

1 Introduction
Date palm (Phoenix dactylifera L.) is a monocotyledonous, dioecious and diploid (
A fungal disease called Bayoud threatens the date palm plantations in North Africa. Bayoud, the fungal vascular wilt of date palm, is caused by Fusarium oxysporum fsp albedinis. Genetic improvement is the most important and efficient tool to produce resistant plant material with good date quality. Classical cross breeding takes a long time (15–30 years) for the production of new varieties. Somatic hybridisation can be an alternative. Recently, somatic hybridisation has been successfully used in vegetative propagated banana (Musa spp.) [3], in the Solanaceae to introduce disease-resistance traits, by transferring resistance from wild relatives to cultivated varieties [4,5], in rice (Oryza sativa L.), in barley (Hordeum vulgare L.) [6], and in citrus (Citrus spp.) [7].
Totipotent protoplasts are considered as a very important experimental material for genetic engineering [8]. Somatic hybridisation by protoplast fusion enables nuclear and cytoplasmic genomes to be combined, fully or partially, at the interspecific and intergeneric levels to circumvent naturally occurring sexual incompatibility barriers. Isolated protoplasts are also exploited in numerous miscellaneous studies involving membrane function, cell structure, synthesis of pharmaceutical products, and toxicological assessments [9–12].
Protoplasts can be isolated from leaves, cotyledons, shoots, roots, and flowers. In monocotyledonous species, the best material for protoplast isolation is embryogenic cell suspension as in cereals and especially rice [13]. Likewise, in rye, Ma et al. [14] used fast-growing friable callus initiated from immature inflorescences to establish embryogenic cell suspensions as a source of totipotent protoplasts. In other monocotyledonous species such as banana, the best source material for protoplast isolation is embryogenic callus [15] and/or cell suspensions [16] because of their totipotency. Protoplasts from leaf mesophyll cells/tissues and callus were recalcitrant for regeneration [16,17].
To our knowledge, no report exists on protoplast technology in date palm. In the present investigation, we report first the induction of callus formation from protoplasts in two genotypes of date palm. To optimize the production of protoplasts and cell division frequency, factors such as donor material, composition of enzyme solutions, duration of incubation period, and culture system were studied.
2 Material and methods
2.1 Plant material
Two genotypes, Deglet nour from Biskra, in the Southeast of Algeria, and Takerboucht from Adrar, in the Southwest, were studied. Deglet nour is sensitive to fusariose and has a good fruit quality; in contrast, Takerboucht is resistant to this pathogen and has a lower fruit quality. The offshoots grown in open fields (20-cm diameter, 3- to 6-kg weight) were taken from adult female plants. Young leaves and the shoot apical tip from offshoots were used for our investigation.
2.2 Initiation and maintenance of callus cultures
The offshoots (2–4 years old) from field grown adult female date palm were used for callus initiation. The shoot tip of offshoots were sterilized with 0.3% benlate (methyl [1-(butylcarbamoyl)benzoimidazol-2-yl] aminoformate) (DuPont, France) for 30 min, followed by transferring to a 30% aqueous solution of 5.4% (v/v) sodium hypochlorite in water with two drops of tween 20 per 500 ml for 45 min. The tip was then rinsed three times with distilled water (duration of each rinse: 10 min). Shoot apical tips (about 5-cm length, 3-cm diameter) of offshoots were excised in small pieces (5-mm length) and cultured on Petri dishes (9.5-cm diameter) on solid medium M1 (20 ml, 10 pieces per Petri dish) containing MS salts [18] and supplemented with 9.0 μM 2.4-D, 14.76 μM IPA, Morel vitamins [19], 87 mM sucrose, and 7 g l−1 agar (Sigma, USA). The pH was adjusted to 5.7–5.8 before autoclaving (20 min, 120 °C, 1 bar). The cultures were kept at 27 °C in the dark. The explants were subcultured at 4-week interval on the same medium. After 3–6 months of culture, friable white and yellow nodular calli were formed.
