

1 Introduction
Leishmaniasis is caused by Protozoans that belong to the order Kinetoplastida, family Trypanosomatidae [1]. Leishmania are injected in vertebrate epidermis and dermis by female phlenotomine sand flies when taking their blood meal. These organisms can parasitize a wide range of vertebrate hosts, from lizards to humans. Leishmania are obligate intracellular parasites that develop inside macrophage phagolysosome. They cause a broad range of clinical diseases in humans ranging from simple skin ulcers, to defigurating lesions of the face, or lethal visceral disease. Leishmaniasis, involving superficial tissues, is referred to as cutaneous leishmaniasis (CL), whereas leishmaniasis, caused by parasite multiplication in deep organs such as spleen and liver, is referred to as visceral leishmaniasis or kala azar (KA). Leshmaniasis represents an important health problem in many countries [2,3].
CL and KA are caused by different Leishmania species that have characteristic insect vectors, geographic distributions and associated pathologic manifestations [2,3]. Based on these characteristics and on enzymatic and DNA markers, the Leishmania classification has been divided into old world species, distributed over the regions of the Mediterranean Basin, East Africa, Middle East, and Asia, and new world species, found in Central and South America. Both new and old world strains can cause cutaneous and visceral disease, CL is divided into localized CL (LCL), diffuse CL (DCL), and mucocutaneous CL (MCL). LCL refers to a few well-delimited ulcers; MCL is a severe chronic inflammation of the mucocutaneous tissues of the face that causes progressive destruction of palatine and nasal septum. DCL is characterized by a large number of acneiform and nodular skin lesions, with large numbers of parasites in the skin tissue. The host immunological response and the host genetic background is critical in the evolution of the infection into either asymptomatic disease, LCL, MCL or DCL.
Experimental work has provided the first evidence for the regulation of infection by host genetic factors. Experimental studies on infections by L. donovani [4–7] have analyzed the control of early parasite multiplication, in the progeny of a cross of resistant x susceptible congenic mice infected by L. donovani. The results indicated a control by a single locus lsh on chromosome 1, whereas further studies identified two additional loci (H-2 and H-11) that are involved later in the infection [8–10]. Lsh was linked to other loci controlling susceptibility to Mycobacterium bovis and M. lepaemurium (locus Bcg) and Salmonella typhimurium (locus Ity) infections. The fact that these pathogens reside in macrophage phagosome and neutralize macrophage anti-microbicidal activity suggested that Ity, Lsh and Bcg could be a single locus that controls pathogens developing in the phagolysosome. Positional cloning identified NRAMP1 as a possible candidate for the control of infection, and nucleotide sequence analysis showed that a single mutation (a G to A substitution at nucleotide position 783) resulted in the non-conservative replacement of Gly105 to Asp105 within the second trans-membrane domain of the NRAMP1 encoded protein. The association was found in 20 resistant and seven susceptible mouse strains, reviewed in [11]. Genetic disruption [12] and use of transgenic animals [13] established that NRAMP1 was indeed the gene controlling infection at the bcg/lsh/ity locus.
NRAMP1 encodes a 90-to-100-kDa integral membrane protein (NRAMP1) containing 12 hydrophobic transmembrane regions and a heavily glycosylated extra cellular loop. The NRAMP1 is expressed in late endosome and lysosome membranes in macrophages. NRAMP1 is thought to act as a pH-dependent divalent cation carrier. NRAMP1-transported iron could stimulate, at low pHs, the generation of ROI (radical oxygen intermediates) through the Fenton reaction in the phagolysosome [14,15]. NRAMP1 may also increase microbicidal activity of the macrophage by transporting cations (Mn2+) outside the phagosome into the cytoplasm, thus depriving the pathogen from a nutriment required for essential enzymatic activity. Furthermore, regulation of cation (Fe2+) concentration in the mitochondrial and cytosol compartments probably affects a number of enzymatic activities such as PKC or act on iron-mediated deactivation of critical transcription factors: these effects could explain NRAMP1 pro-inflammatory effects.
