

1 Introduction
In recent years, obesity has become a major public health problem owing to its prevalence, which stands at more than 25% in certain countries, and its alarming increase in children. The molecular mechanisms responsible for fat mass accumulation and maintenance are still to be elucidated. Obesity results from the interaction of environmental factors (overeating and/or reduction in physical activity) and hereditary factors. This has been shown by numerous epidemiological studies carried out in large and different populations (twins brought up together or separately, adopted children, nuclear families, etc.) [1]. Obesity has a very heterogeneous phenotypic expression and the molecular mechanisms involved in its development are more than diverse. The role of environmental, behavioural and socioeconomic factors in individuals with different biologic susceptibilities is recognized. According to several studies, 30 to 80% of weight variation might be determined by genetic factors. Today, the contribution of genetic factors to obesity can be summarized as below:
- • single mutations contribute to the development of obesity (monogenic obesity). These forms of obesity are rare, very severe and generally start in childhood [2];
- • several genetic variants interact with an ‘at-risk’ environment in common obesities (polygenic obesities). Here each susceptibility gene, taken individually, would only have a slight effect on weight. The cumulative contribution of these genes would become significant when there is an interaction with environmental factors predisposing to their phenotypic expression (overeating, reduction in physical activity, hormonal changes, and socioeconomic factors). The common disease/common variant hypothesis has been proposed [3]. It suggests that the genetic risk for obesity is due to disease-producing common alleles. As a consequence, the percent-age of obesity attributed to them (attributable risk) could be high. This hypothesis is a current one in multifactorial diseases. However, even the strong believers of this hypothesis agree that in certain circumstances the role of rare variant alleles may intervene. The risk for common obesity could be due to large number of loci, each with multiple disease-predisposing alleles but of low frequency [4]. At the present time, these two hypotheses are still valid because the genetic approach has not allowed one to be confirmed more than the other.
2 Genetic obesity
2.1 Mendelian diseases
Obesity is associated with many genetic syndromes [5]. Initially thought as monogenic diseases, molecular researches are leading more and more to the idea of a contribution of different genes in the determinism of these syndromes. Few examples are given in Table 1. To identify new pathways involved in the control of weight, research teams are attempting to identify the genes and mutations responsible for these diseases. The OMIM database http://www.ncbi.nlm.nih.gov/entrez/Query.fcgi?db=OMIM lists them and provides access to their clinical descriptions. The most well-known are the Prader–Willi, Cohen, Alström, and Bardet–Biedl syndromes, but many others have been reported [5]. Tremendous progresses were made after the whole genome screening of affected families and lessons are to be learned from these observations. For example, genes responsible for the Bardet–Biedl (BBS) and Alström syndromes have been characterized. The Bardet–Biedl syndrome is characterized by early onset obesity associated with rod-cone dystrophy, morphological finger abnormalities, learning difficulties and renal disease, amongst other clinical traits. Although a homogeneous entity was initially described, BBS has been associated with at least nine different chromosomal locations, with several mutations identified within some of them (BBS1 on 11q13; BBS2 on 16q21; BBS3 on 3p13; BBS4 on 15q22.3; BBS5 on 2q31; BBS6 on 20p12; BBS7 on 4q27; BBS8 on 14q32.11 and BBS9 on 7q14) [6]. While BBS has for a long time been considered as a syndrome with autosomal recessive transmission, it has been shown that the clinical symptoms of certain forms of BBS were related to recessive mutations on one of the BBS locus associated with a heterozygous mutation on a second locus. The hypothesis of a triallelic transmission was raised [6]. At least eight genes were characterized in BBS, without their function being necessarily known. For BBS6, the positional cloning approach implicated the MKKS gene, situated on chromosome 20, which codes for a chaperone protein. Chaperone proteins facilitate the three-dimensional folding of other proteins. The mutations identified in MKKS resulted in a shortened chaperone protein and represent 5 to 7% of BBS cases, indicating the molecular heterogeneity. The links between MKKS, its eventual target proteins and the BBS clinical traits are largely unknown. Unlike BBS6, the genes implicated in BBS1, BBS2 and BBS4 are very different from MKKS genes, but it is possible that they code for MKKS protein substrates [7]. Fascinating studies performed in single cell organisms have shown that certain BBS genes are specific to ciliated cells [8]. These cells play a fundamental role in mammals' development, contributing to right/left symmetry enabling the organs (heart, liver, lungs) to be correctly positioned. Such dysfunction in the processes affecting the ciliated cells may contribute to alterations in pigmentary epithelia and to structural anomalies in certain organs.
Example of syndromic obesities
| Name/reference | Clinical features | Transmission | Loci/genes |
| Prader–Willi | Neonatal Hypotony | Autosomic dominant | 15q11–13 |
| Mental retardation | Paternal deletion 75% | ||
| Hyperphagia | Imprinting | Maternal uniparental disomy | |
| Hypogonadism | Microdeletion at the SNURF-SNRPN locus | ||
| Short stature | |||
| Bardet–Biedl | Hypogonadism | Autosomic recessive | BBS 1 to 9 and others |
| Pigmentary retinopathy | Chaperonin Protein MKKS (Chr. 20) | ||
| Polydactyly | Triallelic inheritence | ||
| Mental retardation | Expressed in ciliary cells | ||
| Alström | Myocardiopathy | Autosomic recessive | 2p14 ALMS1 |
| Sensory deficit (retinopathy, deafness) | Ubiquitously expressed at low level | ||
| Dyslipidemia, diabetes | |||
| Börjson–Forssman–Lehman | Morbid obesity, epilepsy | X-linked | Xq26.3/Plant homeodomain like finger gene |
| Hypogonadism, facial dysmorphy | |||
| Cohen | Obesity, delayed mental development, characteristic facies, and slender hands and feet | Autosomic recessive | COH1 |
| Ubiquitously expressed | |||
| Vesicle-mediated sorting in transport |
Similar observations might derive from the study of the Alström disease. The Alström syndrome is an autosomal recessive disease of childhood that, apart from obesity, associates retinal cone dystrophy, dilated cardiomyopathy, type-2 diabetes and other clinical traits of variable severity such as hypothyroidism, small size, hypogonadism, anomalies of hepatic function and sometimes mental retardation. Mutations of the ALMS1 gene were implicated. This gene encodes a protein with ubiquitous expression, the function of which is also unknown.