2.3 Protoplast isolation from young leaves of offshoots
Offshoot leaves around the shoot tip (10–15 cm) from field grown adult female date palm were used for protoplast isolation. Leaf pieces of approximately 1 cm in length were excised and surface sterilized with a mercryl lauryle solution (about 5 drops in 100 ml distilled water) for 15 min and followed by washing three times with a domestic detergent (Domestos, Sunlight, France), followed by a repeat surface sterilization with a 30% aqueous solution of 5.4% (v/v) sodium hypochlorite solution for 20 min and rinsing in sterile distilled water three times during 15 min. About 1 g f.wt. of leaf explants were scarified on the lower surfaces and placed in 15 ml of enzyme solution with their abaxial surface downward. Three different enzyme solutions EC1, EC2 and EC3 (pH 5.6) were tested (Table 1). The enzyme solutions were sterilized using a 0.2-μm Millipore filter (Millipore, Billerica, MA, USA).
Composition of enzyme solutions
| Enzyme concentration (%, w/v) | EC1 | EC2 | EC3 |
| Cellulase RS (Yakult Pharmaceutical Ind. Co., Ltd, Tokyo Japan) | 1.5 | 1.5 | 1.5 |
| Pectolyase (Kyowa Chemical Products Co., Ltd, Osaka Japan) | 0.15 | 0.2 | – |
| Hemicellulase (Sigma, USA) | 0.2 | – | – |
| Macerozyme (Sigma, USA) | |||
| Pectinase (Sigma, USA) | – | 1 | 1.5 |
| KCl | 3 | 3 | 3 |
| CaCl2 | 0.5 | 0.5 | 0.5 |
| pH adjusted with HCl 0.1N | 5.6 | 5.6 | 5.6 |
The enzyme solution/young leaf mixture was incubated overnight (12–20 h) at 27 °C in the dark. Before the purification step, the mixture was observed under the microscope. In the case the number of protoplasts observed was not important, the protoplast suspension was transferred to a gyratory shaker (30 rpm) for 15 min.
2.4 Isolation of protoplasts from callus
Two types of callus derived from shoot tip (3–7 cm) were tested for protoplast isolation: nodular yellow callus and friable white callus. About 800 mg f.wt. were used for each experiment. The callus was cut into small pieces, put into 15 ml of enzyme solution in Petri dishes (9.5 cm diameter), and placed in the dark at 27 °C for 12–20 h. The same enzyme solutions were used as for leaf explants (Table 1).
2.5 Purification of protoplasts
The protoplasts were purified as described by [20]. After incubation, the digestion mixture was filtered through 100/25-μm metallic mesh combination to remove debris and large cell colonies. Protoplasts were washed three times through centrifugation (65 g for 5 min) with a washing solution that consisted of 204 mM KCl, 67 mM CaCl2 with pH 5.7. In order to minimize the salt content in the protoplast suspension, they were rinsed again with 0.5 M mannitol and 67 mM CaCl2 (centrifugation 65 g for 5 min). The protoplast viability was determined by fluorescein diacetate (FDA) [21].
2.6 Culture of protoplasts
Protoplasts were cultured at a density of 106. Two culture systems were tested: liquid culture and nurse culture.
For liquid culture, protoplasts were suspended in 4 ml of media in small Petri dishes (5.5 cm diameter). The liquid medium (M5) consisted of MS salts, vitamins of Morel, 0.68 mM glutamine, 117 mM sucrose, 0.4 mM glucose, 0.5 mM MES, 1.9 mM KH2PO4, 9.0 μM 2.4 D, 14.76 μM IPA and 250 mg l−1 polyethylene glycol 4000 (PEG). The pH was adjusted to 5.7 with 0.1 N NaOH before filter sterilization (0.22-μm millex GS filters, Millipore, Billerica, MA, USA). The Petri dishes were sealed with parafilm and transferred into the culture room.
For the feeder layer, embryogenic offshoot-tip-derived calli of Deglet nour were used as nurse cells. The nurse culture was prepared the same day when the protoplasts were isolated. The PCM liquid medium (double strength) contained double strength of MS salts, 9.0 μM of 2.4 D, vitamins of Morel, 2.8 mM glucose, 278 mM maltose, 170 mM sucrose and 2.5 mM Myo inositol (pH 5.7). Callus suspension was made by cutting friable calli into small pieces (0.2 mm) and by adding it to 100 ml of PCM culture medium, in order to obtain a final cell concentration of 2% in PCM/agarose mixture.
