

1 Introduction
Over the past decade, there has been a huge improvement in the understanding of the peripheral and neuronal circuitry mechanisms that regulate feeding behavior and energy homeostasis in mammals. Many ‘anabolic’ hormones/neurotransmitters (i.e. promoting food intake, insulin release and decreased energy expenditure) and ‘catabolic’ hormones/neurotransmitters (doing exactly the opposite actions to the anabolic signaling molecules) have been documented (reviewed in [1–4]). In particular, many neuropeptides that originate and act in the central nervous system (CNS) have been identified that participate to a various extent and with distinct roles in the regulation of food intake/energy metabolism (reviewed in [1,2,5–7]). Furthermore, the critical role of specific hypothalamic areas in the regulation of feeding behavior and energy balance is now firmly established [8]. The concept of ‘feeding’ and ‘satiety’ centers was first coined in the 1950s by Anand and Brobeck [9] and Stellar [10], when they demonstrated that electrolytic lesions of the ventromedial hypothalamus (VMH) or the lateral hypothalamus (LHA) led to hyperphagia and obesity, or hypophagia and weight loss, respectively. Subsequent studies indicated that this dual feeding center hypothesis was oversimplistic [11], but, nevertheless it allowed one to specify the arcuate nucleus (ARC) and LHA peptidergic neuronal populations that play essential roles in the control of feeding and body weight. For instance, neurones co-expressing alpha-melanocyte stimulating hormone (α-MSH) and ‘cocaine and amphetamine regulated transcript’ (CART) are confined to ARC. Both peptides reduce food intake and energy storage by inhibiting locally Neuropeptide Y (NPY)/agouti-gene related peptide (AgRP) neuronal activity [12] or acting throughout other hypothalamic regions including the paraventricular nucleus (PVN), the dorsomedial hypothalamus (DMH) and LHA [13,14]. Indeed, two distinct neuronal populations containing orexigenic peptides were characterized in the LHA, namely the melanin-concentrating hormone (MCH) [15,16] and the hypocretins/orexins (ORX) [17,18] neurones that represent targets for neuronal cell bodies of the ARC [11].
Since the discovery and further characterization of the obesity syndrome in agouti mouse [19,20] there was a growing interest to investigate the role of the central melanocortins in the regulation of energy. These studies were enforced with the molecular cloning of AgRP [21], a brain-expressed α-MSH antagonist [22] and the recognition of the prominent role of the brain α-MSH receptors, MC3-R and MC4-R, in mediating the effects of α-MSH on food intake and thermogenesis [23–27]. Furthermore, the melanocortins form part of a larger peptide family, the melanotropins whose members all exhibit strong action on pigmentation, mainly in lower vertebrates [28]. Among these, there is melanin-concentrating hormone (MCH), a functional antagonist of α-MSH on melanophores, which has a very particular history at the early times, punctuated by long periods of limited studies, interrupted by short phases of intense investigations, when peptide purification or precursor-encoding mRNA cloning data were reported (reviewed in [29–32]). The interest in MCH rapidly increased after 1996 because of the first demonstration that this peptide may induce food intake after central injections [33] and that its gene expression may be modulated by fasting in normal rats [34] or in hypoleptinemic ob/ob mice [33]. Nowadays, the importance of the MCH neuronal system in regulating feeding behavior and energy homeostasis is shown by the great deal of effort made by pharmaceutical companies in order to produce MCH antagonists to treat obesity and metabolic disorders (reviewed in [35–38]). Therefore, this review article discusses the structural and functional links between the α-MSH (and related melanocortins) and MCH peptidergic systems in the central control of feeding and energy homeostasis with a particular emphasis on the recent characterization of pharmacological agents acting on their cognate receptors that may translate into treatments for feeding and weight gain disorders.
2 The pro-opiomelanocortin/alpha-melanocyte stimulating hormone system
Pro-opiomelanocortin (POMC) was the first example of a precursor generating functionally distinct peptides reported in the literature [28]. This polypeptide encoded the 13 amidated amino-acid α-MSH and structurally-related peptides including β-MSH, γ-MSH and adrenocorticotrophic hormone (ACTH), all forming part of the melanocortin family, and the opioid peptide β-endorphin, generated as well as β-MSH after cleavage of the β-lipotropin. The processing of POMC is complex and highly dependant of the considered species due to precursor sequence differences and tissues-specific because of different prohormone convertases (PC) content [39]. Indeed, in birds and rodents, but not in other mammals, the β-lipotropin could not be processed into β-MSH due to the replacement of basic residues at the cleavage site [40]. Furthermore, only POMC-derived peptides β-lipotropin and ACTH are produced by the single PC1 in the pars distalis of the pituitary gland whereas combined cleavages of POMC by PC1 and PC2 in pars intermedia of the pituitary gland or hypothalamus generate several peptides including α-MSH and β-endorphin [39]. In the rat brain, POMC synthesis appears restricted to neurones of the lateral part of ARC and the nucleus of the solitary tract (NST) (Fig. 1A). In the hypothalamic POMC-expressing neurones, the des-Nα-acetylated α-MSH peptide predominates while the acetylated α-MSH forms, i.e. the most potent bioactive peptides, are found in the projections [39].
