the codon were moderate, and inferior to those measured on the rhodopsin gene. Absolute mutational saturation in the MLL data was calculated according to standard methods [27,28] for transitions and transversions and each codon position separately. Results confirmed those of preliminary studies [5] for MLL1, and were comparable for MLL2, the later exhibiting negligible saturation, except for 3rd-position transitions (plots available upon request).
3.1 Analysis of repeatability
Table 4 summarizes the presence of the nodes that were detected across 13 analyses, among which the five separate present datasets analysed by two methods. Previous analyses [8,10] comprising no data in common with the datasets used here are also included. The results of Smith and Wheeler [11] are discussed but not included as their analysis starts from data overlapping with ours (mitochondrial ribosomal sequences and 28S sequences) and therefore cannot be considered as an independent assessment of reliability. Results from the combined analysis of Chen et al. [6] are also presented in Table 4 to compare with results including the added MLL dataset and the added taxa. Only clades repeated in different trees using the same optimality criterion (either BPIM or MP) have been considered. To maximize the descriptive power of the repeatability analysis, partial repeatabilities were also scored with a precise indication of the missing (escaping) taxa or the single occurrences of insertions of additional taxa. This notion of escaping taxon has been discussed already [29]: a ‘repeated clade’ is the sum of the taxa repeatedly present in it, with no repeated contradictory clade; i.e. with no escaping/intruding taxon with repeated position. The single occurrence of non-integration of such an escaping taxon in the clade is provisionally considered to be due to dataset and taxon-specific artefacts, but this hypothesis will be questioned for each dataset studied in the future.
Table of repeated clades. X represents groups present in a given analysis, no marks represents groups contradicted by an analysis. For the MP analyses, x: groups present in majority rule consensus only; X: groups present in strict consensus, X: bootstrap value above 80%. For the BPIM analyses, x: posterior probability between 0.50 and 0.59, x: posterior probability between 0.60 and 0.69, X: posterior probability between 0.70 and 0.89, X: posterior probability between 0.90 and 1. +: taxon intruding in repeated group. −: taxon escaping from repeated group. /: inserting or escaping taxa form a clade. In the column ‘supertree’, clades present in the strict consensus supertree are marked ‘X’. Question marks mean that the corresponding clade is collapsed in that strict consensus. The taxon name abbreviations are presented in the left hand column and in the following list: Ah, Atherina; Ai, Antigonia; As, Astronotus; Au, Austrolycus; B, Bothidae; Bo, Bothus; Ce, Cetostoma; Ci, Chelidonichthys; Cr, Carapus; Cs, Coryphaenoides; Cu, Citharus; Dc, Dicentrarchus; Dr, Drepane; El, Elassoma; Fi, Fistularia; Ga, Gadus; Gs, Gasterosteus; Hi, Hippocampus; Lg, Lagocephalus; Me, Merlangius; Mo, Mora; My, Myripristis; Os, Ostichthys; Ot, Ostracion; Oy, Oryzias; Pd, Pomadasys; Ps, Psettodes; Pt, Pomatoschistus; Sn, Sargocentron; Sr, Serranus; Su, Syacium; Sy, Syngnathus; Tet, Tetraodontidae; Tr, Trachinus; Ve, Metavelifer
Many putative new clades first found in the earliest molecular studies of acanthomorph phylogeny [4,6,8] are also supported by the new MLL datasets: the Gadiform–Zeoidei group (clade A); the Gobiesocoidei–Blennioidei group (clade D); the clade Q (the previous one plus the Atherinomorpha plus Liza plus the Cichlidae); the clade E, grouping aulostomids, dactylopterids, and macrorhamphosids, the association of Channoidei and Anabantoidei with the symbranchiform representatives Monopterus and Mastacembelus (clade F); the clade I, grouping Cottoidei and Zoarcoidei, the association of the Gasterosteidae with or within the former clade (clade Is); the clade K, grouping the Percidae and the Notothenioidei; the clade G, grouping parts of the Trachinoidei (Ammodytes, Cheimarrichthys); the clade L of Chen et al. [6] (comprising Centropomidae, Carangidae, Echeneidae, Spyraenidae, Polynemidae and Menidae), which at last shed some light on the long-sought-for sister taxa of Pleuronectiformes. This clade L is very poorly supported by robustness indices, but is present in most analyses except 28S whatever the method and MLL1 in BPIM, with its composition almost constant, with the exception of some ‘escaping’ taxa in MP and constant in BPIM.