Agarose sea plaque 1.2 g (Sigma, USA) was dissolved separately in 100 ml of distilled water (pH was adjusted to 5.7) and then autoclaved. When the temperature of the agarose solution decreased to 30–35 °C, it was carefully mixed with a 100-ml PCM medium containing nurse cells. Aliquots of 10 to 12 ml of this mixture were poured into small Petri dishes (5.5-cm diameter). After solidification, the medium was covered with sterilized nitrocellulose filter (AA type millipores), and 1 ml of protoplast suspension in M5 medium (see above). All cultures were maintained at 27 °C in the dark. Cell-wall regeneration was observed with calcofluor white brightener stain [22].
The microcalli formed were transferred onto a callus induction medium containing MS salts and supplemented with 13.5 μM 2.4 D and 14.76 μM IPA, Morel vitamins, and 3 g l−1 gelrite. The calli were transferred into the regeneration medium, which consisted of MS salts supplemented with the same level of IPA of the callus induction medium (see above) and 1.4 μM 2.4 D. The cultures were kept in the dark at 27 °C.
2.7 Data collection and statistics
Only viable protoplasts were counted. Protoplast yield was estimated with a Nageotte hematocytometer. At least three replicate counts were used. Results were expressed as yield per g f.wt. for leaves or calli. The protoplast yield per milliliter 0.5 M mannitol and 0.75 mM CaCl2 was calculated using the following formula:
3 Results
3.1 Production of protoplasts
Embryogenic callus (Fig. 1A and B) is the material of choice for protoplast isolation in date palm. In this study, viable protoplasts were isolated from both genotypes Deglet nour and Takerboucht (Fig. 1C and D). Generally, young leaves gave less viable protoplasts than those from calli. In young leaves, the viability rate was 8% in Deglet nour and 9% in Takerboucht; in calli it was 65% in Deglet nour and 57% in Takerboucht. The viability of freshly isolated protoplasts (immediately after isolation) was about 80% in both leaf- and callus-derived protoplasts.

(A) Development of nodular callus from meristematic shoots of cultivar Deglet nour after 4 months of culture on induction medium. Bar=6 mm. (B) Friable and granular callus development of cultivar Deglet nour after 6 months of culture on induction medium. Bar=0.2 mm. (C) Protoplasts isolated from embryogenic callus of cultivar Deglet nour. Bar=30 μm. (D) Protoplasts isolated from leaves of cultivar Deglet nour. Bar=30 μm. (E) Dividing cell 7 days after protoplast isolation from callus in Deglet nour. Bar=5 μm. (F) Second cell division 10 days after protoplast isolation from callus in Deglet nour. Bar=5 μm. (G) Microcalli of Deglet nour on feeder layer 6 weeks after protoplast isolation. Bar=1 cm. (H) Microcalli of Takerboucht on feeder layer 8 weeks after protoplast isolation. Bar=1 cm. (I) Callus formation from callus-derived protoplasts in Deglet nour after 4 months of protoplast culture. Bar=6 mm. Masquer
(A) Development of nodular callus from meristematic shoots of cultivar Deglet nour after 4 months of culture on induction medium. Bar=6 mm. (B) Friable and granular callus development of cultivar Deglet nour after 6 months of culture on induction medium. Bar=0.2 mm. ... Lire la suite
3.2 Factors affecting protoplast isolation
The viability rate of freshly isolated protoplasts was dependent on the composition of the enzyme solution tested. EC1, the combination of cellulase RS and pectolyase with hemicellulase (Table 2) was more efficient for the isolation of protoplasts in date palm.