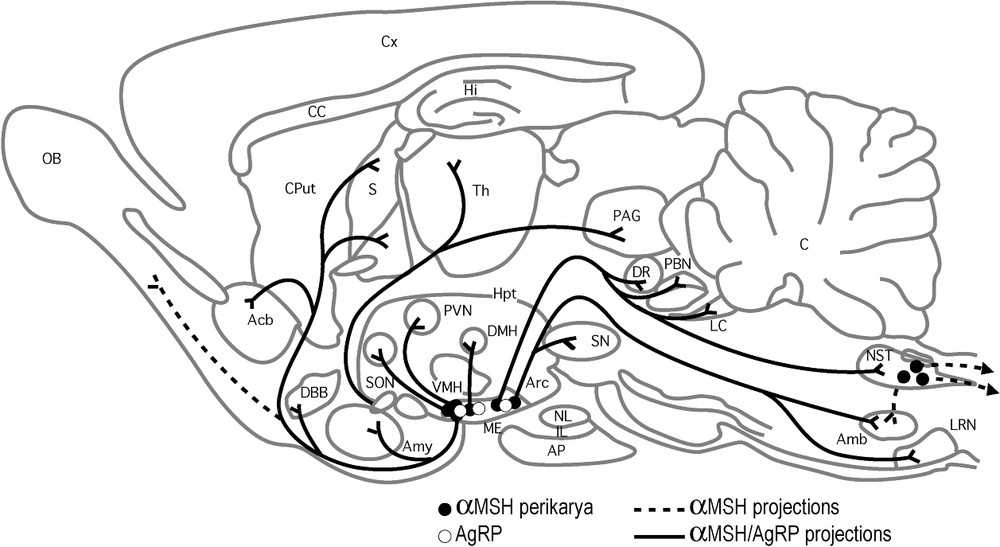
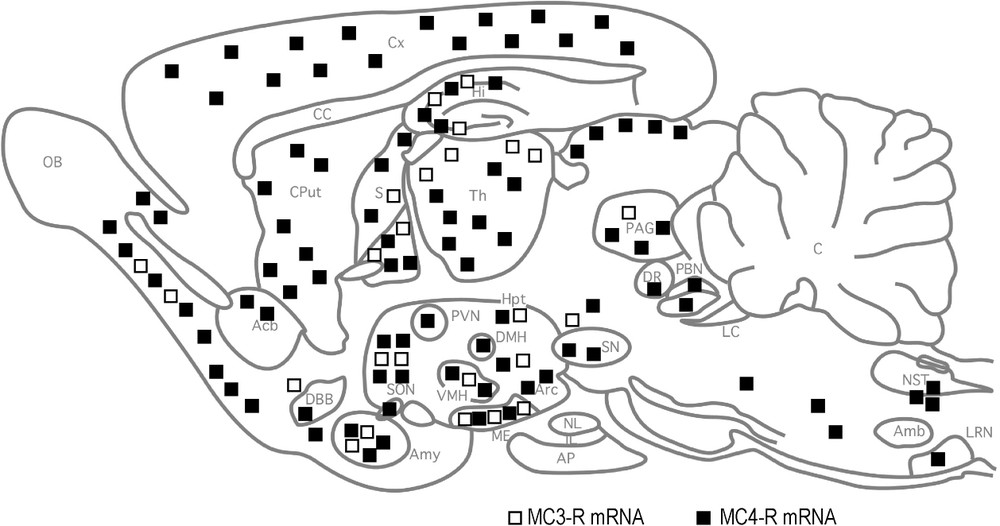
Distribution of α-MSH and AgRP neurons and projections (upper panel A) and MC3-R and MC4-R mRNA expression (lower panel B) in the rat brain. In (A) black and open points indicate α-MSH and AgRP perikarya, respectively; solid lines correspond to similar α-MSH and AgRP projections, while dashed lines indicate specific α-MSH fibers; (B) open and black squares correspond to MC3-R and MC4-R, respectively (from [39]).
As illustrated in Fig. 1A, α-MSH-producing neurons of the lateral ARC innervate several regions in the rat brain, including hypothalamic areas well known to regulate feeding behavior, such as ARC itself (NPY/AgRP neuronal subpopulations), PVN, DMH and LHA. All of these areas expressed either MC3 or MC4 receptors (Fig. 1B) and represent obvious targets for α-MSH to exert its catabolic effects [8]. In addition, neurons that co-express α-MSH and CART in the lateral ARC project to the preganglionic neurons in the spinal cord, suggesting a direct action of α-MSH on the sympathetic system and subsequent regulation of energy homeostasis and fat storage in the adipose tissues [39].
The central effects of α-MSH on feeding behavior and energy balance were tested by acute intracerebroventricular (ICV) injections of the peptide or synthetic analogues with higher potency such as MTII and demonstrated a strong reduction in food intake and adiposity in wild type rodents [41] but also in obesity-prone genetically modified ob/ob mice [42]. These peptides also enhanced metabolic rate and sympathetic outflow [43] and partially compensated for alteration in body temperature regulation in leptin deficient ob/ob mice [44]. Chronic infusion of either the α-MSH analogues led to a reduction in body weight [41], likely resulting from a decrease in fat mass and improved glucose uptake recovery.
Further evidence for a key role of POMC-derived peptides in the central control of feeding and energy balance came from genetic studies in rodents and humans. Humans with a mutation in the POMC sequence that leads to α-MSH deficiency display hypopigmentation, adrenal insufficiency and obesity [45–47]. Targeted deletion of the POMC gene in mice leads also to obesity, resulting mainly from reduced metabolic rate and secondarily from increased feeding [42,48]. Interestingly, the obese phenotype in POMC-mutant mice resulted mainly from altered lipid metabolism as the weight gain, particularly in fat tissues, was much greater than the increase in feeding behavior. Injection of α-MSH in these mutants further indicates a major role for α-MSH in mobilizing peripheral fat stores [42]. The POMC−/− as well as POMC−/+ mice were found also highly sensitive to high-fat diet and developed increased obesity [48] suggesting that under certain environmental conditions, e.g., unlimited access to high energy food, even a single POMC gene is not sufficient to maintain normal energy balance. This phenotype is reminiscent of the common human obesity and supports the hypothesis that haploinsufficiency of POMC gene may contribute to obesity in conjunction with dietary factors.