Evidence for a genetic control of KA in human populations was obtained recently: it was observed that KA seems to occur more frequently in certain families [16]. Statistical evidence for a major genetic locus controlling KA was obtained by segregation analysis [17]. This locus, however, has not been confirmed by linkage analysis.
Epidemiological and clinical data suggested that a KA outbreak was developing in a population of a village (Barbar) of the Atbara River region on the Sudan–Ethiopian border (Fig. 1 and [18]). We initiated a longitudinal study in that village in order to determine which factors might be important in KA spreading. The village comprised large huts grouped in clusters of three to ten huts. Clusters were delimitated by fences and were less than 50 m apart. Trees (Acacia, Balanites, and Azadirachta) were frequent in the village. A number of large termite hills (putative breeding sites for Phlebotomine sandflies) were frequent and scattered throughout the village. Domestic animals (cows, donkeys, goats, and sheep) were reared near the huts. A seasonal river crossed the village and a deep well was the main water source for domestic activities and drinking. The whole Atbara River region is endemic for KA, which occurs as outbreaks. Clinical symptoms associated with L. donovani infections are splenomegaly, lymphadenopathy, recurrent fever and severe anaemia; in the present study, confirmation of KA was sought by detection of Leishmania parasites in lymph node aspirate. Nevertheless, all patients who exhibited KA symptoms and were serologically positive (by Direct Agglutination Test [19]) were given specific treatment. The first indication that cases were increasing in Barbara was recorded in 1995–1996 and indeed a large number of inhabitants of the village were subsequently found to be infected with Leishmania. After six years, around 30% of the village population had presented severe clinical signs of KA and was parasitologically positive, and around 50% had became serologically positive (by ELISA), with no detectable parasites in lymph node, whereas 15 to 20% of the villagers had remained serologically and parasitologically negative [20]. Then, most residents of the village had been exposed and infected with Leishmania, only a third of them however presented clinical disease (KA).
Map of the Sudan region where KA is endemic. The village where the present study was carried out is indicated.
2 How important were environmental factors to determine KA in that village?
Since young and elder children, males and females were infected during the outbreak, transmission was likely to occur in the village around the huts. This conclusion was further supported by the high number of phlebotomine sand flies that were trapped in the huts. Among them, Phlebotomus orientalis, which represented 5% of the collected sand flies, was likely responsible for the indoor transmission of L. donovani [21]. Since phlebotomine sandflies require a specific environment for their reproduction and nutrition and do not fly far, the distribution of phlebotomines breeding sites could have well influenced KA spreading in the village. It was found that a few environmental variables that could have influenced vector distribution, were associated with KA [22]: these include the presence of Acacia trees (which produce a gum on which sandflies feed), of neems (Azadirachta), which are known as insect repellents, and the presence of domestic animals near the house probably because they attract sandflies and because their faeces support the development of sandfly larvae. Interestingly, these factors have influenced the risk of KA in the first two to three years of the outbreak; later, these environmental factors were no longer associated with KA, probably because the disease had spread throughout the whole village, allowing a high infection rate in sandflies (transmission is mostly anthroponotic). The clustering of the huts had also probably contributed to the homogenization of parasite transmission.
A most interesting observation was made in the analysis of the parasite population that was collected from subjects with active disease and from dogs. Isoenzyme analysis revealed a large heterogeneity in the collected Leishmania strains [23,24]. No less than seven different zymodemes were found in human cases, all being closely related and holding ancestral position on the phylogenetic tree, which places Sudan as a possible original focus of VL, from which the disease has spread throughout the world [24]. Unfortunately, the virulence/pathogenicity of the different isolates was not evaluated; nevertheless this observation indicates that the Leishmania population had evolved during the outbreak, possibly allowing the emergence of parasites with enhanced capabilities to escape immune destruction and to bypass certain host barriers.
