

1 Introduction
Surfaces exposed to non-sterile and wet environments can be colonized by adherent microorganisms that encage themselves in hydrated exopolymeric substances (EPS) containing mainly polysaccharides, proteins and nucleic acids, forming a slimy layer known as a biofilm [1]. In medical medium, bacterial biofilms are able to colonize various surfaces (walls, floors, surgical tables, etc.) or implanted medical devices (catheters, endoscopes, pacemakers...), and are responsible for about 65% of nosocomial infections by direct infection or transmission [2,3].
Biofilms are characterized by a high level of structuration, most likely correlated to their specific function and their stability. Indeed, bacteria embedded in biofilms present specific physiological and genetic features that differentiate them from their planktonic counterparts [4]. Furthermore, biofilms are highly heterogeneous, with structures such as streamers and water channels, depending on the prevailing conditions, environmental stresses and EPS nature, which create a vascularization system similar to animal tissue. The biofilm depth can vary from a few micrometres up to centimetres depending on the strain and the environment.
The physicochemical and structural natures of EPS influence the bacterial physiology [5], but also may act to various degrees as a protection against environmental changes, such as desiccation or action of antimicrobials (antibiotics and biocides). Moreover, these exopolymers have been frequently invoked to account for the development of anaerobic micro-zones and structural heterogeneity in biofilms and thus may also act as a reservoir for pathogenic viral particles [6], leading to persistent viral infection.
Thus measuring the diffusive capabilities of various chemical species within the biofilm matrix is of importance to understand the dynamics of these microbial communities and to identify the role and function of EPS. Such studies have been first approached by means of scanning confocal laser microscopy (SCLM) combined with fluorescence recovery after photobleaching measurements (FRAP) [5]. However, FRAP does not allow a direct visualization of the fluorophore diffusion inside the excited volume and requires high fluorophore concentrations that can lead to inaccuracy in the diffusion coefficient determination [7]. More recently, we have demonstrated for the first time that fluorescence correlation spectroscopy (FCS) under two-photon excitation (TPE) can be successfully applied to characterize the penetration and diffusion capabilities of fluorescent tracer particles in monomicrobial biofilms [8]. FCS, based on the analysis of fluctuation intensities in the fluorescence signals emitted by a small number of molecules in a microvolume, is ideally suited for non-invasive in vivo measurements [9]. By comparison with conventional one-photon FCS, TPE minimizes photobleaching in spatially restricted volumes (∼1 femtolitre), thereby preventing long-term signal acquisition and measurement with single molecule sensitivity. Furthermore, the use of infrared wavelengths ensures deeper penetration than SCLM.
In this work we analyse the role of the Stenotrophonas maltophilia biofilm structure and specially the influence of EPS towards the diffusion and reactivity of bacteriophages. These viral particles also named phages are considered as favoured model for human enteric virus due to their similarity of structure, size, composition and morphology as well as behaviour toward conventional water treatment processes. The model viral particle used in this study was a C2 bacteriophage known to recognize specific surface sites of Lactococcus lactis subsp. lactis bacteria lacking on Stenotrophonas maltophilia cells. Stenotrophonas maltophilia is an aerobic bacillus of growing clinical importance [10] because it is the third bacteria involved in nosocomial infections after Pseudomonas aeruginosa and Staphylococcus aureus. This microorganism, when grown as a biofilm, contains mucoid mushroom-like structure typical of many gram negative bacteria [11], and is known to be particularly resistant to antimicrobials.
2 Materials and methods
2.1 Bacterial biofilms in continuous flow cells
This study was carried out with the bacterial strain Stenotrophonas maltophilia 114N-Sm provided by UNIR association (http://www.unir.org). The strain was stored at −20 °C, transferred twice in TSB (Trypcase Soy Broth, BioMérieux) before being cultivated overnight in 100 ml of TSB in continuous flow slide culture at 30 °C under oxygenated conditions.
Bacterial cells in stationary phase were washed by a succession of three centrifugations (

Confocal imaging of Stenotrophonas maltophilia biofilm showing (zone 1) unstructured and (zone 2) mushroom-like structure.
2.2 Prolate bacteriophages
The model viral of particle used in this study was a C2 bacteriophage infecting Lactococcus lactis subsp. lactis. The 22163-bp genome of this lactococal phage was fully sequenced [12]. The C2 phage exhibits a prolate structure with a 100-nm head and a 200-nm non-contractile tail. Lactococcus lactis was cultivated when necessary in M17 growth medium (Oxoid, France) supplemented with 0.5% of glucose and incubated at 30 °C overnight. Phage C2 and the C2-sensitive strain Lactococcus lactis, subsp. lactis were provided by S. Kulakauskas (INRA URLGA, Jouy-en Josas).