The role of the central nervous system (CNS) POMC neuronal network in the response to leptin is now firmly demonstrated (reviewed in [6]). Leptin, the product of the ob gene in mouse, is produced mainly by adipocytes, circulates to the hypothalamus and activates cells expressing the long form of leptin receptor to provoke finally suppression of feeding. POMC neurons in ARC express the functional leptin receptor [7,49,50] and the binding of this hormone stimulates the expression of c-Fos and Stat-3 that secondarily activate α-MSH gene transcription [51]. POMC gene expression is drastically reduced in ob/ob mice, whereas leptin injections restore its normal level [52]. States of hyperleptinemy, such as diet-induced obesity, increase POMC mRNA expression [53], while states of hypoleptinemy, such as food deprivation or lactation, are associated with decreased POMC gene activity [54,55]. At the cellular level, electrophysiological studies indicate that leptin increases the frequency of action potentials (AP) in POMC neurons through two distinct mechanisms, a direct depolarization involving a nonspecific cation channel and a reduction of GABA and NPY secretion from presynaptic terminals [12]. Interestingly, ob/ob mice display higher excitatory tone onto NPY neurons and reciprocally a net increase in inhibitory tone onto POMC neurons, in agreement with the corresponding effects of these peptides on feeding behavior. Furthermore, leptin replacement in ob/ob mice induces profound synaptic rearrangements onto the NPY and POMC neurons that restores the original content of excitatory and inhibitory synapses found in the wild-type mice [56]. These elegant studies combining transgenic mice models expressing distinct fluorescent proteins in specific neuronal populations and patch-clamp recordings in ARC slice preparations strongly suggest that leptin acts by rewiring and remodeling the synaptic inputs to the two key peptidergic neuronal populations of ARC that regulate feeding behavior and maintain energy homeostasis.
3 The MC1-5 receptor family
Five melanocortin receptors, named MC1-R through MC5-R, have been characterized so far [57]. All belong to the G protein-coupled receptor (GPCR) superfamily and signal through adenylyl cyclase-proteine kinase A activation by coupling to Gs [57]. The MC-R subtypes differ by their species-specificity, tissue distribution and ligand selectivity (reviewed in [39]). Indeed, MC1-R is expressed in melanocytes, melanoma cells and macrophages, and mediates melanocortin effects on hair pigmentation in rodents and likely skin color in humans. MC2-R is present in the adrenal cortex and adipocytes and appears essential for ACTH-mediated adrenal steroidogenesis and corticosteroid secretion. As shown in Fig. 1B MC3-R and MC4-R are highly expressed in the brain: the MC3-R is localized in discrete regions of the diencephalon including ARC and PVN and MC4-R more widely distributed in the CNS with high expression in the hypothalamic areas that participate in feeding regulation (e.g., DMH, PVN, LHA) [39,58]. The MC5-R is expressed mainly in exocrine glands and skeletal muscle but also at low levels in restricted brain regions (cortex, striatum, medulla).
Several lines of evidence argue for a major role of MC4-R and to a lesser extent of MC3-R in mediating the effect of melanocortins, mainly α-MSH, on food intake and energy homeostasis. Inhibition of MC4-R with specific antagonists induces hyperphagia, increases fat mass and hyperinsulinemia in rodents [45,59]. Similarly, targeted deletion of MC4-R gene in mice is associated with an increased food intake, late-onset weight gain and hyperinsulinemia [24]. In addition to mediating the anorexic effect of melanocortins the MC4-R may also mediate high-fat diet-induced thermogenesis and enhanced muscular activity [25,26]. Recent studies suggests that the magnitude of these changes in MC4-R−/− mice is partially compensated by an increase in POMC mRNA and a decrease in NPYmRNA expression [60]. Furthermore, MC4-R−/− mice do not respond to MTII, an α-MSH agonist, further indicating that α-MSH acts through MC4-R to regulate feeding [61]. Autosomal dominant mutations of MC4-R in humans are associated with morbid obesity but not adrenal insufficiency or hair pigmentation modifications, suggesting that MC4-R is the key melanocortin receptor that subserve the central regulation of weight gain also in humans [62,63]. On the other hand, MC3-R may also contribute like MC4-R to the regulation of thermogenesis and fat storage since null mice for this receptor also exhibit reduced activity and increased adiposity under high fat diet [26]. However, MC3-R−/− mice are hypophagic and treatment with MTII induces anorexia like in wild type mice [23]. This contrasts with the hyperphagic phenotype observed in MC4-R−/− and strongly suggests that the role of MC3-R in the brain appears restricted to the regulation of energy balance. Interestingly, double knock-out mice (MC3-R−/−/MC4-R−/−) eat excessively, owing to the loss of MC4-R, and store energy more efficiently, owing to the lack of both MC-R [27]. This indicates that both MC3-R and MC4-R participate in regulating energy homeostasis but with distinct and non-redundant roles.
The CNS circuits that contribute to cachexia and anorexia are less studied than these involved in obesity, but there is accumulated evidence that the melanocortin pathway may play an important role in mediating chronic disease-associated cachexia (reviewed in [64]). ICV injection of melanocortins mimics most of the effects induced by intraperitoneal lipopolysaccharide (LPS) administration on feeding behavior. Conversely, ICV injection of melanocortin antagonists suppresses LPS or tumor development-induced cachexia. Anorexia and cachexia provoked by LPS injection or chronic tumors are blunted in MC4-R−/− mice while MC3-R−/− mice exhibit higher response to the same treatments [65,66]. This paradox may be explained when considering the differential distribution and role of the MC3-R (presynaptic inhibitory autoreceptor) and MC4-R (post-synaptic receptor). Together, these data suggest that the melanocortin circuitry is required to mediate the anorectic/cachectic actions of inflammatory response factors. However, the anorectic effect of cytokines such as Il1 and Il6 persists and may be exacerbated in ARC-lesioned mice [64].
