

1 Introduction
Aluminium is the third most common and the most abundant element in the Earth's crust. Its toxicity becomes acute in acid soils that comprise almost 40% of arable land, limiting crop productivity. Aluminium ion has been shown to inhibit root growth by affecting cell elongation, especially in the distal part of the transition zone of the root apex [1,2]. Though Al stress primarily affects root growth inhibition, in spite of much research on aluminium toxicity in plants, the exact mechanism of aluminium stress signalling remains unclear.
Any imbalance in the cellular redox homeostasis results in oxidative stress. In the atmosphere, molecular oxygen is mainly utilized as a hydrogen acceptor, yielding water in aerobic organisms [3]. An inevitable result of electron transports in mitochondrial inner membrane, chloroplast thylakoid membrane and plasma membrane is the univalent reduction of molecular oxygen in plant cells with the resultant production of toxic reactive oxygen species (ROS). ROS include superoxide radical (), hydroxyl radical (), alkoxyl radical (), hydrogen peroxide (H2O2), singlet oxygen (1O2), etc. These ROS have the capacity of oxidising lipids, proteins, nucleic acids of the cell, leading to cell death [4]. Mitochondria are unique organelles that carry out important oxidation–reduction reactions, resulting in the production of energy in the form of ATP. Mitochondria have been considered major ROS producers in animal cells and in plant cells without chloroplasts, such as root cells, through univalent reduction of oxygen, especially at complex I and III sites, which results in the production of . undergoes an enzymatic and non-enzymatic dismutation reaction, immediately producing cytotoxic H2O2 [5]. Toxic hydrogen peroxide is a product of organellar oxidative reactions and can act both as oxidant and reductant. It is the most stable form of the ROS and is capable of rapid diffusion across cell membrane [6]. The plant mitochondria are different from animal ones in having a branch point at the level of ubiquinone into a unique alternative pathway. This is represented by a carrier named alternative oxidase (AOX), which helps in a reduction of oxygen that is not associated with the generation of energy, which in turn reduces the ROS burden and the more general cytochrome pathway (CYT pathway) represented by complex III and complex IV [49]. Aluminium ion, though it is not a transition metal, has been shown to induce oxidative stress in plant cells and also in root and leaf tissues [7–10]. An Al-induced and Fe-dependent lipid peroxidation with a loss of membrane integrity has also been shown to cause an apoptosis like cell death in tobacco cells [9,11]. Al-induced cell death was similarly observed in barley roots [12].
The mitochondrial permeability transition pore is thought to be a multiprotein pore consisting of voltage-dependent anion channel (VDAC), the adenine nucleotide translocator (AdNT) and cyclophilin-D. In animal cells, it is well known that mitochondria undergo a permeability transition of the outer membrane in response to calcium (Ca), ROS and various other apoptogenic factors, which opens up the mitochondrial permeability transition (MPT) pore that can allow solute of 1.5 kDa to be leaked out; this opening is followed by subsequent collapsing of the inner membrane potential [13]. The similar existence of MPT pores in plants has been recently reported in potato, wheat, and Arabidopsis [5,14,15]. Like animal cells, plant MPT pore opening has also been found to be induced by Ca and oxidative stress; this in turn sometimes released cytochrome c and other matrix proteins that may cause a programmed cell death. Programmed cell death in plants has got some similarities with animal apoptosis [5,14].
We isolated mitochondria from Al-treated tobacco cells and investigated directly Al effects on the mitochondrial functions (e.g., respiration, ATP synthesis, ROS production) and also whether Al does induce opening of MPT pore, which in turn causes high-amplitude swelling and release of cytochrome c of mitochondria, resulting in an irreversible dysfunction of mitochondria, followed by cell death. In our earlier work, we have shown that the cultured tobacco cells at logarithmic phase of growth are well comparable to the root meristematic cells, and that Al has been found to cause a mitochondrial dysfunction studied in whole cell with a concomitant loss of growth capability in a simple Ca medium [10]. In the present investigation, we will report the details of aluminium effects on mitochondrial functions in mitochondria isolated from tobacco cells under Al stress and propose a new intracellular mechanism of aluminium toxicity in plants cells, where Al imposes an oxidative stress situation, resulting in an irreversible dysfunction of mitochondria and cell death in tobacco cells.