The clade I (Cottoidei–Zoarcoidei) found by independent studies [6,8] was recovered again, but the presence of Spinachia (Gasterosteidae), either as a sister-group of the clade, or as a sister-group of Zoarcoids only, is now confirmed by repeatability: both coding genes supported it, whatever the optimality criterion. The clade Q, grouping Liza (Mugilidae), Haplochromis (Cichlidae), Atherinomorpha (represented by Poecillia, Belone and Bedotia), and the clade D were present in the combined tree (Fig. 3) of Chen et al. [6], but were not repeated in their separate analyses. That group was recovered by all trees in BPIM with some escaping/inserted taxa and by all but the rhodopsin tree under the MP criterion. An equivalent group is present (Fig. 2) in the work by Miya et al. [8].
Some other groups that were present but did not appear as repeated in Chen et al. [6] have received some support through the present new dataset. The clade M grouping Labrus and Scarus, present until now only in the rhodopsin and combined datasets, confirmed the monophyly of the Labroidei, but only in its most restricted meaning (Labridae–Odacidae–Scaridae, though the Odacidae have not been sampled). Two different publications [30,31] proposed to extend the group to Cichlidae, Embiotocidae and Pomacentridae, however warning [31] that the synapomorphies supporting the clade were almost all characters of the highly-specialized pharyngeal region, and possibly subject to function-related convergence. A cichlid has been added to the previous datasets [6,9]. Unexpectedly, it grouped with the Atherinomorpha, Mugiloidei and clade D within a wider clade called ‘clade Q’. However, this result is not so surprising. In a comparative study of model fishes based on 20 nuclear protein-coding genes, Chen et al. [32] recently found that the Cichlidae were closer to the medaka (Atherinomorpha) than to the pufferfish (Tetraodontiformes). The monophyly of the wider Labroidei stands therefore to question, and representatives of Embiotocidae and Pomacentridae need to be added to resolve the position of these groups.
3.2 Monophyly of the main acanthomorph groups
Monophyly and taxonomic content of some of the acanthomorph groups had never really been questioned, because of the sizeable amount of morphological data supporting them (e.g., Tetraodontiformes, Pleuronectiformes). The monophyly of others, like Beryciformes (considered here as comprising Trachichthyoidei, Berycoidei and Holocentroidei), Scorpaeniformes or Zeiformes, has been questioned repeatedly, as the characters supporting them are few and sometimes ambiguous [33,34].
Some groups that have traditionally been considered as monophyletic do not appear as such in most molecular analyses: Scorpaenoidei, Pleuronectiformes, Tetraodontiformes, and Serranidae are especially problematic. The monophyly of Scorpaenoidei (represented by Chelidonichthys and Scorpaena) was never recovered by our analyses, although Miya et al. [8] found Triglidae with Scorpaenidae. Recently, Smith and Wheeler [11], with a study including a very large sampling of Scorpaeniformes, inferred a tree where the Scorpaenoid lineage was rendered paraphyletic by the inclusion of many non-Scorpaenoidei (Cottoidei and Hexagrammoidei), but also many non-Scorpaeniformes taxa (Notothenioidei, Grammatidae, Blennioidei, and even Atherinidae). Scorpaeniformes as a whole do probably not represent a monophyletic group, but complementary studies are necessary to determine which of the families or subgroups can still be considered as valid.
Monophyly of flatfishes is hard to recover with a wide sampling, whatever the molecular marker. But the taxa ‘escaping’ from the group were not the same depending on the gene and reconstruction method that were used, and that should be interpreted as the result of marker-specific artefacts rather than as a hint of some non-monophyly of the group. It might be interesting to draw attention to the fact that most groups, even those well-supported by morphological data, are hard to recover as monophyletic as soon as a consequent sampling is used. The monophyly of Tetraodontiformes that was first recovered with a wide sampling with the RAG1 dataset [10], was also recovered with the new MLL datasets. The group was represented here by six species chosen for their diversity. In trees from MLL1, they formed a clade; however, Siganus (not available for the other part of MLL) was inserted among them in MP, although not in BPIM. In trees from the 12S–16S dataset, Siganus was also grouped with a partial Tetraodontiformes.
Serranid monophyly was never recovered [9,11]. The group in its present composition was supported by several apomorphic features, including the presence of three opercular spines, and several reductive specializations [35], but in our analyses Serranus was generally not associated with the other Serranids (Rypticus, Pogonoperca, Epinephelus, Holanthias).