These examples emphasize the necessity for multicentre studies grouping together the families affected by these syndromes in order to characterize the genes responsible for these rare diseases. Although certain genes have been cloned, the physiopathological links between their protein products and the development of diseases characterized by multiple clinical traits that sometimes overlap (retinal disease, mental retardation, insulin resistance) are to be identified [9]. Why these dysfunctions lead to obesity is an open question. The possibility that these genes may contribute in a minor way to the determinism of common obesity should also be explored.
2.2 Monogenic obesity by alteration of the leptin and melanocortin pathways
A significant success derived from the study of candidate genes implicated in rodent monogenic obesity (Fig. 1). The researches were conducted among individuals meticulously explored and characterized by specific biochemical or hormonal anomalies [2]. Unlike the previous examples, the genetic anomalies affect key factors of body weight regulation linked to the leptin action and the melanocortin pathway, the target of leptin in the hypothalamus (Fig. 1). Linkage studies, mouse models and spontaneous human mutations as well as pharmacological studies have uncovered the primary role of the leptin/melanocortin pathway in regulating energy homeostasis [10].
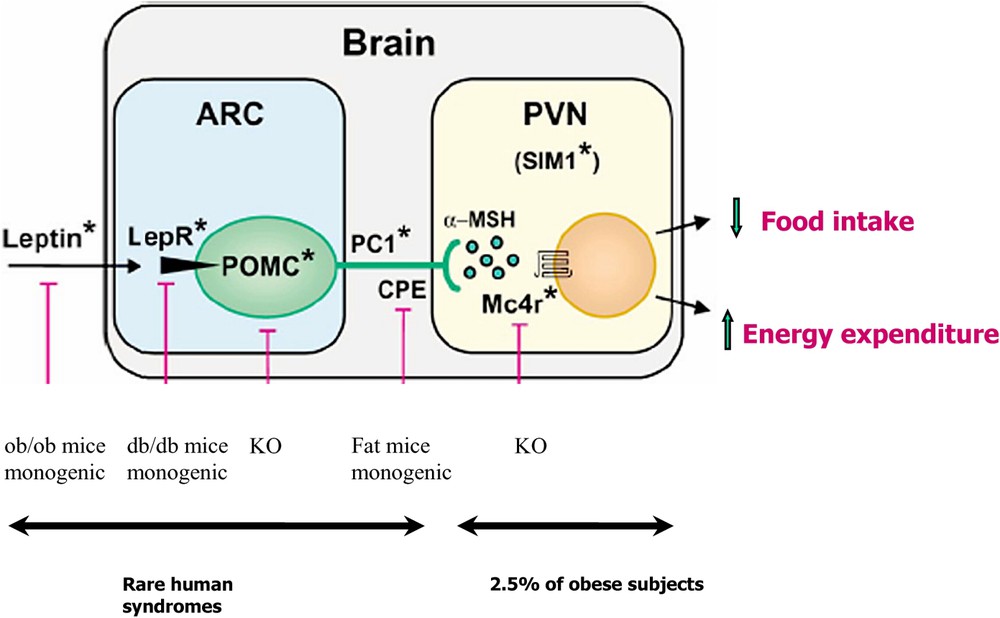
Human mutation affecting the leptin and melanocortin axis. The figure is adapted from D. Cummings' review in [68]. LepR leptin receptor, POMC proopiomelanocortin, aMSH (alpha melanin stimulating hormone), PC1 proconvertase 1, CPE carboxypeptidase E, MC4R melanocortin-4 receptor. The pink bars represent either animal models of obesity or monogenic natural models (leptin receptor deficient ob/ob, leptin receptor deficient db/db, CPE deficient mice) or transgenics. When the gene is invalidated, obesity occurs (POMC or MC4R). Stars are human counterpart gene anomalies associated with syndromic obesities or commoner obesities (MC4R). Masquer
Human mutation affecting the leptin and melanocortin axis. The figure is adapted from D. Cummings' review in [68]. LepR leptin receptor, POMC proopiomelanocortin, aMSH (alpha melanin stimulating hormone), PC1 proconvertase 1, CPE carboxypeptidase E, MC4R melanocortin-4 receptor. The pink ... Lire la suite
The proopiomelanocortin (POMC) protein participates to the transmission of the leptin signal from the periphery in a central homeostatic response. The production of POMC in the central nervous system is stimulated by leptin and the post-translational process of the protein give rise to the production of different peptides, harbouring many functional properties [11]. The nature of the POMC-derived peptides depends on the type of endoproteolytic enzyme present in specific brain localizations. In anterior pituitary, the presence of PC1 enzyme allows the production of ACTH and β-lipotropin peptides, while the contemporary presence of PC1 and PC2 in hypothalamus determines the production of α-, β-, γ-MSH and β-endorphins.
Rare mutations in leptin, leptin receptor (LEPR), POMC, and proconvertase 1 (PC1) genes lead to severe obesity. The mode of transmission of the disease is autosomal recessive and its penetrance is complete (Table 2). Several families carrying leptin gene mutations have been recognized [12–14], a family with three patients affected by the leptin receptor mutation [15], five families carrying a POMC mutation [16] and two patients carrying a proconvertase 1 (PC1) mutation [17,18]. These mutations are responsible for severe early onset obesity and many endocrine anomalies. The weight curve of the affected patients attracts attention. Patient mutation's carriers show an exponential increase in weight with severe obesities that develops from the very first years of life.