Phage titration was performed using the serial dilution technique and the formation of lytic plaques on a layer of the sensitive bacteria on a Petri dish, as described previously [13]. Experimentally, 0.1 ml of the phage suspension was added to 0.2 ml of a mid-log-phase bacterial culture of the C2-sensitive bacteria L. lactis. 30 μl of CaCl2 1 M were added to favour phage infection, and the mixture was incubated 10 min at 30 °C. Each mixture was added to 3 ml of soft agar (M17 Oxoid supplemented with 8 g l−1 of agar and 0.5% of glucose) in order to create a soft double layer on agar plates (M17 Oxoid supplemented with 15 g l−1 of agar and 0.5% of glucose). Petri dishes were incubated overnight at 30 °C in order to enumerate the visible lytic plaques.
For phage amplification, 108 PFU (Plaque Forming Units) of phage C2 were added to 400 ml of a L. lactis culture (DO600 nm = 0.1). Infected bacterial cells were incubated at 30 °C until clearance of the suspension. The mixture was then centrifuged to eliminate bacterial debris for 10 min at
2.3 Fluorescent probes
The anionic carboxylate-modified fluorescent latex beads of 110-nm diameter (range size of bacteriophage head), obtained from Molecular Probes were used as reference. The beads were used in suspension in deionised water (Purit, France). The solutions were filtered and sonicated before each experiment in order to eliminate most of the aggregates and to obtain a final concentration from 1 nM (in solution) to 10 nM (in biofilms). The latex beads could be considered to have a globular structure, enabling an estimation of their diffusion coefficient using the Stokes equation.
The selection criteria for bacteriophage fluorescent labelling were imposed by the experimental conditions required for FCS under TPE: (i) the fluorophore must be excited by two photons in the wavelength range 700–900 nm; (ii) detection of the fluorescence signal emitted during the counting interval needed to be significant despite the low excitation energy and the low fluorophore concentration used, which required to use a fluorophore with a significant fluorescence quantum yield; (iii) the fluorophore must be able to label the bacteriophage and not the living cells inside the biofilms. Because of these constraints, the fluorescent probe used to label the C2 bacteriophage was Sytox green (Molecular probes). The maximal concentration of labelled bacteriophages was obtained by adding 30 μl of Sytox green (5 mM solution) to 5 ml of C2 bacteriophage suspension titrated at 1010 PFU ml−1. The excess of free Sytox green was removed by careful rinsing. The labelled bacteriophage solution was used as it was for FCS measurements in biofilms and diluted by a factor 10 for experiments in solution. Before the FCS measurements inside biofilms, the growth medium flow was stopped and 2 ml of fluorophores were injected aseptically through the flow cell.
2.4 FCS set-up
The TPE experimental system used in this study has been described in detail elsewhere [14]. Briefly, the fluorescence excitation source was a femtosecond Ti:Sapphire laser (MIRA 900, Coherent Inc., Santa Clara, CA) pumped in the green by a continuous-wave diode-pumped solid-state laser (VERDI, Coherent Inc.) generating 100-fs pulses at a 76-MHz repetition rate with about 700-mW average power output. An excitation wavelength of 890 nm was fixed for all of the TPE experiments described here. The laser beam is expanded 3 times at the entrance of the microscope in order to reach a diameter of approximately 5 mm in diameter to fill the back aperture of the objective. After the beam expander, parallel laser light epi-illuminates a Zeiss
A general concern in fluorescence microscopy measurements and especially for applications with slow moving particles through biofilm depth is the risk of photobleaching the fluorophores at high laser power values. For this reason, we always used less than 1 mW at the sample and thus no cellular degradation was observed during the time of experiments as verified by confocal microscopy with the live/dead bacterial viability kit (Molecular probes). Furthermore, while IR excitation wavelength was used (890 nm), no significant temperature increase due to water absorption was observed.