3 Candidate gene testing indicates linkage of KA to the NRAMP1 region
The village comprised Aringa (2/3), Fellata (1/6) and Haousa (1/6), who were living close to each others. The risk of KA was found to be 4.7 (1.9–11.7) and 2.2 (0.9–5.5) fold higher in Aringa than in Haoussa and Fellata, respectively [22]. In contrast, the prevalence of infections (with or without overt clinical disease) at the end of the outbreak was comparable in all ethnic groups. There was also a family clustering of KA cases [22] that could not be related to ecological factors (Fig. 2). KA distribution in families was not, however, consistent with a single major gene control and testing the hypothesis of a major locus by segregation analysis yielded negative results [25]. This could have several explanations, including clinical heterogeneity, unappreciated differences in environmental risk factors or a polygenic control.
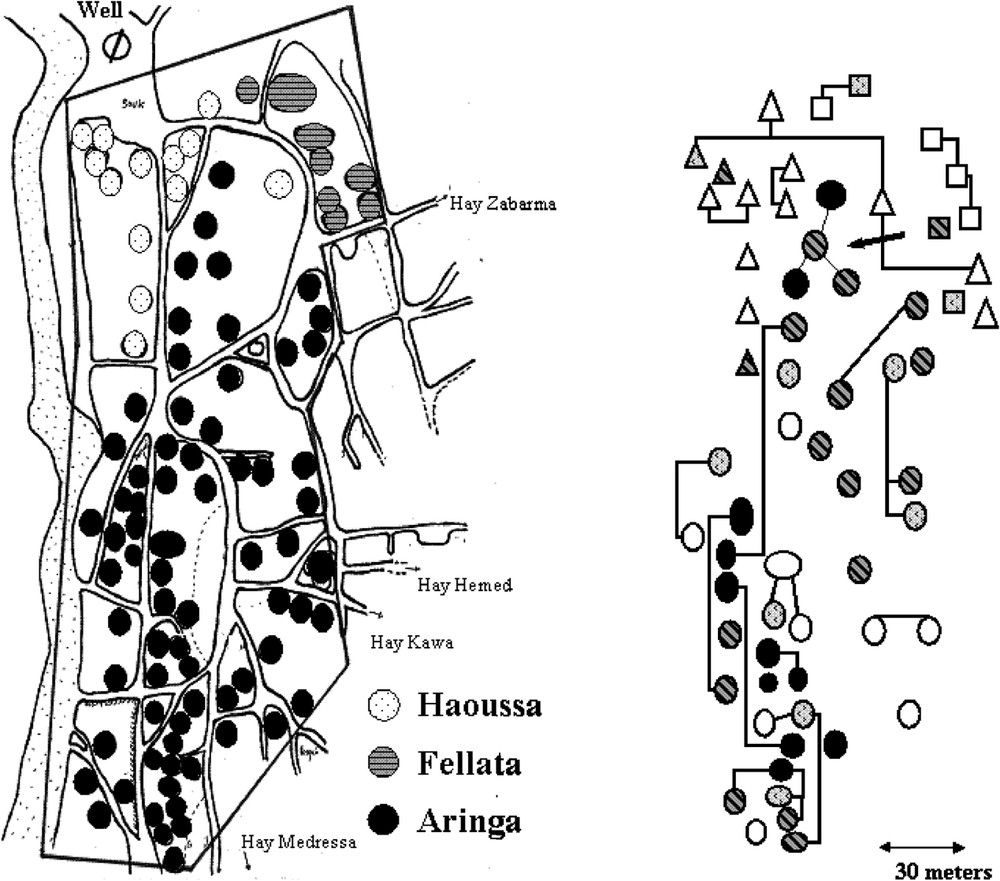
Map of the central part of the village showing the distribution of KA cases in different households. The ethnic origin of the household is indicated. Familial relationships are indicated by lines on the right part of the figure. Only household with young (20 years) subjects are shown.