Mutations in human obesity affecting the leptin and the melanocortin pathways
| Gene | Transmission | Obesity | Associated phenotypes |
| Leptin | Recessive | Severe, from the first days of life | Gonadotropic, thyrotropic insufficiency |
| Leptin receptor | Recessive | Severe, from the first days of life | Gonadotropic, thyrotropic and somatotropic insufficiency |
| Proopiomelanocortin (POMC) | Recessive | Severe, from the first month of life | ACTH insufficiency |
| Mild hypothyroidy and ginger hairs | |||
| Proconvertase 1 (PC1) | Recessive | Severe, from the first month of life | Gonadotropic and corticotropic insufficiency |
| Hyperproinsulinemia | |||
| Other dysfunction of gut peptides | |||
| Melanocortin-4 receptor (MC4R) | Dominant | Early onset, variable severity, large size | No other phenotype |
In the individuals with a mutation of leptin- and its receptor-gene, there is complete failure of puberty through hypogonadotrophic hypogonadism and thyrotropic insufficiency. Insufficient somatotropic secretion is seen in patients with a leptin receptor mutation. High rate infection associated with a deficiency of T cell number and function was described [12]. In some individuals with leptin deficiency either due to leptin gene or leptin receptor mutations (K. Clément, unpublished observation), there is evidence of spontaneous pubertal development. The follow-up of LEPR deficient sisters revealed also the normalization of thyroid mild dysfunction at adult age (K. Clément, unpublished observation). An important contribution in the understanding of the role of leptin in the reproductive function was brought after endocrine investigation of the subjects with leptin and leptin receptor mutations.
Obese children with a POMC deficiency have an ACTH deficiency that can lead to acute adrenal insufficiency from birth. Five patients originated from Germany, Slovenia, the Netherlands and Switzerland are homozygous or compound heterozygous for POMC gene mutation. Children with a dysfunctional POMC have a resting metabolic rate in agreement with their corpulence as well as a mild central hypothyroidism that necessitates hormonal replacement [16]. The reason of hypothyroidism is unknown, even if the role of melanocortin peptides in influencing the hypothalamic pituitary axis is proposed. These children have ginger hair due to the absence of α-melanocyte stimulating hormone (αMSH) on the peripheral melanocortin receptors involved in pigmentation. Several observations including one unpublished from our group suggest that the skin and hair phenotype might vary according to the ethnic origin of POMC mutation carriers. More clarification on the skin phenotype would be needed. No other biological or hormonal phenotype has been described in these children. The first patient carrier of PC1 mutation is severely obese and has postprandial hypoglycaemic malaises and fertility disorders. The delayed postprandial malaises are explained by the accumulation of proinsulin through lack of PC1, which is also involved in insulin maturation. The absence of POMC maturation due to the PC1 mutation causes blocking of the melanocortin pathway and explains the obese phenotype [17]. The discovery of a second case of PC1 deficiency revealed new features associated with this syndrome [18]. A congenital PC1 deficiency leading to severe diarrhoea due to severe small intestinal dysfunction was found in a young obese girl, suggesting the role of PC1 on the absorptive function of the intestine. A milder intestinal phenotype was found after the reinvestigation of the first PC1 deficient patient. The processing of prohormones – progastrin and proglucagon – was altered, explaining, at least in part, the intestine phenotype.
These studies have participated to validate a crucial role of the leptin and melanocortin pathways, in controlling food intake and energy expenditure, that have been individualized within a redundant system of food intake control. Their implication in endocrine pathways was also enlightened in humans.
Leptin-deficient children and adults benefited from subcutaneous injection of leptin, resulting in weight loss, mainly of fat mass, with a major effect on reducing food intake and on other dysfunctions including immunity [19]. A detailed microanalysis of eating behaviour of three leptin-deficient adults before and after leptin treatment, revealed reduced overall food consumption, a slower rate of eating and decreased duration of eating of every meal given to the three subjects after leptin therapy. This study supports the role of leptin in influencing the motivation to eat before each meal [20]. Hormonal and metabolic changes were evaluated before and after leptin treatment [21]. Leptin treatment is able to induce features of puberty even at adult age. In POMC deficient children, a three-month trial using MC4R agonist but with a low affinity was inefficient on weight or food intake [16]. POMC deficient families might benefit from the development of new MC4R agonists if such drug become available.
2.3 New ‘monogenic’ obesities
Recent clinical cases with a strong genetic influence have been described in humans. A severely obese young patient of 6 months who has a de novo balanced translocation between chromosomes 1p22.1 and 6q16.2 was identified. She had a rate of early weight gain comparable to what found in leptin or leptin receptor deficient subjects and voracious appetite but increased linear growth. This translocation causes a disruption of the SIM1 gene on chromosome 6 (Fig. 1). This gene codes for a transcription factor involved in the development of the supraoptic and paraventricular nuclei of the hypothalamus, in the mouse [22].
A de novo heterozygous mutation in NTRK2 gene was also described in a eight-year-old boy with early onset obesity and a mental retardation, developmental delay and anomalies of higher neurological functions like impairment of early memory, learning and nociception [23]. This gene codes for TrkB, the receptor for the brain-derived neurotrophic factor (BDNF), itself involved in the regulation of food intake [24]. It is expressed throughout the central nervous system. This mutation leads to the impairment of the receptor autophosphorylation and alters the signalling to MAP kinase. Several other TrkB variants were found in patients with early onset obesity, but their functional effects and their involvement in obesity remains to be demonstrated.
These studies emphasize the interest in precisely exploring subphenotypes of obese patients to discover the role of new genes – and their subsequent proteins – in the etiopathogenicity of obesity. In future genetic screening, the role of gene encoding transcription factors playing a role in hypothalamus nuclei formation might be considered as candidates in patients with obesity and hypothalamic hormonal dysfunction. Whether or not these genes, including SIM1, might interact with the capacities of the leptin and melanocortin axis in controlling food intake and energy expenses is probably a question investigated by the specialists of the field.