2.5 FCS diffusion analysis
The conceptual and theoretical basis of FCS has been well established [7,9,15,16]. Assuming that the excitation intensity profile could be approximated using a three-dimensional Gaussian distribution, the fluorescence correlation curves in the case of free Brownian motion of molecules can be fitted using the following normalized autocorrelation function:
| (1) |
Under biphotonic excitation, the translational diffusion time can be related to the beam waist
| (2) |
For approximately spherical particles (such as latex beads) of hydrodynamic radius (R), the local viscosity η of the medium can be obtained via the Stokes–Einstein equation:
| (3) |
In the presence of obstacles that lead to deviation of the molecules from Brownian motion (called anomal diffusion) [7], the normalized autocorrelation function is transformed into:
| (4) |
The parameter
3 Results and discussion
The main goal of this work was to demonstrate the possibility of investigating the influence of biofilm structure on viral particle diffusion using FCS under TPE.
The diffusion processes were characterized by comparing the fluorescence correlation curves measured for C2 bacteriophages in solution and integrated into the Stenotrophonas maltophilia biofilm. As a reference, FCS measurements were also performed using reference anionic latex beads.
3.1 Solution measurements
Typical experimental fluorescence correlation curves obtained in aqueous solutions at room temperature (water viscosity
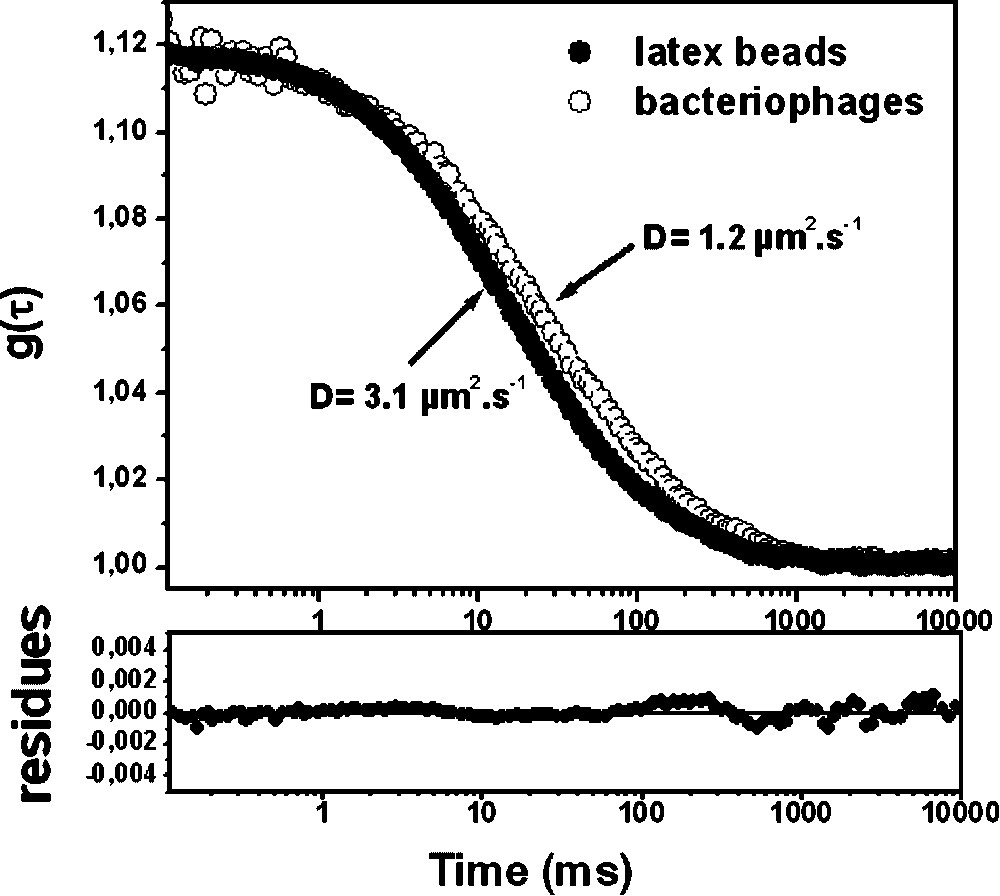