The monophyly of Moronidae, represented in our data by Dicentrarchus, Lateolabrax and Morone was not recovered in some of our trees, but none of the taxa associated with them were repeatedly found and therefore no conclusion can be drawn on their monophyly or paraphyly. The Anabantoidei–Channoidei group, questioned by Lauder and Liem [36], was recovered as proposed in Chen et al. [6]. Scombroidei sensu Johnson [37] did not appear as monophyletic, as sphyraenids were repeatedly within the clade L. The other Scombroidei components (Centrolophidae, Stromateidae, and Scombridae) grouped together, with the addition of Kali (Chiasmodontidae), that Pietsch and Zabetian [38] considered as a member of the Trachinoidei. The split of the Zeiformes was also corroborated, the Zeioidei being repeatedly grouped with Gadiformes, while Capros with Tetraodontiformes, Lophiiformes, Acanthuroidei and other perciform groups.
Trachichthyoidei, Holocentroidei and Berycoidei (Clade B of Chen et al. [6]) were never recovered as a group. Additionally, Beryx was repeatedly associated with the two Stephanoberyciformes representatives, Rondeletia and Barbourisia. Holocentroidei were associated to Beryx and the Stephanoberyciformes in trees from the 12S–16S dataset only, in agreement with Miya et al. [8]. While these two datasets contain no data in common, they both originate from the mitochondrial genome, and therefore caution must be used before considering them as evolving independently. Additional data are needed.
Gasterosteiformes appeared polyphyletic. Gasterosteids were associated with clade I (Zoarcoidei, Cottoidei); either as a sister-group of the clade (rhodopsin in MP but no support in BPIM) or inside it as a sister-group of Zoarcidae (in MP analyses of 12S–16S, both MLL, and Miya et al. [8], and in BPIM analyses of 12S–16S and MLL1). As the first of these two hypotheses is not present repeatedly while the second is, it seems safe to consider the second hypothesis as the more reliable. Aulostomidae and Macroramphosidae were associated with Dactylopteridae (clade E). No position is repeated for Syngnathidae, Mullidae, Callionymidae, most probably because they all have long branches whatever the dataset.
Trachinoidei were not monophyletic: Kali repeatedly joined some scombroid components in clade H as already pointed out by Chen et al. [6], but a partial monophyly was consistently recovered, grouping Ammodytes, Cheimarrichthys, and Uranoscopus. However, a wider sampling of the group is necessary before any general conclusion can be drawn.
These results illustrate the need for wide taxonomic samples in future acanthomorph molecular phylogenetic investigations, particularly for the groups considered as dubious on a morphological basis (percoids, scorpaenoids, trachinoids, ophidiiforms...).
4 Discussion
4.1 Congruence
The current practice of ‘total evidence’ emphasizes character congruence and measures reliability from robustness indicators (bootstrap proportions, Bremer supports, etc.). Far from that alleged ‘Popperian’ view of systematics [39–41] other systematists, along with discussions about abductive and non-Popperian notion of ‘testability’ in phylogenetic inference [13,42–51], reconciled with fully acknowledged background knowledge (if explicit and justified). This reconciliation in a foundationalist point of view [52] legitimates arguments for naturalness of data partitions and the use of models in phylogenetic reconstruction [51]. The present work interprets the degree of confidence one should give to a clade by qualitatively assessing taxonomic congruence between trees based on independent markers. Congruence is analysed at the level of statements on relationship hypotheses, not at the level of characters. The present approach therefore entails no Popperian predictive test.