2.4 Obesity related to mutation of the melanocortin-4 receptor (MC4R)
One important actor in the melanocortin pathway is the melanocortin-4 receptor (MC4R). MC4R is a G-protein coupled receptor with seven transmembrane domains [25,26] expressed in the hypothalamus. MC4R plays an important role in controlling weight homeostasis. Mice with a genetic invalidation of MC4R (knockout) develop morbid obesity and increased linear growth. Heterozygous mice present intermediate obesity with various degrees of severity [27]. The use of pharmacological agonists of MC4R in rodents reduces food intake, while antagonists of this receptor increase it [25]. Considering the pivotal role of the melanocortin pathway in the control of food intake and the discovery of syndromic obesities associated with melanocortin dysfunction, the MC4R gene became a candidate of choice for the genetic study of human obesities.
In 1998, we contributed to report one of the first two frameshift MC4R mutations resulting in a truncation of the MC4R protein [28,29]. Since then, more than 90 different mutations resulting in a change of amino-acid in the protein have been described in different populations of German, French, English, Danish and American children and adults [30]. They include frameshift, inframe deletion nonsense and missense mutations, located throughout the MC4R. The frequency of these mutations has been assessed as being between 0.5 to 2% of moderate obesities and could reach 4–6% in severer forms [31].
Arguments for the pathogenicity of MC4R mutations remain solid. In the families in whom segregation has been studied, the obesity has a usually autosomal codominant mode of transmission. Penetrance of the disease is incomplete and the clinical expression varies. Subjects with homozygous MC4R mutation develop severer obesity [32,33] than heterozygous carriers. Subjects with heterozygous MC4R mutations are not always obese [34] and if they are, the severity of the condition is variable in most studies. A recent study has shown that MC4R mutations are strong predictors for obesity and also suggests that the role of the environment is not negligible. Other potentially modulating genetic factors may intervene. A V103I common variant studied in more than 7500 subjects was negatively associated with obesity, but no functional consequence of this variant on MC4R function was described [35].
The phenotype of subjects with MC4R mutations is debated. Many authors agreed for a role of MC4R mutation in the facilitation of very early onset of obesity. One study performed in English children has suggested that bone mineral density and size increase in MC4R mutation carriers [36]. The potential increased bone density observed in MC4R-deficient patients may be caused, at least in part, by a decrease in bone resorption. A decrease of bone resorption markers was indeed found in serum of patients with MC4R homozygous mutations [37]. An association between feeding behaviour disorders like ‘binge eating’ and MC4R gene sequence changes has been suggested [38]. This remains very controversial [39,40]. In the Branson study, MC4R mutations with functional consequences were mixed together with common non-functional variations.
As a general rule, clinical analysis, even if carefully conducted, does not allow detection of MC4R mutation linked-obesity that resembles early onset obesity [39]. They are known as non-syndromic obesity that can be placed between the exceptional forms of monogenic obesities with complete penetrance and the common obesities of polygenic origin.
Functional studies of MC4R mutations have confirmed the role of these mutations in causing obesity in patient carriers. The response to melanocortin agonists of mutant MC4R was assayed. After ligand binding, MC4R activation stimulates Gs protein with a subsequent increase of cAMP levels. The production of intracellular cAMP in response to αMSH demonstrated a broad heterogeneity in the activation of the different MC4R mutants in response to αMSH, ranging from normal or partial activation to the total absence of activation [30]. Deeper functional investigations were performed for many, but unfortunately not all, mutations. An intracellular transport defect of the mutated receptor, by intracytoplasmic retention, has been described for the majority of MC4R mutations found in childhood obesity. This mechanism explains the impaired response to agonists. In addition, MC4R has a constitutive activity, meaning a basal activity not necessitating the presence of a ligand, for which agouti-related peptide (AGRP) acts as an inverse agonist [41]. In the absence of the ligand, MC4R therefore has an inhibitory action on food intake. The systematic study of basal activity of some mutations has shown that an alteration in this activity may be the only functional anomaly found, in particular for mutations located in the N-terminal extra-cytoplasmic part of the receptor [42]. A tonic satiety signal, provided by the constitutive activity of MC4R could be required in the long-term regulation of energy balance.
It is admitted that MC4R mutation causes obesity by a haploinsufficiency mechanism rather than a dominant negative activity. While the role homo and hetero-dimerization in G-protein synthesis and maturation is emphasized, some dominant negative effect of MC4R mutations might not be excluded. Previous studies reported a dominant negative activity of one MC4R mutation (D90N). There are efforts toward a classification of the MC4R mutations based on their functional consequences and association with the subphenotypes of obesity [34,40]. It will be essential to pursue the precise functional characterization of naturally occurring MC4R mutations carried by obese subjects in view of therapeutic intervention aiming at improving melanocortin action in the control of body-weight homeostasis. The search for MC4R mutations in large general populations is also necessary to estimate their global frequency in populations not specially recruited for obesity gene studies, as recently performed in a German population [34].
2.5 MC3R mutations in humans
MC3R gene has been the source of genetic investigation in obese and diabetic subjects because its role in the control of body-weight homeostasis was recognized among its multiple actions [43]. MC3R is expressed in various tissues including the arcuate nucleus, but different functions of MC3R and MC4R in controlling body weight homeostasis were suggested. MC3R appeared more involved in increasing feed efficiency with less effect on the control of food intake itself. MC3R KO mice are obese with an increased fat mass, but with reduced lean body mass. They are not hyperphagic while compared with MC4R KO mice phenotype [27,44] and are prone to obesity after a high fat diet. Rare genetic data have been produced regarding naturally occurring mutations in MC3R gene [43]. Common polymorphisms have been reported in obese and diabetic subjects, but only one study reported the occurrence of a MC3R mutation (Ile183Asn) in a small obese family originated from India. This mutation was not found in a small number of lean subjects. The Ile183Asn mutation was observed in a 13-year-old obese girl and in his obese father, but the other two members of the family carrying the wild-type allele were also obese or overweight. This case report is insufficient to conclude about the role of the mutation in the pathogenesis of obesity in the father and his daughter, but the functional analysis of this mutation revealed a defect in MC3R receptor activation by the agonist. Further genetic and functional studies, are necessary to clarify the role of MC3R in the pathogenesis of severe obesity or abnormalities of fat partitioning.