Influence of genotype, donor material and enzyme solution on protoplast yield (×105)
| Genotype | Donor material | EC1 | EC2 | EC3 |
| Deglet nour | nodular callus | 5.6 ± 0.1 a | 2.95 ± 0.1 a | 0.95 ± 0.09 a |
| friable callus | 3.9 ± 0.1 c | 2.25 ± 0.2 b | 0.68 ± 0.08 c b | |
| leaf | 1.97 ± 0.05 e | 0.72 ± 0.03 c | 0.21 ± 0.03 d | |
| Takerboucht | nodular callus | 4.95 ± 0.2 b | 2.05 ± 0.2 b | 0.78 ± 0.03 b |
| friable callus | 3.25 ± 0.3 d | 1.85 ± 0.06 b | 0.62 ± 0.04 c | |
| leaf | 1.00 ± 0.06 f | 0.53 ± 0.03 c | 0.05 ± 0.03 e |
The protoplast yield was also dependent on donor material, genotype, and incubation time. Regarding the donor material, the best response was obtained with nodular callus; the protoplast yields were
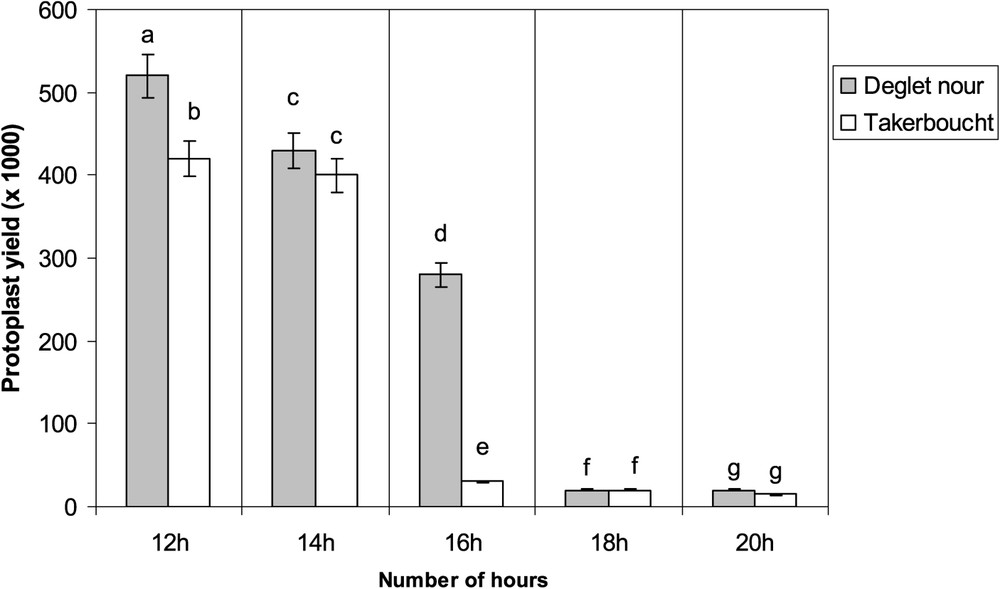
Impact of incubation time on protoplast yields from nodular callus. Data are means of three independent experiments. Differences between means were assessed using the Student–Newman–Keuls test; means with the same letter are not significantly different at α=0.05.
3.3 Protoplast response in culture
The protoplast viability rate was 80% at day 0 (immediately after isolation) before transferring in culture medium; however, the cell viability diminished with increasing culture duration, depending on the explant, culture system, and genotype; regarding the cell viability, callus-derived protoplasts showed the best responsiveness as compared to leaf-derived protoplasts. In Deglet nour, the cell viability rate of callus-derived protoplasts at day 4 was 65% on the feeder layer and 60% in the liquid medium (Table 3). In Takerboucht, the frequency of cell viability of callus-derived protoplasts was 57% on feeder layer and 49% in liquid medium (Table 3). Three days after the protoplast culture, the cells began to regenerate the cell wall and became oval. In terms of cell-wall regeneration, the best responsiveness was obtained with callus-derived protoplasts, and the best culture system was the feeder layer. At day 4, the rate of cell-wall regeneration of callus-derived protoplasts in Deglet nour was 54% on feeder layer and 25% in liquid medium; in Takerboucht, the frequency of cell-wall regeneration of callus-derived protoplasts was 38% on feeder layer and 14% in liquid medium (Table 3).