2 Materials and methods
2.1 Plant and cell culture, growth conditions and Al treatment
A tobacco (Nicotiana tabacum L. cv. Samsun) cell line SL (non-chlorophyllic) was used. Cells were grown in a modified Murashige and Skoog medium and subculturing was done after every 7 days. Al treatment was administered to exponentially growing logarithmic phase cells four days after subculture with AlCl3 in a Ca medium (3 mM CaCl2 and 3% [w/v] Suc), pH 4.5, containing 10 mg fresh weight cells ml−1 for 18 h and kept on a rotary shaker at 100 rev min−1 at 25 °C in darkness. The growth capability of Al-treated cells was determined from the extent of growth of Al-treated cells as compared to the untreated control cells after post-Al-treatment culture in the growth medium for six days [16].
2.2 Isolation of mitochondria from tobacco cells
Isolation of mitochondria was done according to the method of Millar et al. [17], with some modifications. Cells of 10–20 g fresh weight cells were collected by centrifugation at 750 g for 3 min, washed with Ca medium (pH 5.0). Tobacco cells were ground in standard homogenization buffer (0.3 M mannitol, 1 mM EGTA, 25 mM MOPS–KOH (pH 7.8), 10 mM tricine, 8 mM cysteine, 0.1% (w/v) BSA and 1% (w/v) PVP-40) in the presence of 1–2 g of sea sand. The homogenate was centrifuged at 2500 g for 9 min and the supernatant was collected. Supernatant was centrifuged at 15,000 g for 15 min and the resulting supernatant was discarded. The pellet was resuspended in standard mannitol wash buffer (0.4 M mannitol, 1 mM EGTA, 10 mM MOPS–KOH, pH 7.2 and 0.1% (w/v) BSA) and the two previous centrifugation steps were repeated. Crude mitochondria in the pellet were washed by the suspension buffer (0.4 M mannitol, 1 mM EGTA, 10 mM tricine, pH 7.2) for 15 min at 15,000 g. The final pellet containing mitochondria was made to an appropriate volume with suspension buffer and used immediately for all experimental purposes without storage. The washed mitochondria were layered on a percoll gradient (2 ml 40%, 15 ml 28%, 5 ml 20% in washing buffer). After centrifugation for 45 min at 40,000 g, mitochondria were removed from the 28% and 40% interface and washed twice with washing buffer and finally suspended in suspension buffer before use.
2.3 Respiratory measurements
Oxygen consumption by isolated mitochondria was measured in a Clark-type oxygen electrode in 1 ml of reaction medium containing 0.3 M mannitol, 10 mM TES–KOH (pH 7.5), 5 mM KH2PO4, 10 mM NaCl, 2 mM MgSO4 and 0.1% (w/v) BSA. Pyruvate (5 mM), malate (0.5 mM), succinate (10 mM), NADH (1 mM), ADP (0.5 mM), KCN (0.5 mM) and n-propyl gallate (nPG) (20 μM) were added to modulate oxygen consumption rates. Under these conditions, mitochondrial respiration was defined as the O2 uptake in the absence of any inhibitor minus any residual respiration (O2 uptake in the presence of both KCN and nPG). State-III respiration was studied with the addition of ADP along with other substrates. State-IV respiration included measurement after all ADP had been exhausted. CYT pathway capacity is defined as the O2 uptake sensitive to KCN in the presence of 20 μM nPG, whereas AOX pathway capacity was defined as the O2 uptake sensitive to 20 μM nPG in the presence of 0.5 mM KCN [18]. Cytochrome c oxidase activity was measured as ascorbate (5 mM) and cytochrome c (25 μM)-dependent oxygen consumption in the presence of 0.05% (w/v) Triton X-100. Mitochondrial intactness was measured in terms of outer membrane intactness, which was determined with the method of Neuberger [19] by measuring Cyt. c oxidase activity in the absence or presence of Triton X-100 [0.05% (v/v)]. Intactness (%) is calculated as [100 – (Cyt. c oxidase activity without Triton/Cyt. c oxidase activity with Triton) × 100], whereas purity against peroxisomal contamination was checked by assaying catalase. All respiratory measurements were done at 20 °C.