4.2 New clades
In this study, Siganus was the closest to the Tetraodontiformes or was within the complete group (in trees from MLL1) or within partial tetraodontiform groups (in trees from 12S–16S), but not for the trees from the rhodopsin dataset and the MLL2 dataset. The association of Siganus with Tetraodontiformes revives the hypothesis of a relationship between Acanthuroidei and Tetraodontiformes proposed (among others) by Mok and Shen [53]. In Miya et al. [8], a caproid appears as the closest, with Lophiiformes as a sister-group of both. In Holcroft [10], a clade formed by Drepanidae and Ephippidae is the sister-group of the Tetraodontiformes, with Moronidae and Acanthuroidei and a Caproidae–Lophiiformes–Siganidae clade as the sister-group of all. Our study showed that all those taxa alternatively appeared as the closest to Tetraodontiformes, depending on the dataset and the optimality criterion, with some irresolution in several BPIM trees (both MLL, combined tree). Rosen [54] placed Zeioidei and Tetraodontiformes together, with Caproidei as the sister-group of both. According to all the available sequence data, Zeioidei were best separated from caproids (making Zeiformes polyphyletic, as suggested by Johnson [55]) and placed with Gadiformes (clade A), but caproids seemed indeed related to Tetraodontiformes, as hinted by Winterbottom [56]. The difficulty in recovering the monophyly of acanthuroids when the sampling of the group is more complete might also have played a role in the difficulties to recover monophyletic Tetraodontiformes and to recover the wider group of their relatives. The results of this work and previous publications, while not bringing a definitive answer, allowed us to identify a group of tetraodontiform relatives (clade N) that considerably reduced the list of potential sister-groups of the Tetraodontiformes among the whole Acanthomorph diversity: Caproidae, Lophiiformes, Acanthuroidei, Drepanidae, Pomacanthidae Chaetodontidae, and possibly partial Moronidae.
4.3 Clades proposed by previous molecular studies
A number of new clades for systematics of teleosts that were proposed by Chen et al. [6] have already been discussed in the original publication. Some new elements need to be reported. The clade X (first reported in Dettai and Lecointre [9] with a different methodology), comprising Cottoidei, Zoarcoidei, Gasterosteidae, Notothenioidei, Percidae, Serranidae, Trachinidae, and scattered Scorpaeniformes components, was supported only by MP tree from MLL1 and by BPIM trees from both MLL datasets. It was also present, but with very reduced samplings, in Holcroft [10] (a triglid and a percid) and Miya et al. [8] (no Percidae, Notothenioidei, Serranidae or Trachinidae). This group can therefore provisionally be considered as repeated, as it was supported by three independent datasets, even without using the partial combination methodology described by Dettai and Lecointre [9]. It is interesting to discuss the clade X in the light of the study by Smith and Wheeler [11]. The comparison is somewhat complicated to interpret, as their taxonomic sampling is widely different and, more importantly, part of their and our datasets are overlapping (12S and 16S, 28S). Their results therefore cannot be considered as fully independent from ours. In their tree, partial serranids (Epinephelinae) constitute a sister-group of the inclusive clade S comprising all Scorpaeniformes plus a clade grouping Trachinidae and Cheilodactylidae. The clade S contains several non-Scorpaeniformes groups. Many of these had been detected as members of the clade X [9]: Percidae, Notothenioidei, Zoarcoidei, Gasterosteidae, while some had not been included in previous studies: Grammatidae and Congiopodidae. The difference in location of these taxa when compared with the present study can probably be partially attributed to the difference in datasets and taxonomic sampling. Nonetheless, one of the clades included in clade S groups the Atherinidae and the Blennioidei, clearly contradicting previous studies as well as ours: atherinids and blennioids had been repeatedly associated with other groups in previous studies ([6,8], this study).
Imamura and Yabe [57] discussed several of the relationships within the clade X. They found a unique combination of 13 morphological characters uniting Zoarcoidei and Cottoidei, both reassessed and found to be monophyletic. Due to the presence of several characters shared by the Notothenioidei and the group Cottoidei–Zoarcoidei, they proposed to make them sister-groups. They also proposed a clade grouping the ‘scorpaenoid lineage’ and serranids, based on two character states previously described as synapomorphies of the scorpaenoid lineage and three reductive synapomorphies, first described as serranid synapomorphies. As no Percidae is included in their taxonomic sampling, it is not possible to say whether the Percidae + Notothenioidei group repeatedly found in molecular analyses, including this one, is supported by those morphological characters. This interesting study would need to be coded into a matrix and reanalysed, as it takes into account most of the members of the clade X and brings hope on the finding of morphological characters to support this clade.