3 “Polygenic” obesity
3.1 General and theoretical approach
The genetic study of common obesity is based on the analysis of variations in genomic DNA (genetic polymorphism or SNP, Single Nucleotide Polymorphism) situated within or near candidate genes. Genetic studies of different types, depending on whether they are carried out in family members or unrelated and depending on the statistical methods employed (linkage family studies, association studies in unrelated obese subjects) are aiming to determine whether an association exists between an allele of a gene and obesity traits [45,46]. DNA and clinical data banks have been constituted in Europe and in the United States. The results of these genetic studies, that concern a large number of genes and chromosomal regions, are reported each year in the international journal Obesity Research [45]. We will not give details on all studies, but will provide illustrative examples.
3.2 The genome regions linked to obesity
Knowledge of genetic polymorphism and development of powerful molecular tools have made feasible to ‘quickly’ explore the genome of the families with common obesity. The objective is a systematic examination of all chromosomes in the families of obese subjects thanks to highly polymorphic markers to detect increased allelic sharing in obese sibpairs. This task is performed without preconceptions about the functions of the genes and aims at identifying known or unknown genes predisposing to obesity. The use of powerful molecular and statistical tools allows the genes to be positioned and eventually cloned. It is sometimes arduous to find the right gene when genomic linked regions encompass thousand of bases. These approaches have been applied to different cohorts worldwide: families with extreme obesities occurring during adulthood or childhood (European and American studies), families recruited from the general population (Quebec family study), and particular ethnic groups (Pima Indians, Mexican Americans, African Americans and Amish), among others [45]. This strategy has brought to light several chromosomal locations linked to obesity. At least seven genes located on chromosomes 2, 5, 6, 10, 11, 19 and 20 have been significantly implicated in common obesity (all references in [45]). Some loci might be more specific of morbid obesity or of obesity occurring in childhood [47,48]. A few regions have been confirmed in different populations; for example the 2p21 region seems to play a role in the variability of circulating leptin levels in French, Mexican Americans and African Americans. The region on chromosome 10 is a region linked to obesity in French, German Caucasians and in the Amish among others. The global picture of chromosomal regions linked to obesity is even more complex when looking at all the results produced. Virtually no chromosome, except Y, is spared and more than 200 chromosomal regions have been linked to different phenotypes such as fat mass, the distribution of adipose tissue, the occurrence of a metabolic syndrome, resting energy expenditure, energy and macronutrient intake, weight variation, the levels of circulating leptin and insulin. Most studies are nevertheless based on body mass index which is a crude phenotype. Interactions between several chromosomal regions such as on chromosome 10 and 20 were shown, and more recently on chromosome 13 and 2. These regions may interact to influence extreme obesity [49]. This is a sysiphean task to keep an updated view of all regions linked to obesity, particularly because a large number of non-significant and unreplicated results are produced. A further challenge is to identify the genes explaining the increased polymorphic allele sharing in linked regions. When this has been done and potential target gene suspected, the predisposing allelic variations must be determined. The confirmation of the gene implication in the pathogenesis of the disease is another issue. Although several interesting candidates have been identified in these regions, the general rule is that the role of these genes in the etiopathogenicity of human obesity is to be established.
3.3 Candidate genes
The choice of a candidate gene in obesity research is based on several arguments including the physiological role of its encoded protein in the mechanism of obesity, its chromosomal location in a region linked to obesity in human or animal models (regions known as QTL or Quantitative Trait Loci), the phenotypic consequences of its genetic manipulation (Knockout, transgenesis, floxed) in rodent models and eventually the in vitro functional characteristics of gene mutations or variations studied. The pattern of expression of the gene transcripts in key tissues for weight control, or even its modified expression in response to the environment can be used. All criteria are rarely taken into account. To briefly summarize the 10 to 15 years of genetic analysis in obese populations in different countries, the ‘candidate gene’ approach has not revealed the preponderant and unambiguous role of candidates in the etiopathogenicity of common obesity and even recent and promising studies are uncertain as illustrated by the GAD2 study (discussed below). The reasons of the lack of replication of most association and linkage results performed in different populations are numerous and classically include lack of statistical power to detect modest effect, lack of control over type-1 error rate, population stratification and overinterpretation of marginal data. Other factors may also include non-genetic factors, i.e. environmental or ethnic factors that may be rather different in different populations and are known to strongly influence the development of obesity. Similarly to the overall picture of linkage studies, it is rather difficult to isolate substantial results from sets of noisy data.
A very large number of genes and polymorphisms have been tested (more than 50 in 250 studies in heterogeneous populations). These genes are implicated in controlling food intake, energy expenditure, lipid and glucose metabolism and more recently in inflammation processes. The obesity gene map published each year extensively summarizes all the genes and variants screened [45], http://obesitygene.pbrc.edu/. Table 3 shows examples of the most studied genes in independent populations. Noteworthy, positive but also negative associations were found for all gene variants. For a given polymorphism, the effects reported are sometimes uncertain, conflicting even, and illustrate the complexity of obesity as well as statistical bias sometimes due to the lack of power of analyses. Certain genetic variants are associated with different phenotypes of obesity that include obesity history, aggravation with time, associated metabolic and cardiovascular complications, feeding behaviour characteristics, corpulence interacting with amount or quality of food and degree of physical activity.