2.4 Measurement of mitochondrial ATP
Mitochondria (0.1 mg) isolated from control and Al-treated cells were added to a reaction medium containing 0.3 M mannitol, 10 mM TES–KOH (pH 7.5), 5 mM KH2PO4, 10 mM NaCl, 2 mM MgSO4 and 0.1% (w/v) BSA. Pyruvate (5 mM), malate (0.5 mM) and ADP (0.5 mM) were added to this reaction medium. To this assay mixture were added 25 mM Glycyl–Gly–NaOH, pH 7.8, 15 mM MgSO4 and luciferase–luciferin (Wako Pure Chemicals, Osaka) and after thorough mixing, ATP content was measured after 5 min of incubation in a luminometer (model LB9506; EG&G Berthold, Germany), as described earlier [20].
2.5 Detection of mitochondrial ROS ( and H2O2)
The mitochondrial superoxide radical was quantified by following the method of Purvis [21] using percoll-purified mitochondria. NADH-dependent superoxide generation was assayed at 25 °C by the superoxide dismutase (SOD)-sensitive rate of oxidation of epinephrine to adrenochrome with an increase in absorbance at 480 nm using an extinction coefficient of 4.0 mM−1 cm−1. A non-enzymatic assay according to Snell and Snell [48] was used for measuring H2O2 production in isolated mitochondria. To 880 μl of double-distilled water, 20 μl of mitochondrial suspension was added, followed by addition of 100 μl of titanium sulfate. The reaction mixture was incubated for 15 min at room temperature and the oxidation of titanium sulfate was observed at 410 nm (UV160, Shimadzu, Japan). Absorbance was converted into H2O2 concentrations by using a H2O2 standard curve.
2.6 Mitochondrial protease assay
Assay of protease activity was done by measuring the rate of degradation of resourfin-labelled casein. Isolated mitochondria were incubated with 200 μl of 0.2 M Tris-HCl, 10 mM MgCl2, 10 mM CaCl2, 50 mM ATP, 1% (w/v) resourfin-labelled casein, pH 7.5 at 25 °C for 16 h by adding 480 μl of 5% (w/v) trichloroacetic acid, then the reaction was terminated. The mixture was centrifuged at 20,000 g for 5 min. The supernatant (400 μl) was taken and added to 600 μl of 0.5 M Tris-HCl (pH 8.8) to precipitate undigested casein, and the absorbance was measured at 574 nm. Protease inhibitors, 1 mM AEBSF (4-(2-aminoethyl)-benzenesulfonylfluoride hydrochloride) or 1 μM pepstatin were added respectively to the reaction mixture separately.
2.7 Inner membrane potential () measurement in isolated mitochondria
Mitochondria (0.1 mg protein/ml) were incubated in a buffer (220 mM sucrose, 68 mM mannitol, 10 mM KCl, 5 mM KH2PO4, 2 mM MgCl2, 500 μM EGTA, 5 mM succinate, 2 μM rotenone and 10 mM HEPES, pH 7.2) supplemented with 5 μM of rhodamine 123 for 5 min and the -dependent quenching of rhodamine fluorescence (excitation 490 nm, emission 535 nm) was measured continuously in a spectrofluorometer.