4 A natural antagonist to MC4 receptor: the Agouti related protein (AgRP)
Characterization of the monogenic mouse obesity model named Ay (Agouti) led to the discovery of the first natural α-MSH antagonist, namely the Agouti protein, that promotes yellow hair color when normally expressed in the hair follicle and a morbid obesity syndrome when ectopically expressed in the brain (reviewed in [67]). The antagonism of α-MSH by Agouti at the MC1-R blunts the production of black hair pigment (eumelanin) and allows for accumulation of the yellow pigment (pheomelanin) that results in the yellow hair color of Ay mice [68]. The most obvious interest in the Agouti mouse model is related to the dominant pleiotrophic phenotypes associated with the ectopic expression of the Agouti protein. Indeed, many of these phenotypes are related to alterations in feeding behavior and energy balance, suggesting the interaction of the ectopic Agouti protein with other melanocortin receptors that are involved in the regulation of energy homeostasis (reviewed in [67,69]). Early studies suggested that both central and peripheral actions of Agouti could contribute to the obesity syndrome. For instance, Agouti increases intracellular calcium in skeletal muscle and adipocytes and enhances fatty acid synthase activity [70,71]. Furthermore human Agouti appears widely expressed in skin, heart, reproductive track and fat tissue suggesting that this protein exerts a larger spectrum of functions in humans than in rodents [72]. Further studies [22,68] using partially purified mouse Agouti revealed that this protein was a potent α-MSH antagonist at MC1-R and MC4-R and does not bind to MC3-R and MC5-R. Interaction with MC2-R was also ruled out based on the persistence of the obese phenotype in adrenalectomized Agouti mice. The strong anabolic effects of Agouti, and the direct link with MC4-R, led to the hypothesis that an Agouti agonist may be synthesized in the brain and could be involved in the regulation of feeding behavior and energy balance by acting through the MC4-R signaling pathways. The molecular cloning and expression analysis of the ART gene by Shutter et al. [21], renamed later AgRP for Agouti gene-related protein, definitively confirmed this hypothesis.
The AgRP gene encodes a 132 amino acid (AA) protein in human and a 131 AA protein in rodents [21,73]. The full AgRP mRNA is expressed predominantly in the hypothalamus and the subthalamic nucleus while a truncated transcript lacking the 5′ non-coding exon is found in peripheral organs such as the adrenal gland, testis or lung [21]. Different promoter activity may explain the brain- or periphery-specific expression of the two AgRP mRNA species [73].
AgRP influences many neuroendocrine systems, in particular the hypothalamic-pituitary-thyroid axis and brain functions related to immune challenge (reviewed in [67]). However, the best characterized role of AgRP is its potent stimulatory effect on feeding behavior. AgRP exhibits a very long-lasting effect on food intake (up to 7 days after a single injection) that may not be related to a sustained blockade of MC-R [74]. One possible mechanism to explain this unique persistent orexigenic effect could be its action as inverse agonist at the MC4-R [75], i.e. independent of its melanocortin antagonist effect. Alternatively, we can also propose a potential interaction with co-receptors independent of the melanocortin receptor family, such as the cell surface heparin sulfate proteoglycan syndecan-3 that enhances AgRP action at the MC4-R and could mediate the effects on energy expenditure (but not food intake) of the N-terminal part of AgRP [76,77].
A striking feature of the AgRP neuronal distribution is its co-expression with NPY, another potent orexigenic peptide, in the ARC neurons (Fig. 2) [78]. This neuronal subpopulation also expresses most of the receptors (long form of the leptin receptor, GHS-R...) and intracellular signaling proteins (SOCS3, c-Fos...) that allow for an integrated response to peripheral satiety or feeding-initiation hormones (reviewed in [67]). The NPY/AgRP neurons represent therefore a key element, together with the α-MSH/CART neuronal subpopulation (see above), of the hypothalamic neuronal circuitry that regulates food intake and energy balance. Many hormonal factors that participate in the control of feeding behavior and energy balance regulate AgRP production (see Fig. 2). Satiety signals, like leptin or insulin, decrease AgRP expression and subsequently appetite by reducing the firing of AgRP/NPY neurons [12,78]. Conversely, AgRP expression is enhanced in mice lacking leptin or insulin receptors [59] as well as in leptin-deficient (ob/ob) mice [21]. These results suggest that leptin and insulin convey their satiety signals by inhibiting (at least in part) the activity of the AgRP/NPY neurons. On the other hand, hunger-mediated hormones, such as the gut-produced ghrelin and glucocorticoids, activate AgRP/NPY neurons [67]. Ghrelin is an endogenous ligand for the GHS-R and stimulates feeding and body weight gain by central mechanisms independent from its action on GH secretion [79,80]. Pharmacological and genetic evidence argues for a crucial role of the AgRP/NPY neurons in mediating ghrelin action in food intake (Fig. 2). Indeed, acute ghrelin injections enhance c-Fos, NPY and AgRP expression in these neurons [81,82] and ghrelin-induced food intake is fully abolished in AGRP−/−/NPY−/− double mutant mice [83]. However, as discussed below, neither NPY−/−mice nor AGRP−/−mice displayed this ghrelin resistant phenotype [84].
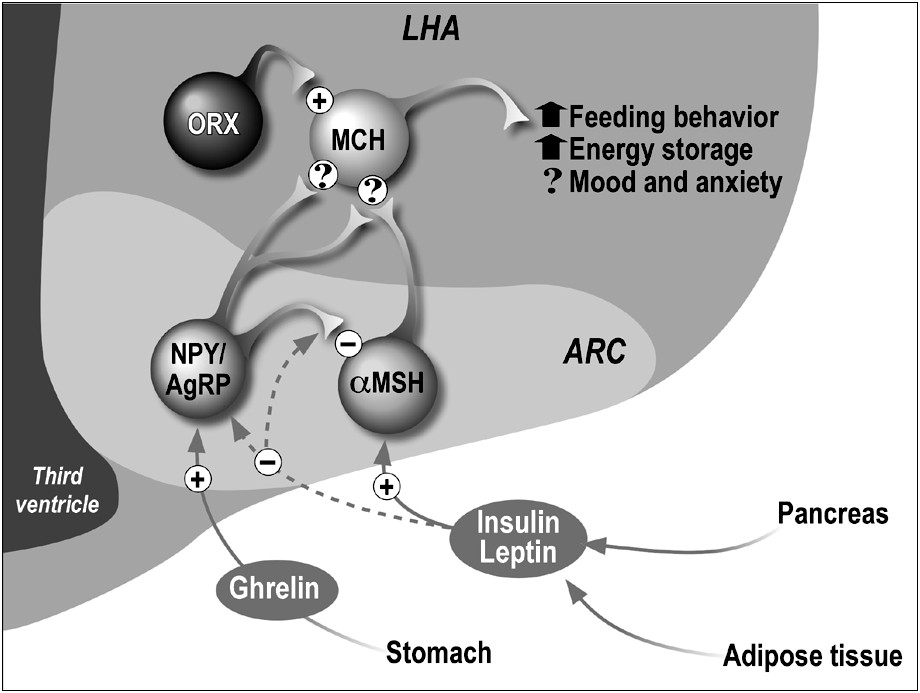
Schematic representation of the melanotropin-related peptidic systems in the hypothalamus involved in feeding behavior and energy homeostasis. Peripheral hormones are indicated that regulate mainly AgRP/NPY and α-MSH/CART neurones in ARC; solid and dashed lines indicate stimulatory and inhibitory effects, respectively. These two neuronal populations interact directly in ARC and project to ORX and/or MCH neurons in LHA. Peptides exert stimulatory (+) or inhibitory (−) actions on the target cells. Some behavioral and physiological actions of MCH are noted. ? indicates controversial data.