Influence of genotype, donor material and culture system on frequency of cell viability cell wall regeneration four days after protoplast culture
| % of cell viability | % of cell wall regeneration | ||||
| Liquid medium | Feeder layer | Liquid medium | Feeder layer | ||
| Deglet nour | callus | 60 ± 2.0 a | 65 ± 2.0 a | 25 ± 3.0 a | 54 ± 3.6 a |
| leaf | 5 ± 2.0 c | 8 ± 2.0 c | 3 ± 1.0 c | 5 ± 1.0 c | |
| Takerboucht | callus | 49 ± 2.6 b | 57 ± 2.6 b | 14 ± 1.7 b | 38 ± 3.6 b |
| leaf | 4 ± 1.0 c | 9 ± 1.0 c | 1 ± 0.3 c | 2 ± 1.0 c |
Cell division was obtained in both callus and leaf-derived protoplasts; however, the callus-derived protoplasts showed a better response. The higher division rate of callus-derived protoplasts was achieved on feeder layer; in contrast, leaf-derived protoplasts did not show any significant difference in both culture systems. The cell division rate of callus-derived protoplasts (Fig. 1E and F), related to the number of isolated protoplasts at day 0 (immediately after isolation), was 30% on feeder layer and 20% in liquid medium in Deglet nour at day 10; in Takerboucht, the division frequency was 15% on feeder layer and 7% in liquid medium (Table 4).
Influence of genotype, donor material, and culture system on cell division rate (%) 10 day after protoplast isolation
| Genotype | Donor material | Liquid medium | Feeder layer |
| Deglet nour | callus | 20 ± 2.6 a | 30 ± 3.6 c |
| leaf | 5 ± 2.0 be | 6 ± 2.5 e | |
| Takerboucht | callus | 7 ± 1.7 b | 15 ± 2.0 d |
| leaf | 4 ± 1.0 be | 5 ± 1.5 e |
The dividing cells continued to grow and developed into microcalli on feeder layer 8 weeks after protoplast plating, as shown in Fig. 1G and H. The number of microcalli was 14 000 per Petri dish in Deglet nour and 9000 per Petri dish in Takerboucht. In both genotypes, about 25% of the microcalli developed to callus (Fig. 1I) on the callus induction medium. The calli that were transferred onto the regeneration medium failed to regenerate into shoots or roots.
4 Discussion
In this investigation, we have established a protocol that allowed callus formation from protoplasts in date palm. Out of 800 genotypes of date palm identified in Algeria [23], the cultivars Deglet nour and Takerboucht were selected for the current study. Deglet nour produces dates with excellent fruit quality (soft, red color, perfumed, small-medium fruit size with medium ripening), making it an important commercial cultivar in northern Africa; nevertheless this genotype is susceptible to bayoud, the most important date palm disease in northern Africa. Takerboucht has a lower economical value because of the medium quality of its fruit (semidry to dry, yellow orange color, small fruit size, medium ripening); however this cultivar is known to be resistant against bayoud disease. A combination of the two genomes by protoplast fusion may contribute to produce a superior date palm variety with excellent fruit quality and resistant to bayoud.
To date, there is no report on the production of protoplasts in this species. The isolation of protoplasts depended on the choice of genotype, the donor material, the enzyme solution, and the incubation time. The embryogenic callus was a suitable donor material for protoplast isolation. In monocotyledonous trees, like banana, it was shown that the appropriate donor materials for efficient protoplast isolation were callus and embryogenic cell suspension [17]. But the induction of embryogenic suspension cultures of banana requires more than one year [24], and the number of genotypes that can be used to produce suspension cultures is limited [25,26]. In other monocotyledonous species, such as in rice [27], maize [28], and barley [29], suspension cultures were the most suitable donor material for protoplast isolation. Cell-suspension cultures established in date palm in our lab were not stable (data not shown); therefore, calli were used as donor material for protoplast isolation in the present study.
The efficiency of protoplast isolation depended on the environmental conditions, particularly the composition of the enzyme solution. The combination of cellulase RS–pectolyase–hemicellulase gave a higher number of protoplasts. Using the same approach in banana protoplast isolation [17], the mixture of cellulase–pectolyase–hemicellulase was more efficient for protoplast isolation from cell suspensions after 15–17 h in darkness. The protoplast isolation involved the treatment of tobacco leaves with pectinase to separate the cells, followed by cellulase to remove their wall [30]. The procedure was simplified by a single treatment with a mixture of enzymes [31].