Nevertheless, familial clustering of KA and differences in disease prevalence in ethnic groups suggested an influence of host genetics on susceptibility to KA. Then linkage analysis was carried out on affected siblings with genetic markers of regions containing obvious candidate genes. Analysis of the data was performed using the Maximum Likelihood Binomial (MLB) model-free method [26,27] that accommodates several cases in the same families. The study was concentrated on the Aringas to reduce the genetic heterogeneity of the study sample.
The genotypes of 172 individuals from 37 families containing two to five affected sibs (97 children) were determined. The mean age of onset was 10 years (3 to 27 years). No evidence of linkage was obtained with the micro satellite markers of the IFNGRI, or HLA/TNF or 5q31–q33 region (LOD scores 0.14). A suggestive LOD-score was obtained with D12S92 (LOD score = 0.94;
This finding was the first observation suggesting that NRAMP1 alleles were involved in the control of human KA. The different alleles at this polymorphic site have different effects on the transcription of NRAMP1 in vitro [28]. In our family-based association analysis, no distinct NRAMP1 polymorphisms were associated with an increased risk of VL. A much larger sample would have been necessary to reveal such an association. Together with the results observed in human tuberculosis [29,30] leprosy [31,32] and HIV [33], our data support the view that NRAMP1 is implicated in the control of intracellular pathogens in humans and in mice. A recent study in Sudan further supports our conclusions: that study has analyzed the genetic control of KA in a village that is close to our study population. This study replicated the linkage with polymorphisms in NRAMP1 and demonstrated that the main effects was contributed by a SNP in intron 4 [34]. These polymorphisms had been previously associated with susceptibility to tuberculosis [29]. Since the extent of linkage disequilibrium upstream the promoter region has not been determined, it cannot be totally excluded that a polymorphism outside the tested region could be responsible for the observed association. This is however unlikely, given the large body of literature on the role of NRAMP1 in both mouse and human susceptibility to intra cellular pathogens including L. donovani. These results altogether indicate a contribution of NRAMP1 to the susceptibility of Sudanese (Aringa and Massalit, which are close ethnic groups) subjects to KA.