AgRP neurons project to various regions in the brain outside the ARC, with major inputs to the PVN (mainly on neurons co-expressing TRH and MC4-R), LHA (putative MCH or orexins-containing neurons also expressing MC4-R) and areas of the limbic system (nucleus accumbens, lateral septum, amygdala) (Fig. 1A) [78]. An interesting observation is the long-term activation of c-Fos in some of these brain areas that may contribute to the long-lasting hyperphagia induced by AgRP [85,86]. However, the precise neuronal sites and signaling mechanisms that mediate AgRP effects in these regions remain to be clarified.
Besides the ARC neurons, the site where AgRP is the most abundant is the cortex of the adrenal gland [87]. Although it is out of the scope of this review that mainly focuses on the central control of feeding, it is worth mentioning that peripheral AgRP, and particularly the pool expressed in the adrenal gland, may play an important role in the regulation of energy balance. Indeed, fasting in rats induced AgRP up-regulation in the adrenal cortex that paralleled that observed in the hypothalamus. In addition, plasma AgRP levels also rise after fasting in rats [88] as well as in obese humans [89]. This data is particularly relevant when considering that the active isoform of human AgRP may cross the blood-brain barrier (BBB) [90] and could therefore target directly MCR-4 within the brain.
Genetically-modified mice that overexpress or lack the AgRP gene also provide important clues about the physiological relevance of AgRP in appetite regulation and energy homeostasis. Transgenic mice that overexpress AgRP under the control of the actin promoter are hyperphagic, obese and display a large spectrum of alterations in hormonal readouts (hyperinsulinemia, hyperglycemia, decreased corticosterone...), but do not exhibit any change in their coat pigmentation [91]. These findings are in close agreement with the pharmaceutical studies and further support a role of AgRP after fasting in promoting enhanced food intake. In great contrast, AgRP−/− mice do not display any modifications in feeding behavior, body weight and hormonal parameters when compared with the wild-type mice under ad libitum feeding conditions [92]. This is highly reminiscent of the phenotype reported for NPY−/− mice as well as in AGRP−/−/NPY−/− double mutant mice when food were provided freely [92]. This data strongly suggests that compensatory mechanisms operate during mouse development to prevent deleterious effect of the loss of either of these orexigenic peptides and/or that none of these peptidic systems function tonically under conditions of free access to food. In agreement with the last hypothesis, AgRP−/− and NPY−/− mice display impaired hyperphagia induced by a 24-h fasting, suggesting that AgRP (and NPY) may be stimulated only under conditions of energy deficiency (e.g., after food deprivation, reduced glucose utilization or on low-energy diets) [6].
Very recently, two studies [93,94] have addressed the critical role of the AgRP/NPY neuronal system in the regulation of energy balance by using a new strategy named ‘toxin receptor-mediated cell knockout’. In summary, transgenic mouse lines were generated that expressed the diphteria toxin receptor (DTR) only in the AgRP-expressing neurons of ARC, then diphteria toxin (DT) was injected to provoke selective ablation of the AgRP/NPY neurons in neonates [94] or in adult mice [93,94] and feeding behavior was monitored. Under these conditions, neonatal ablation of these neurons did not induce significant change in feeding and body weight, whereas removal of the AgRP/NPY neurons in adult mice produced a drastic reduction in food intake and body weight that appeared associated with POMC overexpression. The loss of feeding behavior resulted directly from a central effect of DT and was not associated with a decrease in water drinking, indicating the specificity of the observed modifications in adult mice. These very elegant studies demonstrated for the first time that the AgRP/NPY neurons were critical components of the neuronal networks controling feeding in adults and that ablation of this ARC neuronal system early after birth led to compensation during post-natal development to allow for normal growth and feeding at adulthood.
In humans, seric AgRP levels were found elevated under resting condition in obese [89] and also after fasting in obese [88] or lean individuals [95] in agreement with the data of animal models. Interestingly, genetic studies have identified several functional single nucleotide polymorphisms (SNPs) at the AgRP locus that might be related to obesity or anorexia (reviewed in [67]). For instance, two SNPs located in the promoter region of the AgRP gene and with opposite action on transcriptional activity were found associated with leanness and reduced type 2 diabete incidence only in Blacks of Africa or America [96,97]. Another SNP in the coding region of the gene that lead to AgRP protein conformation changes was linked to anorexia in Whites only [98]. These data indicate that rare alleles at SNPs in the AgRP gene may predispose humans to obesity and diabete-resistance in an obesigenic environment.
5 A natural modulator of Agouti protein signaling: the Mahogany protein
The mahogany (mg) mutation was initially defined as a negative modifier of agouti: original obese yellow Ay mice that also carry mg mutation display a lean phenotype and black (eumelanin) pigmentation [99] reviewed in [100]. This suggests that mg mutations may interfere with Agouti protein signaling through both MC1-R in skin and MC4-R in hypothalamus, and possibly with the AgRP signaling. However, independently of interaction with the Agouti-induced signaling pathway, mg mutations increase energy expenditure and compensate for hyperphagia of the Ay mice [101]. Moreover, mg mutations have no effect on other monogenic obesity mouse models such as tubby (Tub), db/db (Lep-R) and fat (Cpe fat) mutants, suggesting a specific interaction with the Agouti system [101]. Three alleles of mahogany, Atrnmg, Atrnm1, AtrnmgJ3 have been reported so far that are coisogenic on mouse strains LDJ/Le, C3H/HeJ and C3HeB/FeJ, respectively [102,103]. The first two alleles result from retrovirus insertion in introns that change the normal splicing of the primary transcript while the last allele corresponds to a short deletion that introduces a stop codon leading to a truncated protein.