Experimental fluorescence correlation curves g(τ) corresponding to (●) anionic latex beads of 55-nm radius and (○) fluorescent labelled C2 bacteriophages in water. Continuous lines correspond to the fitting of the curves using Eq. (1). Parameters obtained from fit are: N = 1.5,
Experimental fluorescence correlation curves g(τ) corresponding to (●) anionic latex beads of 55-nm radius and (○) fluorescent labelled C2 bacteriophages in water. Continuous lines correspond to the fitting of the curves using Eq. (1). Parameters obtained from ... Lire la suite
Moreover, the experimental diffusion coefficient measured for the fluorescent microspheres is in good agreement with the theoretical value (
3.2 Application of FCS to microbial biofilms
We have observed that, in our experimental conditions, the bacterial autofluorescence as well as the overall biofilm movement were able to create a correlation signal (Fig. 3, inset). However, the translational diffusion coefficient associated to this background movement (
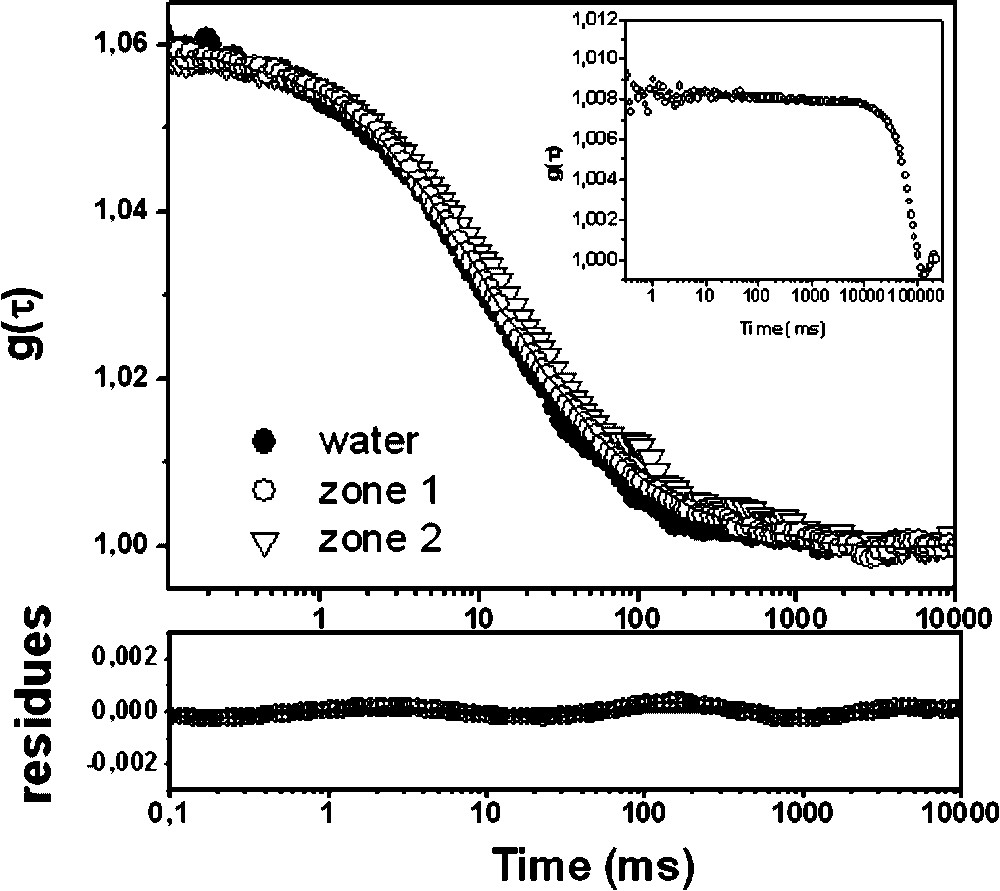
Fluorescence correlation curves for the anionic latex beads obtained in water and in the two distinct structural zones of Stenotrophonas maltophilia biofilm. The fitting of the curves was obtained using Eq. (1) in water (●) and zone 1 (○) and with Eq. (4) for the more distorted curve in zone 2 (▿).
3.2.1 Latex beads
The differences of mobility of the anionic latex beads at different horizontal positions within the S. maltophilia biofilm are apparent in Fig. 3: the FCS measured in zone 1 (more homogenous structure) can be fitted using Eq. (1), while the FCS recorded in zone 2 (mushroom-like structure) requires an anomal diffusion model (Eq. (4)) for interpretation.