4.4 Supertrees versus a tree based on simultaneous analysis
Fig. 1 shows that repeatability is our main criterion to assess reliability [6,12], and robustness is merely a technical information about the structure of the data. From a single gene, an artefact like unequal base composition among distantly related taxa can lead to a robust ‘compositional’ clustering. Such an unexpected clade is not recovered from other genes, so it is not repeated (Fig. 1, bottom left). In the simultaneous analysis, such a false and robust grouping can be the one found in the tree based on all available data, if alternative ‘signals’ from other markers are not strong enough to overwhelm the artefact. To summarize, the clades considered as reliable, supertrees [58] seemed suitable because a given clade is present only if the number of times it occurs among source trees exceeds the number of times alternative clades occur. In supertrees, the relative strength (data amount and structuration) of the internal ‘signal’ of each dataset has no influence on the outcome, so only the occurrence of clades in separate analyses is taken into account, not their robustness. Comparing the tree obtained from the combined analysis (Fig. 3) with the supertree (available upon request, however the clades recovered by the strict consensus supertree are listed in Table 4), distal nodes are generally congruent with the combined tree, while the resolution of the supertree is considerably less in deeper nodes. This is not surprising, as those deep nodes change from one tree to another. As we do not claim any phylogenetic conclusion from these unstable nodes, supertrees could be suitable to summarize repeatability. Supertrees do not handle correctly escaping/inserting terminals, and therefore lose information compared to repeatability tables like Table 4.
4.5 The tree based on all available data cannot be trusted alone
One could argue that the tree inferred from the combined dataset contains most of the repeated clades, and therefore could have been used on its own. Such a belief has its pitfalls. First, the tree based on simultaneous analysis also presents clades that contradict repeated clades. For instance, the Bothidae (Pleuronectiformes) are repeatedly associated with the other Pleuronectiformes representatives in clade L in separate analyses, but not in the tree resulting from the simultaneous analysis. Second, the tree based on the simultaneous analysis sometimes contains clades that agree with none of the topologies obtained in separate analyses. For instance, the position of Bothidae in the MP tree based on the combined data reflects none of the hypotheses in separate trees. When the tree inferred from the combined dataset is used alone, there is no way to make a difference between these cases, and to have an idea of the reliability of the clades.
4.6 The need for other genes
The number of independent genes previously available was rather small with regard to potential artefacts. A previous work [9] has shown that dataset combinations distinctly ameliorate the recovery of repeated clades. This study included one more dataset (MLL1). It showed that even with markers presenting potentially good properties as to saturation, two different parts of the same gene can lead to different trees, so pinpointing the danger of ‘magic-bullet’ markers [10]. Each dataset is a limited sampling of a mix of similarities both from common descent and homoplasy that can hide the phylogenetic relationships in some parts of the corresponding tree. The signal shared among markers (which is considered to be due to common descent as the markers underwent the same history), is therefore hidden by marker-specific biases. While the method of scoring repeatability is interesting because it is probably the best way to detect the shared signal, it is often too conservative, because the repeated clades are ‘lost’ in some of the markers due to homoplasy. An example of this is the grouping of Spinachia with zoarcids and cottids. Among the three datasets presented by Chen et al. [6], this group only appeared in the tree based on the rhodopsin data and in the combined tree. It was therefore not possible to consider it as reliable. It could only be regarded as a typical example of a grouping in the combined tree forced by the sole rhodopsin dataset. However, this group is now also present in the new MLL trees and the trees built using long mitochondrial sequences [8]. This example shows that three datasets might not be enough to detect repeated clades: increasing the number of datasets offers new opportunities to unveil repeated clades. But not all markers are equally efficient. Mitochondrial markers in general present high levels of saturation at those divergence times, and even protein coding genes can be subject to numerous biases, as exemplified by rhodopsin [6,59]. A carefully chosen marker brings better results [9,10], as shown by the higher efficiency of both MLL fragments to recover repeated clades compared to previously used markers of similar length (28S, 12S–16S, rhodopsin).
Some stress must also be put on the importance of wide taxonomic samplings. Several groups that had never been proposed before have emerged from the molecular results of the recent years (i.e., clade A: Gadiformes with Zeoidei), just because they had never been compared in a common matrix. The monophyly of many previously-described groups remains to be assessed with a wider sampling, and the surprises brought by the recent molecular studies [6–11] are probably far from coming to an end, promising years of exciting research on acanthomorph relationships.
Acknowledgements
We thank Wei-Jen Chen, Pascal Deynat, Cécile Fischer, Samuel Iglesias, Guillermo Orti, Leo Smith, Natalia Tchernova for recent tissue samples, and Blaise Li for help in tree calculation. We warmly thank Tony North, Gael Lancelot, Régis Debruyne, and Francesco Santini for readings and comments on the manuscript. We thank the ‘Service de systématique moléculaire’ (IFR CNRS 101) of the ‘Muséum national d'histoire naturelle’, Paris, France, for support.