Positional cloning identified a candidate gene for mahogany, named initially Mgca [102,103]. This gene encoded a predicted 1428 AA type I transmembrane protein that contained three EGF domains, two laminin-like EGF repeats, a CUB domain, two plexinlike repeats, a C-type lectin, and seven Kelch repeats in tandem. The Mgca-derived protein shares 93–97% sequence identity with a soluble protein named attractin in its extracellular domain and the intracellular domain results likely from alternative splicing of the same gene. Therefore, the mouse mahogany protein was renamed attractin and mutation referred as Atrnmg.
Attractin mRNA was found expressed in many organs, including the brain, skin, heart, lung, liver and kidney [102,103]. In the CNS, attractin mRNA was also widely distributed with high levels in the primary olfactory cortex, limbic structures, many regions of the brainstem and most of the areas involved in the motor control [104]. In the hypothalamus, expression of the attractin mRNA was localized in nuclei involved in circadian rhythm and neuroendocrine regulation such as the suprachiasmatic nucleus, the supraoptic nucleus and PVN as well as in nuclei deeply implicated in the control of feeding behavior and energy balance such as the preoptic nucleus, VMH and ARC [104]. Based on the wide distribution of attractin gene products, it was difficult to assign a precise function to this protein but the strong expression in some brain areas suggested potential roles in the control of metabolic rate and higher complex behavior, such as motor control, cognition and learning.
The structure of attractin predicts that it functions as a receptor-like protein: potential roles in Agouti-dependent MC-R desensitization, in interactions of cells expressing MC-R and in MC-R signaling have been proposed (reviewed in [100]). Using a wide range of biochemical, transgenic rescue and genetic-interaction experiments, He et al. [105] provided evidence that attractin displays multiple functions, some related and other independent of the Agouti-mediated signaling. First, attractin is a low-affinity receptor for Agouti protein but not for AgRP in vitro. Second, in vivo AgRP-induced obesity still persists in the absence of attractin whereas Agouti-induced obesity is lacking. Third, the absence of the transmembrane-form of attractin provokes spongy degeneration throughout the brain that appears independent of Agouti protein signaling. This phenotype results likely from abnormalities of myelination since a mutant of the attractin ortholog in rat, named zitter (zi) rat, as well as mg mice exhibit hypomyelination and vacuolation associated with body tremor [106]. In addition, comparison of the attractin sequence among worms, flies and mammals predicted conserved functions that are clearly unrelated to Agouti-binding receptor but still remain to be defined.
The final question remains: can we establish a direct role for attractin in energy metabolism control that allows for the production of anti-obesity drugs useful in humans? Tremors resulting likely from brain demyenilation were conspicuously found associated with attractin mutants in rodents (mg and zi) and contribute to increased energy expenditure that may compensate, at least partly, for the hyperphagia. It is therefore tempting to speculate that drugs designed to compete with attractin in order to reverse the obese phenotype in humans have also good chance to interfere with myelination, a risk that raises some doubt about the successful development of inhibitors of attractin to treat obesity.
6 A functional antagonist of α-MSH: the melanin-concentrating hormone
Melanin-concentrating hormone (MCH) is a cyclic peptide that was found expressed in the brain of many vertebrate taxa, the most well-established structural, expression and functional studies being in teleosts and mammals (reviewed in [29–32]). The peptide was initially discovered, and incidently named, on the basis of its telling pigment aggregation property in salmon fish skin, opposing the pigmentary dispersing effects of α-MSH. In fishes, MCH was characterized as a prominent neurohypophysial 17 AA-long peptide [107]. The ability of MCH to aggregate melanin was not found confined to teleost fishes since holostean fishes also respond to this peptide. However, there is no evidence that MCH plays a major role in color change other than in neopterygians, suggesting therefore that its skin paling function evolved later and uniquely in a group ancestrals to teleosts and holosteans [108,109].
In mammals, MCH is a nonadecapeptide that displays strong sequence conservation in the ring structure with the salmon counterpart [110]. In the rat brain, most MCH perikarya are located in sub-zona incerta (ZI) and LHA and project widely throughout the brain, from the olfactory bulb to the spinal cord and with particular abundance in hypothalamic areas, parts of the extrapyramidal system and the reticular formation of the brainstem [15,16]. This broad distribution of MCH projections suggested early on that the peptide could be involved in many brain functions [16]. Indeed, recent studies using intracerebroventricular injections of MCH agonists/antagonists and MCH transgenic mouse models support a major role for MCH in the central regulation of feeding behavior and energy homeostasis (see below). However, MCH also exerts an effect on many different goal-oriented behaviors, such as drinking [111,112] sleeping [113] reproduction [114–116] sensorimotor-integration [117,118] and general arousal [119]. Interestingly, two MCH cell groups were clearly identified in the LHA, based on co-expression of MCH, CART and neurokinin 3 (NK3) receptor (type B neurons) or only MCH (type A neurons) [120]. These MCH-expressing subpopulations display very different projection pathways with type A neurons having quite exclusively descending inputs to reach the spinal cord, whereas type B neurons project massively to all known telencephalic areas receiving ascending MCH inputs. Furthermore, at least four MCH neuronal sub-populations can be distinguished according to their times of genesis during rat diencephalon development, two of them overlapping with type A and type B MCH neurons [121]. It might be attractive to associate either of these MCH cell groups with defined MCH functions but this hypothesis appears likely to be oversimplistic in view of the complex interactions with local neuronal circuitry (in particular ORX-expressing cell bodies) [122].