Examples of genes frequently associated with obesity phenotypes in humans
| Genes (Code) | Locus | Animal model/phenotype | Human obesity locus (N) | Functional genetic variant |
| Food intake | ||||
| Leptin (LEP) | 7q31 | ob/ob mice/severe obesity | yes | no |
| Leptin-R (LEPR) | 1p31 | db/db mice/severe obesity | yes | no |
| Agouti related peptide (AGRP) | 16q22 | transgenic/obese | yes | yes (promoter activity) |
| KO/non-obese | ||||
| Dopamine D2 receptor (DRD2) | 11q23.2 | KO/non-obese | no | ⇓ nb receptor |
| Energy metabolism | ||||
| Uncoupling protein 1 (UCP1) | 4q28-q31 | KO and transgenics/reduced adiposity | no | no |
| Uncoupling protein (UCP2) | 11q13 | KO non-obese | yes | yes (change in UCP2 mRNA) |
| Uncoupling protein (UCP3) | 11q13 | KO non-obese | yes | no |
| β3 adrenergic receptor (B3-AR) | 9p12 | KO/Increased adiposity | yes | ⇓ activity |
| G protein beta 3 subunit gene (GNB3) | 12p13.3 | no | Yes | yes (modified G protein activation) |
| Adipose tissue metabolism | ||||
| Adiponectin (ACDC) | 3q27 | KO diet-induced insulin resistance | yes | yes |
| Peroxisome proliferator activated receptor γ (PPARγ) | 3p25 | KO/decreased brown adipose tissue | no | ⇓ activity |
| Tumor necrosis factor α (TNFα) | 6p21.3 | KO/increased adiposity under high fat diet | yes | yes (transcriptional activity) |
| β2 adrenergic receptor (β2-AR ) | 5q31-q32 | KO: increased adiposity under high fat diet | yes | ⇓ activity |
| Interleukin 6 (IL6) | 7p21 | KO/mature onset obesity | no | yes (transcriptional activity) |
| Lipasehormone sensitive (LIPE) | 19q13.2 | KO/reduced abdominal fat mass/resistance to diet induced obesity | yes | yes in 1 study |
| (decreased adipocyte lipolysis) | ||||
| Glucocorticoid receptor (NCR3C1) | 5q31 | floxed/age-dependent modified adiposity | yes | differential response to glucocorticoids |
| Lipid and glucose metabolism | ||||
| Insulin (INS) | 11p15.5 | no | yes | yes |
| LDL receptor | 19p13 | no | yes | no |
The genes implicated in the monogenic forms of obesity do not seem to play a more preponderant role in the development of common forms of obesity than others. However, they might not be excluded as potential targets for multi-factorial obesities and have been studied in different population as we reported [50]. Some associations and linkage between obesity phenotypes and the leptin or leptin receptor gene region were found (Table 3 and [50]). POMC was considered as a strong positional candidate and direct gene screening revealed several polymorphisms, generally of low frequency, located in the coding and uncoding regions reviewed in [31]. In Mexican Americans, three variants located in 3′ and 5′ untranslated region, with no functional impact on the protein, and haplotypes revealed association with leptin levels [51]. The reason why these variants have been associated with leptin levels is unknown and another variation located in the POMC gene and or another nearby gene might be involved. How genetic variation in POMC (or another gene) could contribute to leptin serum levels variability is to be discovered. In other populations, most of the polymorphisms located in the POMC gene were not associated with obesity phenotypes, leptin levels, or weight variation and no functional influence could be predicted except for three rare newly discovered mutations [52–54]. In children from the UK, the frequency of the Arg236Gly mutation was mildly increased and functional analysis revealed that the mutation prevents the normal processing of βMSH and B endorphin, resulting in an aberrant fusion protein [52]. Although able to bind MC4R, this aberrant peptide leads to a decreasing activation of the receptor. In a larger recent study comprising 1428 subjects from 248 UK non-obese families, not specifically recruited for the genetic study of obesity, an association was found between variants spanning the POMC gene (variants located in the 3′ and 5′ region) and the waist/hip ratio (WHR) but not with BMI or leptin levels. The proportion of the variance in WHR attributed to the genetic variation was relatively small (1.1%) [55]. More investigation of the POMC gene in large obese and non-obese population is needed to decipher the role of POMC gene variation in the contribution to the polygenic or oligogenic nature of obesity.
The identification of new genes implicated in obesity may also be achieved by the strategy of positional cloning. Three recent studies have enlightened a significant difference in the SNP frequency between populations of obese subjects and controls. These SNPs are located in the gene encoding SLC6A14 (solute carrier family 6 [neurotransmitter transporter], member 14) on chromosome X, [56], GAD2 on chromosome 10 [57] and ecto-nucleotide pyrophosphatase/phosphodiesterase 1 gene (ENPP1/PC-1) ENPP1 on chromosome 6 [47]. The SLC6A14 gene is interesting, because it codes for an amino acid transporter able to regulate the availability of tryptophane during the synthesis of serotonin and may affect the regulation of appetite and mood. The genetic association with the SLC6A14 gene was replicated in an independent French population [58], but other screening in large cohorts collected in Europe and North America is necessary, as is to undertake functional studies of the genetic variation to demonstrate the physiopathological role of SLC6A14 [59].
The role of ENPP1 was identified in a locus on chromosome 6q16.3 associated with childhood obesity and showing linkage in other population of diabetics and obese subjects [47]. ENPP1 was considered as a candidate gene for type-2 diabetes [60]. Genetic analysis of ENPP1 gene revealed association between a three-allele risk haplotype and obesity occurring in children or adults as well as in subjects with type-2 diabetes [47]. The physiological role of ENPP1 is a matter of debate, but it may inhibit the insulin receptor by interacting with a specific region in the α-subunit. The expression levels of ENPP1 are increased in muscle and adipose tissue of insulin-resistant subjects. The role of one known ENPP1 variant, the K121Q change, associated with type-2 diabetes, might be mediated by a functional change induced in insulin-signalling transduction from the α- to the β-subunit of the insulin receptor. In the French study, an ENPP1 haplotype confers a higher risk of glucose intolerance and T2D to obese children and their parents and associates with increased serum levels of soluble ENPP1 protein in children. Variants of ENPP1 could have a role in mediating insulin resistance and in the development of both obesity and T2D. This study provides clues for underlying common molecular mechanism to both conditions [47]. Like SLC6A14, the validation in independent populations and the confirmation of functional studies are necessary.