4 Genome-wide linkage study indicates linkage of KA with the 22q12 region
A two-step genome-wide linkage study was carried out on the same affected siblings. In a first step, 38 families including two or more siblings affected with KA were genotyped for 367 micro satellite markers, covering the whole genome and providing a 10–15-cM primary genetic map. Regions located on chromosomes 22q12, 2q24 and 2q35 yielded maximum multipoint LOD scores of 0.83 (
In the second step of the genetic analysis, the three regions that presented suggestive linkage with KA were further investigated in 25 additional families and typing 13 additional markers at the 22q12, 2q23–2q24 and 2q31 (NRAMP1) loci. The multipoint linkage analysis of chromosomes 2q23–q24 and 2q35 yielded similar maximum LOD scores (1.53 and 0.79, respectively) as in the first step. In contrast, LOD scores in region 22q12 increased to 3.50 (
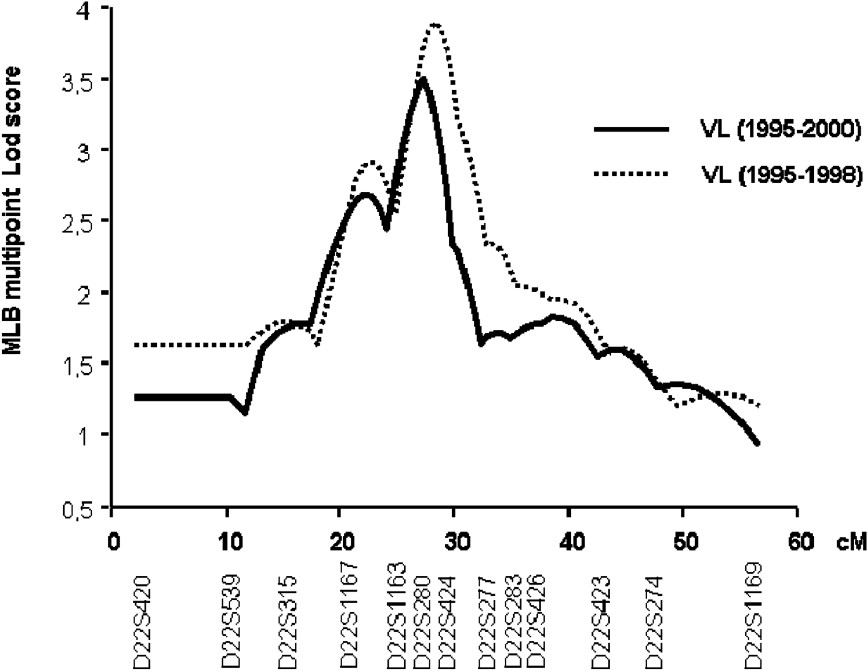
Multipoint analysis localizing the major KA-susceptibility locus on chromosome 22. Results of the multipoint linkage analysis performed on the whole family set with chromosome 22 microsatellite markers. Multipoint LOD scores were computed on all KA cases (1995–2000; 63 families) (solid line). Multipoint LOD scores were also computed on KA cases that occurred before 1999 (dashed line).
We repeated the analysis only on early KA cases, because we had observed that early and late KA cases might be associated with different immunological responses. KA recorded early in the outbreak had occurred rapidly after primo-infection, whereas later in the outbreak, KA developed in subjects who had seroconverted several months before. Furthermore, adult KA was more frequent late in the outbreak, whereas children were affected at early times in the outbreak (Fig. 4). In this analysis, 52 out of the 63 families remained informative (with at least two affected children). An increased maximum LOD score of 3.90 (
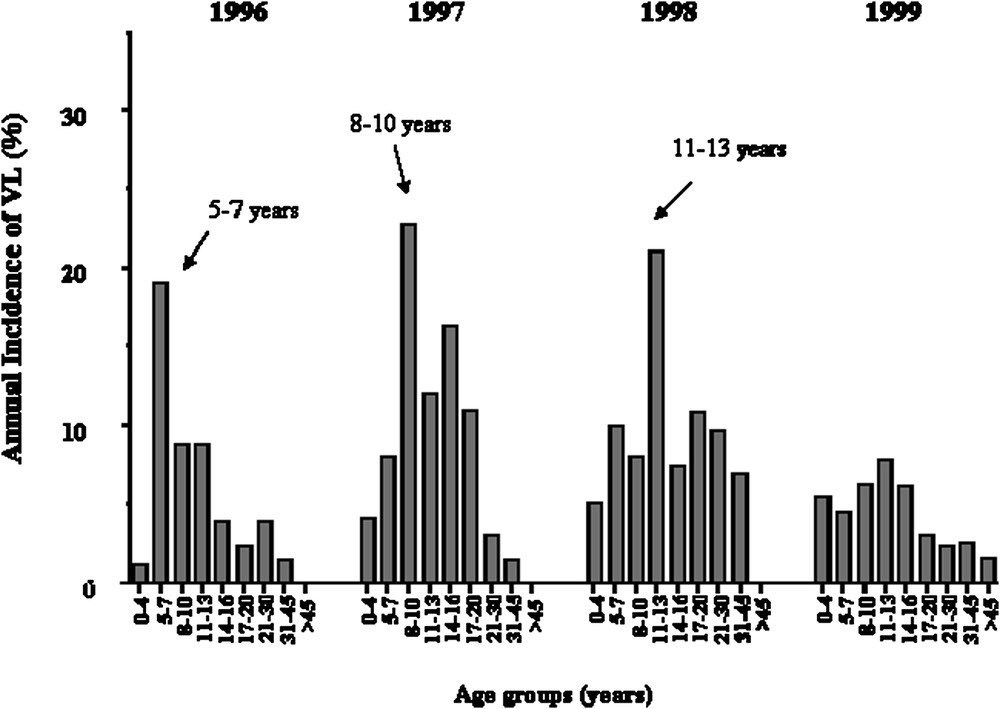
Annual incidence of KA in years 1996, 1997, 1998, 1999 according to age groups, showing that the age of KA cases has increased with time.