In zone 1, we found a mean
Because of the complex nature of biofilms, it is necessary to rule out any artefacts before the diffusion can be termed truly anomalous. In particular, the diffusion rates can be mediated by binding of the probes to the biofilm constituents through physicochemical interactions, essentially electrostatic and Lewis acid–base interactions, as already observed in different eukaryotic living cells [9,15,18]. Micro-electrophoresis measurements of the latex beads revealed their anionic nature with a zeta potential of
Given the higher density of EPS in the mushroom-like structure compared to the homogeneous zone 1, an increased viscosity might have been expected, resulting in a parallel shift of the fluorescence correlation curve toward longer times (Fig. 3). However, the curve measured in zone 2 could not be superposed upon those obtained for the anionic latex beads in media with controlled viscosity [8]. Thus, with regard to the behaviour of the latex beads inside the mushroom structure, its diffusion can be only described as anomalous. The apparent diffusion coefficient obtained in this case was
Translational diffusion time
| Fluorophore | Zone 1 | Zone 2 | ||
|
|
|
|
|
|
| Carboxylated modified latex beads | 13 ± 1 | 2.0 | 23 ± 7 | 2.5–2.9 |
| Fluorescent labelled bacteriophages | 123 ± 15 | 2.5–2.9 | 130 ± 10 | 3.0–3.4 |
Reasons for a non-Brownian diffusion of particles inside a biofilm can be only speculated. However, it should not be surprising that environmental heterogeneities (c.a. rafts, EPS density, water channels...) or local confinement can occur in the mushroom-like biofilm structure.
3.2.2 Fluorescent labelled bacteriophages
Fluorescent correlation curve profiles obtained for the labelled bacteriophages were markedly dependent upon local biofilm structure. Within more homogeneous structure (zone 1), the fluorescence correlation curves were already distorted in comparison with that obtained for free bacteriophages. These distortions increased with the compactness of local biofilm structure. Fig. 4 provides a series of fluorescent correlation curves at different depths within a mushroom-like region (zone 2) of the biofilm, nonetheless revealing any significant difference in all the bulk of the mushroom structure.

Comparison of the fluorescence correlation curves for the labelled bacteriophages in water and at different depths of the mushroom-like structure of the biofilm (zone 2). The depth was expressed in μm from the glass cover slit surface. The fitting of the curves in zone 2 of the biofilm was obtained using Eq. (4).
No inhibition of the viral particle diffusion was observed, but it appears complex in each part of the biofilm and requires to be analysed considering its intrinsic properties. As mentioned previously, the Stenotrophonas maltophilia bacteria are not sensitive to the C2 phages, precluding specific interaction between the two particles. Moreover, a viral particle can be considered as a biocolloid able to exchange physicochemical interactions with the surrounding medium. However, the bacteriophage immobilization inside the biofilm matrix due to electrostatic interactions should be also reasonably excluded as for the reference latex bead. Indeed, the electrophoretic mobility measurement reveals a global negative charge of the phage surfaces with a zeta potential of −29 mV. Thus, the mobility of the C2 bacteriophages can be termed truly anomalous and the correlation curves satisfactorily fitted using Eq. (4). The experimental
4 Biological relevance
From a biological point of view, it has been previously hypothesized that the synergetic action of the bacteria in producing EPS may protect the majority of the biofilm occupant from infectious phage [19,20] or that bacteriophage migration through mucoid biofilms was governed through a reduction in EPS viscosity by enzymatic degradation [6,21]. Our in situ experimental measurements suggest that, even in the absence of specific enzymatic reactions (no local viscosity change detected), viral particles can penetrate inside the EPS structure of mucoid biofilms. After penetration inside the polymeric matrix, the biological particle may then take advantage from the specific ‘biofilm lifestyle’, and in particular benefit from protection against ‘outside’ environmental stresses, such as desiccation or the action of antimicrobial agents. Bacterial cells present in a bacterial biofilm are typically 1000 times more resistant to antibiotics than they planktonic counterparts [2]. During biofilm erosion or sloughing, these protected immobilized viral particles may be released in the environmental fluid, and be in contact with their target hosts for a viral infectious cycle. Previous studies have demonstrated that immobilized viral particles on solid surface most often keep their infectious potential after desorption [22]. Interactions of pathogenic viruses with bacterial biofilm are also consistent with previous studies reporting the persistence of enteric virions or poliovirus-1 in microbial biofilm [23–25].
From a public health point of view, biofilms were already regarded as a common cause of bacterial infections. They should now also be considered as protective reservoirs of pathogenic viral particles, and are probably responsible for numerous persistent viral infections.
Acknowledgements
The authors would like to thank H. Bidnenko and S. Kulakauskas for their expertise in handling lactic acid bacteria bacteriophages and the French department of Essonne for its financial support for the purchase of a laser confocal microscope (ASTRE No. A02137).