The GAD2 gene encodes the 65 kDa subunit of the glutamic acid decarboxylase enzyme (GAD65) that catalyzes the formation of GABA (γ-aminobutyric acid) itself interacting with neuropeptide Y (NPY) in the hypothalamus to stimulate food intake. Three SNPs showed an association with morbid obesity in a French population. In the families, the SNPs segregate with obesity and an association was observed in a case-control study. A protective effect was attributed to the wild-type allele. The role of one SNP located in the GAD2 promoter, was strengthened by a functional study revealing that the allele most frequently found in obese subjects exhibits greater transcriptional activity than the wild allele. This could result in an increase in hypothalamic GABA and a subsequent increase in food intake. This success story deriving from positional cloning strategy is dampened by the recent findings in a very large population of 2359 subjects comprising more than 600 families of German origin, thus four times as many subjects as in the first report. In two independent case-control studies, the authors were not able to confirm the association of GAD2 gene with severe obesity. The functional data for the most important SNP located in the promoter did not support previous findings [61]. No detectable effect of the promoter SNP was seen on transcription of the reporter gene in cell lines. This genetic study illustrates once again the issues raised by genetic studies in humans affected of multifactorial diseases. If the reasons of the GAD2 conflicting results might be attributed to usual caveats complicating the interpretation of genetic studies (as described above), the discrepancies between functional data is more problematic and will probably need clarification. Whether the linkage previously found on chromosome 10 is due to the impact of another candidate gene will also probably be investigated by gene hunters.
3.4 Gene–environment interaction
Although the essential role of gene–gene interaction and gene–environment interaction is underlined in the determinism of obesity, it is amazing to find that these interactions are finally little covered in genetic studies, owing to the complexity of building these studies and processing the data. Some studies explored the effect of candidate gene variants on spontaneous weight gain or on the response to weight loss intervention either induced by dietary changes or gastric surgery in humans. Genetic variations of adrenoceptors, uncoupling proteins and PPARγ were the most investigated but in a still limited number of subjects [45]. A recent book chapter where references can be found reviewed these findings [62]. The combined presence of the two SNPs of UCP1 and the β3-adrenergic receptor in already obese subjects would aggravate weight gain during life. The relationships between the spontaneous change in body weight and body composition and gene variant of adiponectin, β3-adrenergic receptor, interleukin 6 and PPARγ were described [45]. A possible effect of the Bcl 1 restriction polymorphism of the UCP 1 gene on weight loss achieved during four months of calorie restriction was investigated. Carriers of the UCP 1 variant lost more weight than non-carriers. Subjects homozygous for the polymorphism had the greatest weight loss, indicating a dose-response effect of this gene variant on weight loss. A recent study addressing the effects of the Trp64Arg polymorphism of the beta 3 adrenergic receptor, and a promoter mutation of the UCP3 gene reports that subjects carrying the mutation show the lowest reduction in abdominal adipose tissue. The beneficial effects of weight loss on body fat distribution and glycaemic control were more pronounced in carriers of both genes ‘wild-type’ allele and smallest in variant allelic carriers. Other studies have indicated that the Arg allele is associated with an impaired response to weight reduction in Japanese subjects. A German study also indicated that subjects carrying the Arg allele of Trp64Arg, and the Arg allele of the Gly972Arg polymorphism in the insulin receptor substrate 1 (IRS-1) gene, experience low weight reduction in response to calorie restriction (review in [62]). In obese subjects from Finland, the Trp64Arg mutation in the beta 3 adrenergic receptor and the promoter mutation in the UCP-1 genes were associated with faster weight gain after a very low calorie diet. Taken together, the role of polymorphisms in the beta 3 adrenergic receptor and UCPs has been emphasized in the determination of weight loss or weight gain and to some extent confirms the first findings in the French population. Nevertheless, their effects may be highly dependent on the interaction with other specific genes or overall genetic background of the subjects investigated. The study of the Pro12Ala mutation in the PPARγ gene also illustrates this complexity, since discrepancies in the results were found usually when analysing the effect of this mutation in different populations without taking into account environmental factors. The Ala allele is associated with low BMI when subjects eat food enriched with unsaturated fatty acids, while it is associated with higher BMI when food has a low ratio of unsaturated/saturated fatty acids [63]. The metabolic response to weight loss and weight regain following a six-month dietary intervention in post menopausal women was analysed. Weight loss did not differ between women carrying the Ala allele and women homozygous for the Pro allele, but fat oxidation was significantly decreased following the weight loss intervention only in Ala carriers. Carriers of the Ala allele presented a larger reduction in insulin response to an oral glucose tolerance test compared with women homozygous for the Pro allele. Finally, 12-month weight regain was greater in Ala carriers.
Interaction between physical activity and gene variation were also investigated. In French obese patients, it has been shown that a variant of the promoter of the UCP3 gene may contribute to the corpulence of obese subjects and also have effects depending on the degree of physical activity of patients [64]. The presence of a frequent allele of this variant would reduce the beneficial effects of physical activity on corpulence in morbidly obese subjects. Physical activity–genotype interaction was also illustrated by the glutamine glutamic acid common variant of the β2 adrenergic receptor that has been studied in many populations. Glutamine variant allele appeared to be associated with obesity but this effect is seen only in men who are physically unactive [65]. Lindi and colleagues also found relationships between the Pro12Ala PPARγ polymorphism and long-term weight change in response to energy and fat restriction and increased physical activity [66].