We then investigated possible gene–gene interactions between the major 22q12 locus and the other two regions 2q23–q24 and 2q35. A conditional analysis showed no significant positive gene–gene interactions. When looking for negative gene–gene interactions; the single point LOD score increased markedly at the two adjacent 2q23 markers D2S141 and D2S142 (LOD scores = 3.74 and 3.84, respectively), using the 30 families displaying negative LOD scores at D22S280. However, the multipoint analysis yielded rather uniform LOD scores between 1.75 (
5 Looking for the genes in the 22q12 region that could increases susceptibility to KA
The 22q12 region contains several genes whose alleles could modulate susceptibility to KA including the IL-2 receptor β chain gene (IL-2RB), which is located in the 22q12 chromosomal region close to D22S280. IL2RB is a good candidate for the control of KA. IL-2 is critical for T-cell proliferation and differentiation. T-cell responses and IL-2 production are strongly suppressed in VL patients [36,37] and high levels of circulating soluble IL-2 receptor have been observed during the acute phase of the disease [38]. Studies on other protozoan parasites such as Trypanosoma cruzi also support the hypothesis that some parasites evade the immune system by inhibiting the transcription of the IL-2R γ and β chain genes [39]. Interestingly, two other genes, NMI (N-myc interactor) and STAM2 (Signal-Transducing Adapter Molecule 2), located in the 2q22–q23 region, encode proteins involved in the IL-2 signalling transduction pathway.
This led us to sequence the genes in several affected and non-affected cases. The association of the most frequent (frequency 20%) was tested. Only a weak association was observed. Work is ongoing to evaluate further this gene – and other candidate ones – in a larger population.
6 Conclusion
Thus, susceptibility to KA in Barbar's population is determined by a major gene on the chromosome 22q12 and by mutations in NRAMP1. The data also suggest a control by gene(s) in the 2q22–q23 region. The proportion of alleles at the 22q12 locus, shared by affected siblings at the peak LOD score in region 22q12 was 0.63 (all KA cases) and 0.66 (KA before 1999). The locus-specific sibling recurrence-risk ratio (
Such genetic heterogeneity might be a characteristic of Leishmania infections by comparison with Schistosoma infections, which are influenced by a few major loci confirmed in several populations [47]. Several factors might have contributed to such heterogeneity, first the heterogeneity of the studied phenotype (KA is a broad phenotype and alterations in more than one pathways can cause KA); second, the Leishmania population in Barbar was heterogeneous and has evolved with time: various zymodemes were identified during the outbreak and the composition of the parasite population was different at the end as compared to the start of the outbreak [24,48]. Thus, Leishmania virulence may evolve with time, enabling the parasite to infect subjects who resisted in the early stage of the epidemics. So far, however, there has been no clear demonstration of such an evolution of parasite virulence. Studies in experimental models using strains isolated from patients with cutaneous leishmaniasis have shown, however, that a Leishmania isolate collected from patients with a poor response to treatment escape immunological mechanisms that control less pathogenic isolates [49]. No such observations were made on schistosomes and there is no evidence for different pathogenicity of isolates from subjects (from the same region) with different clinical diseases; most studies rather support the view that the schistosoma genome, unlike the leishmania genome, do not evolve rapidly in order to escape immunological or chemotherapeutical pressures.
Vous devez vous connecter pour continuer.
S'authentifier