MCH is generated after cleavage at a dibasic amino acid site in the C-terminal part of a precursor of 132 AA in fish and 165 AA in mammals [15,123]. Apart from MCH, additional peptides could be liberated from pro-MCH. Indeed, cleavage at an Arg99Arg100 site may generate a 13 AA-peptide named Mch-gene-related peptide in trout (Mgrp; [124] or Neuropeptide (N)–glutamic acid (E)–valine (V) in Coho salmon [125]. In mammals a 13 AA amidated peptide named Neuropeptide (N)–glutamic acid (E)–isoleucine (I) is produced together with MCH in hypothalamic neurons [15,16]. While the functions of Mgrp/NEV remained elusive in teleost fishes, the peptide counterpart in mammals, i.e. NEI, has been associated with regulation of stress response and anxiety, and behavioral effects such as grooming, locomotor activities and female sexual receptivity [126,127].
Initial efforts to characterize the MCH receptor(s) either in fish or mammalian systems were largely restrained due to the lack of a suitable binding assay associated with the highly hydrophobic nature of MCH and its high sensitivity to degradation (reviewed in [128]). The use of an MCH analog named [Phe13, Tyr19]-MCH permitted the discovery of binding sites on various mammalian cells lines including melanoma cells [129] and SVK-14 keratinocyte cells [130]. However, using this analog, the low reliability of binding properties and of intracellular signaling associated with its binding, made the overt expression of functional MCH receptors in these cells highly questionable [131,132].
The first MCH receptor (named here MCH-R1) was identified almost simultaneously by several laboratories, using a ‘reverse-pharmacology’ strategy [133,134] reviewed in [36,135,136]. This receptor corresponds to a splice variant of an orphan G protein-coupled-receptor (GPCR) named SLC1/GPR24 [137]. As illustrated in Fig. 3, MCH-R1 mRNA and protein are widely distributed in the rat and mouse brain, in agreement with the major projection sites of MCH neurons [138,139] reviewed in [32]. In the hypothalamus, a very high expression of MCH-R1 is found in ventromedial and dorsomedial nuclei, both major regions that regulate feeding.
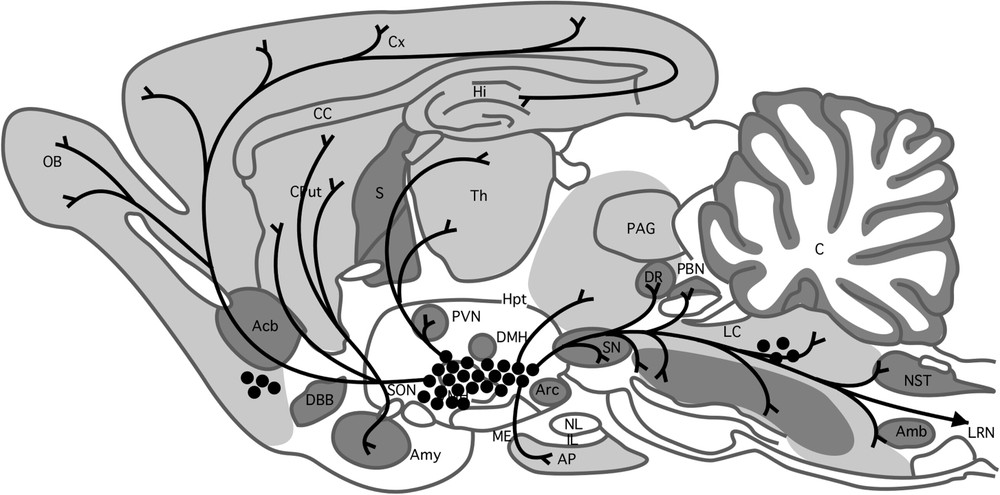
Sagittal section of the rat brain showing the distribution of MCH neurons and projections and MCH-R1 mRNA expression. Black points indicate MCH neuron perikarya and lines correspond to fibers. High, medium and low expressions of MCH-R1 are noted in dark, light grey and white, respectively (from [16,140]).
In transfected cells models, MCH-R1 couples to multiple G proteins including Gi, Go and Gαq; MCH may therefore potentially inhibit cAMP production induced by forskolin, activate MAP kinase activity, and increase intracellular free Ca2+ levels [140]. In mammalian cells that express MCH-R1 endogenously, MCH stimulates mainly the MAPK pathway, but it may also inhibit or potentate forskolin-induced cAMP production (reviewed in [136]).
A second MCH receptor, called MCH-R2, was initially identified in humans by in silico screening (reviewed in [36,135,136]) and more recently in fishes [141] but is apparently lacking in rats and mice [142]. MCH-R2 displays only 38% sequence identity to MCH-R1 and exhibits a partly distinct expression profile in the human brain and peripheral tissues (reviewed in [32]). Furthermore, analysis of the signaling profile of human MCH-R2 in heterologous cell lines suggests that this receptor couples quite exclusively to Gαq protein to increase intracellular Ca2+ levels. The functional importance of MCH-R2 remains elusive due to the lack of an appropriate animal model.