In dicotyledonous, it was shown that the combination of cellulase onozuka RS and pectolyase Y-23 significantly improved the yield and the viability of leaf mesophyll protoplasts in Prunus cerasus L. [32]. The optimum enzyme mixture for protoplast isolation in callus of the same plant material was cellulase onozuka RS and macerozyme R10. It was reported that in Echinacea angustifolia, the yield of protoplasts released also increased when the cellulase concentration was increased to 2.0% (w/v), and the lower cellulase concentration (less than 1%) could not lead to liberation of enough protoplasts [33]. Cellulase at a concentration higher than 2% may cause over digestion of plant material [34,35].
Several factors influence protoplast release, including the extent of cell wall thickening, temperature, duration of enzyme incubation, pH of the enzyme solution [36], agitation, the nature of the osmoticum, and plasmolysis prior to enzyme digestion of source tissues in salts [37]. Protoplast yield and viability can be further enhanced by slicing of source (preplasmolysed) tissues, manual or enzymatic removal of the epidermis, and conditioning of the donor material or its culture on media containing suitable osmoticum [38–40].
During the present study, cell-wall regeneration, cell division, and callus formation were obtained. Among the plant growth regulators that we tested (data not shown), only the combination 2.4D–IPA induced cell division. In earlier studies on rose mesophyll protoplasts, NAA and BA were the most efficient growth regulators for the regeneration of microcalli [41]. In lily protoplasts, the addition of picloram to the culture medium was critical of development of microcalli [42]. Our investigation demonstrates that nurse culture was effective for mitotic activity of date palm protoplasts. Recently nurse culture technique has been successfully used to solve regeneration problems in recalcitrant protoplasts [20,42,43]. The number of microcalli we obtained was close to those obtained in earlier studies in banana [44]; however, the obtained calli did not develop into plants in our study. Shoot organogenesis depends on many parameters, including the genotype, protoplast-derived material, plant growth regulators, culture system, and exposition time of protoplasts on nurse cells.
Previous investigations showed the impact of genotype on plant regeneration from protoplasts in apple and banana [17,45]. Currently, protoplasts can be isolated from almost all plant tissues, calli and cell suspensions; however, the type of the donor material is crucial for shoot organogenesis and somatic embryogenesis; callus tissues contain less embryogenic or dedifferentiated cell aggregates compared to cell suspensions, the most used donor material for protoplast isolation in monocotyledonous. This may be the cause of the lack of plant regeneration. The main plant growth regulators auxin and cytokinin, alone or in combination, are generally essential for efficient protoplast-to-plant systems [8]. Plant growth regulator concentrations and combinations need to be optimized for each protoplast development step. The following plant growth regulators were tested in our preliminary experiments: 2.4 D, IPA, IAA, BA, and NAA. Only the combination 2.4D–IPA induced sustained cell divisions and callus formation. None of the plant growth regulators induced plant regeneration, which may be related to the negative interaction between those plant-growth regulators and some metabolites produced by callus tissues. The nature of positive or negative factors produced by feeder cells is still completely unknown; however, we recently found that the interaction between feeder layer cells and protoplasts is very time-dependent in banana protoplasts [44]. During the present study, both liquid culture and feeder layer systems were used. Alginate and agarose techniques may be a useful alternative to liquid or to the feeder layer method [46]. The conversion frequency of microcalli into somatic embryos was very low when using a feeder layer compared to when microcalli were transferred onto regeneration medium immediately after their formation [44].
5 Conclusion
In the present studies, we induced first sustained cell division, microcallus formation, and callus regeneration from recalcitrant date palm protoplasts by optimizing the protoplast isolation and culture conditions. For optimizing the shoot regeneration from date palm protoplasts, future investigations should focus on the following points: (1) development of stable embryogenic suspension culture as a source for protoplast isolation because of the higher frequency of embryogenic cells, (2) optimizing the combination and concentration of plant growth regulators, (3) optimizing exposition time of microcalli on nurse culture, and (4) application of other culture procedures like alginate and agarose culture.
Acknowledgements
The financial support of the Algerian Ministry of Higher Education and of the ‘Comité mixte d’évaluation et de prospective de coopération interuniversitaire franco-algérienne' (CMEP) are gratefully acknowledged.