Many other genes are currently being studied, particularly in wide European trials in which numerous environmental factors have been characterized. One of the future tasks will be to determine the combinations of genes and their polymorphisms predisposing to the development of obesity and under what environmental pressure this occurs. Well-controlled intervention studies are currently underway in wide populations in Europe such as in the NUGENOB, http://www.nugenob.com or DIOGENES, http://www.diogenes-eu.org European programs. In the context of gene–gene and gene–environment interaction, recommendation given for genetic association studies in complex diseases and especially those related to sample size, multiple testing and replication will be a real challenge. New methods have to be developed to take into account the issues of multiple testing. Progress in the knowledge of the human genome, development of the biocomputing tools and new analysis strategies taking into account hundreds of items of genetic and environmental information will be necessary to tackle these questions.
4 Lesson learned from genetic studies in common obesities
By looking at the complex picture of obesity-related susceptibility genes, one might keep in mind that these genetic studies have mainly produced a large repertoire of predisposing alleles of diverse importance. In contrast to monogenic obesities, these gene allelic variations are neither necessary nor sufficient for the expression of obesity phenotype. To illustrate this statement, Table 4 shows some candidate genes studied in the French population in the last ten years, including recent one. The Pro12Ala mutation of the PPARγ gene was also largely studied in different populations. Apart for issues related to statistics as discussed above, one can note that these studies produced a list of common variants that may modify the risk of obesity occurring, but with a small degree of certainty. Even if confirmed or replicated in independent studies, these susceptibility alleles have to be taken into account in the overall picture of many contributing environment-linked factors that may display strong effects, at least of similar intensity, if not higher. The genetic background (characterized by the combination of multiple allelic variations), epigenetic or environmental factors that may include diet, exercise, stress, hormonal, socio-economical factors and the developmental stage of epigenetic events may intervene in the occurrence, development and maintenance of obesity. It is conceivable that even individuals with a higher genetic susceptibility than others will have a potentiality to react to the environment highly dependent on the moment when the event occurs during the life span. This raises the issue of the appropriateness of future genetic testing in the complex context of common obesities, in which in any case the prevention is not a trivial task.
Risk factors of obesity phenotypes evaluated for common genetic variations in French obese subjects and in metaanalysis (for PPARγ)
| Gene/variant | Phenotype | Odd ratio |
| β3 AR (Trp64Arg) | High weight gain | 1.7 |
| UCP1 (–3826A/G) | High weight gain | 1.4 |
| GAD2 (risk haplotype) | Morbid obesity | 1.3 |
| PTP1B (risk haplotype) | Obesity, dyslipidemia | 1.49 |
| SREBP (risk haplotype) | Obesity, dyslipidemia | 1.53 |
| SLC6A14 (risk haplotype) | Obesity | 1.27 |
| PPARγ (Pro12Ala) | Diabetes | 1.4 (metaanalysis) |
| ENPP1 | Obesity, diabetes | 1.50–1.6 |
Least but not last, regarding the number and type of susceptibility genes described, one key issue is whether or not there are common genetic factors that may intervene specifically in the determinism of obesity. Many susceptibility genes have also been analysed in different common diseases, revealing association or linkage. Genetic linkage map for obesity, diabetes or cardiovascular diseases frequently overlap at some loci. As an example of candidate gene, a recent study suggested that a common polymorphism of interleukin 6 or its receptor was associated with weight gain and insulin sensitivity. The same variant has also been associated with many other complex diseases, such as chronic alcoholic liver disease, asthma, psoriasis or chronic bronchitis among others. As hypothesized by Becker [67], one might ask about the influence of common disease influencing alleles when they are in different genetic backgrounds, in other genetic combinations, influenced by different epigenetic or environmental factors. If these susceptibility genes are not causative and modify obesity risk in a given environment, what are they doing in the mean time? Are they neutral or deleterious for other diseases? It would probably be necessary to deeply analyse the overlapping results across different common disease to get a global view of well-known diseases.
5 Toward new strategies for the genetic analysis of obesity
Among the difficulties in identifying molecular targets, the difficulties of analysis, mostly of a statistical nature, and the necessity to combine several methods in heterogeneous populations are regularly pointed out. Obesity is characterized by a high phenotype heterogeneity linked to differences in stages of evolution. Each stage in the development of obesity (weight gain, weight maintenance and chronicisation, variable response to treatment, comorbidities occurrence) is probably associated with a different molecular mechanism. At present, we do not know of any biological or molecular predictor (biomarkers) of passing from one stage to the other. Study of gene expression in the tissues involved in the different stage of obesity may contribute to revealing the role of certain signals and provide with a better understanding of the mechanisms of energy homeostasis. If obesity is considered as a disease of adaptation to environmental pressures, one hypothesis is that roots of fat mass development could be identified in the groups of genes expressed in the peripheral tissue, which are implicated in responses (or non-response) to changes in the nutritional environment. This is one rational behind works aiming at combining various fields (clinical, biochemical, genetic, transcriptomic and other ‘omics’) and process the mass of information generated, in subjects of diverse phenotypic and genotypic characteristics in different nutritional conditions. The need to tackle multifactorial diseases, integrating all available sources of information is widely recognized. By using ‘omic’ analysis strategies, it is now possible to quantify the expression of a large number of genes/protein in different tissues as well as metabolites under different conditions simultaneously. These strategies complementary to the genetic approach can be used to study the regulation of energy balance. At some stage, overlapping approach between ‘omic’ strategies and knowledge regarding the susceptibility genes available in humans and rodents will be necessary.
6 Conclusion
With the constitution of DNA and clinical databanks of broad cohorts of unrelated patients and of families, decisive progress in the understanding and management of obesity can be foreseen. The combined studies of hereditary characteristics, of the genome and the function, regulation and interaction of genes, are modifying our present approach to diseases with a high impact on public health. The future will tell us whether the choice of new combined strategies was judicious in bringing to light targets for new pathways and therapeutic.
Acknowledgements
Program of genetic research on obesity in French families is supported by the Department of Clinical Research/‘Assistance publique–hôpitaux de Paris’ (Hospital Clinical Research Program), Alfédiam and Inserm “Avenir”.
Vous devez vous connecter pour continuer.
S'authentifier