The relevance of MCH/MCH-R1 expressing neuronal pathway in the regulation of feeding behavior and energy balance is now supported by numerous data as listed below:
- – acute ICV injections of MCH transiently stimulate food-intake in rats [33,143,144] an orexigenic effect which is not mediated by NPY, galanin, or ORX but could be blocked by α-MSH (reviewed in [6,32]). Intrahypothalamic injection studies and electrophysiological characterization further identified the PVN, VMH, and ARC as major targets of MCH action, with differential effects in small-litter and overnurished rats [145–147]. Interestingly, MCH also stimulates water consumption independently from its actions on feeding and water osmolality regulation consistent with the strong adipsia observed in LHA-lesioned rats;
- – chronic ICV infusion of MCH in rats [144] or mice [148,149] induced hyperphagia and body weight gain, especially on a moderate high-fat diet. The development of obesity is accompanied by an increase in liver and fat weights resulting in lipogenic activity stimulation of white adipose tissue (WAT) and concomitant reduction of brown adipose tissue (BAT) functions. Furthermore, plasma glucose, insulin and leptin levels are also raised in these animals, all these parameters being associated with obesity in man;
- – while acute or chronic ICV injections in rodents of MCH-R1 agonists fully recapitulate the feeding and/or obese phenotype observed with MCH itself [149–151] central or oral administration of MCH-R1 antagonists leads to a sustained reduction of appetite, body weight, and fat storage without overt alteration of lean mass [152,153] reviewed in [35,37,154];
- – in agreement with the pharmacological data, genetically modified mouse models also demonstrated an important role of MCH and MCH-R1 genes in appetite and, mainly, energy metabolism. Indeed, transgenic mice overexpressing the MCH gene develop hyperphagia, mild obesity, and insulin-resistance, when maintained on a high-fat diet [155]. Conversely, MCH gene ablation in mice leads to a lean phenotype associated with hypophagia and an increase in oxygen consumption, both effects being highly dependent upon the genetic background and food diet [156,157]. In this regard, the reduction in body fat observed in mice combining MCH and leptin deficiencies, by crossing MCH null mice and ob/ob mice, does not result from feeding behavior changes (the mice remaining hyperphagic) but from an improvement in energy expenditure regulation (increased locomotor activity and resting metabolic rate) [158]. It is worth noting that targeted disruption of the MCH-R1 gene in mice maintained on regular chow also leads to a lean phenotype associated with both hyperphagia on one hand and increased energy metabolism on the other [159,160]. Consistent with the hyperactive phenotype, MCH-R1 null mice are resistant to high fat diet-induced obesity [159].
All these pharmacological and physiological studies have boosted interest in MCH as a suitable target for pharmaceutical intervention in the treatment of the obesity syndrome. Most of the efforts focus on the development of small-molecule antagonists for MCH-R1 reviewed in [35,37,154]. The first orally active MCH antagonist in rats was originally isolated by the Takeda group in 2002 and named T-226296 [152]. This compound binds with high affinity to MCH-R1, suppresses the stimulatory effects of MCH on food intake, and significantly decreases feeding and body weight in rats under high-fat diet by reducing meal size but not meal frequency. The T-226296 molecule does not alter locomotor activity or induce malaise in a one hour test period. These results provide encouraging clues that T-226296 molecule (and derivatives) may be effective in treatment of overeating disorders. The second non-peptide MCH-R1 antagonist, namely SNAP-7941, was characterized in 2002 by the Synaptic group [153]. This molecule is a competitive antagonist of MCH with high affinity for MCH-R1 and displays potent anorectic effects when injected intraperitoneally (ip) alone or in preventing the orexigenic effect of MCH. Chronic ip administration of SNAP-7941 in diet-induced obese (DIO) rats suppresses feeding and provokes a sustained weight loss that is reversible after the termination of drug treatment. This compound does not have apparent toxic or aversive actions. Strikingly, oral administration of SNAP-7941 molecule leads to unexpected anxiolytic and antidepressant effects as demonstrated in a rat forced-swim test, rat social interactions and guinea pig maternal-separation vocalization tests. Interestingly, two new MCH-R1 antagonists synthesized at Taisho Pharmaceutical Company and named ATC0065 and ATC0175 also exhibit antidepressant and anxiolytic effects in rodents after oral administration, without sedation or motor coordination impairment [161]. These results provide further support for an anxiogenic effect of MCH, as found after MCH injection in the medial preoptic area or nucleus accumbens shell in rats, and offer novel approaches to treat depression and/or anxiety. However, conflicting data were reported regarding the role of MCH in the regulation of mood similar to the contrasting results obtained regarding the regulation of the HPA axis (reviewed in [32,38]). Furthermore, the specificity of SNAP-7941 as well as other MCH antagonists needs to be evaluated in a MCH-R1 null mouse model to preclude the possibility that the in vivo activities of these compounds may be mediated by another receptor.
7 Conclusions
As illustrated in this article, compelling evidence has accumulated that peptides of the melanotropin family, including melanocortins and MCH, and cognate receptors are central regulators of appetite and energy balance. The physiological relevance of these peptidic neuronal systems is strongly supported by transgenic animal models and genetic studies in man that all point to a regulatory chain that links the catabolic peptide α-MSH and its receptors (mainly MC4-R) to the anabolic peptides AgRP and MCH (see Fig. 2). It is worth noting that α-MSH has a tonic inhibitory effect on feeding under basal conditions, but not under condition of high-fat diet. Conversely, AgRP does not function tonically and acts mainly under the circumstances when food is scarce and glucose availability reduced by stimulating food-seeking behavior. Similarly, MCH also responds to increased metabolic demand but mainly to states of fatty acid deficiency and restores the energy balance by decreasing energy expenditure (Fig. 2). It is clear, therefore, that despite redundancy in actions, these peptidic systems regulate feeding behavior and energy homeostasis through diverse behavioral and physiological processes.
The most challenging issue now is to understand how these neuronal circuitries may function in man and to what degree the overwhelming accumulation of data in animal models could provide some therapeutic clues to treat obesity and anorexia. In an evolutionary perspective, it is obvious that the complex control system that regulates energy stores was constructed during periods of food scarcity and that the present worldwide obesity epidemy results mainly from environmental factors that favorize hyperphagia of high-energy food. Even although anti-obesity drugs, such as blockers of the MCH systems, could be promising agents, they would be of poor long-term health benefit without parallel modifications in our food consumption behavior.
Acknowledgements
This article is dedicated to the memory of Dr. Guillaume Hervieu, a major contributor in the MCH field who passed away in 2005. I thank very much Drs. Alice Guyon and Enzo Lalli (IPMC, Valbonne, France) for their critical reading of, and helpful comments on this manuscript, as well as Valérie Briet and Franck Aguila for their assistance in the manuscript preparation and artworks. The work carried out in my laboratory at the IPMC was supported, in part, by the CNRS (programme OHLL), the ‘Association pour la recherche sur le cancer’ (ARC: subvention 3375/2004-2005), the ‘Fondation de France’ (‘Comité Parkinson’) and the ‘Convention de collaboration’, chaire de recherche Merck Frosset, Université Laval (Québec)/CNRS UNSA.