2.8 Measurement of Mitochondrial Permeability Transition (MPT)
Membrane Permeability Transition (MPT) was assayed by measuring mitochondrial swelling as described by Pastorino et al. [22], with some modifications. Mitochondria were suspended in a medium containing 220 mM sucrose, 10 mM Tris-MOPS and 10 μM EGTA, pH 7.4. One millimole of pyruvate and 1 mM of malate were added as respiratory substrates for complex I, and MPT was initiated by the addition of 10 μM CaCl2. Mitochondrial swelling was measured as a decrease in absorbance at 546 nm (Shimadzu, Japan). Swelling was further tested for its sensitivity to 1 μM cyclosporine A (CsA).
2.9 SDS-PAGE and western-blot analysis
Mitochondrial proteins (25 μg) were separated in a 10% (w/v) SDS-PAGE. After electrophoresis, proteins were visualized using CBB staining or transferred onto a PVDF membrane in a transfer buffer (Tris-Cl, pH 8.2, glycine, SDS and <15% methanol). Blocking of the membrane was performed by using a solution of Tris-buffered saline containing 0.3% Tween 20. Cytochrome c on the membrane was probed with a primary antibody against mouse cytochrome c (1/500 dilution, Sigma) overnight. After three vigorous washings in the same blocking buffer for 15 min, each membrane was incubated with an anti-mouse conjugated with horseradish peroxidase conjugate (1/200 dilution, Sigma) for 3 h at room temperature. After several washes in the blocking buffer, the membrane was transferred to appropriate assay reagents for chromogenic development.
2.10 Measurement of Ca2+ transport across mitochondrial inner membrane
Transport of Ca2+ into mitochondria was measured as described [23] with a metallochromic indicator Arsenazo III using the dual wavelength 665 nm and 685 nm in a dual beam UV-Visible spectrophotometer (UV160, Shimadzu, Japan). The assay was performed at 25 °C in a reaction mixture containing 0.25 M sucrose, 25 μM EGTA, 25 μM Arsenazo III, 10 mM HEPES-Tris (pH 7.4) and 0.1 mg mitochondrial protein; change in absorbance indicates Ca2+ transport across the inner mitochondrial membrane.
2.11 Transmission Electron Microscopy (TEM)
For transmission electron microscopy, tobacco cells were collected after treatment with or without 50 μM Al for 18 h and fixed in 2.5% glutaraldehyde in 0.05 M PIPES buffer (pH 7.2) for 1 h. Cells were post-fixed for 2 h at 4 °C with 2% osmium tetroxide in 0.1 M cacodylate buffer. After several washes with distilled water, cells were stained en bloc with 2% (w/v) uranyl acetate and kept overnight at 4 °C. After thorough washing with distilled water, cells were dehydrated with graded series of ethanol and embedded in 50% Epon/50% Spurr's resin and kept overnight at 68 °C for polymerization. Ultrathin sections (60 nm) with pale gold to silver reflectance were cut with a diamond knife using an ultramicrotome and mounted on copper grids. Post staining of the sections were done with 2% (w/v) uranyl acetate and Reynold's lead citrate for 7 min each and observed with a Hitachi H-7000B transmission electron microscope (H-7000B, Hitachi, Japan).
2.12 In situ TUNEL assay
Nuclear DNA fragmentation was identified in situ using the TUNEL staining kit as per the manufacturer's instruction. The kit detects free 3′-OH groups of degraded nuclear DNA. Cells were fixed in 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS, pH 7.2) at 4 °C and subsequently transferred to PBS containing 0.5% Tween 20. After washing with distilled water, cells are treated with Proteinase K for 30 min at 37 °C. After washing with PBS, cells are incubated with a TUNEL reaction mixture containing fluorescein-dUTP for 1 h at 37 °C. Cells were counterstained with propidium iodide (0.5 μg/ml) for 20 min. After rinsing with PBS, cells are mounted in glycerine (90% glycerine, 10% PBS) and viewed under a fluorescence microscope, Axiovert S100 (Zeiss, Germany).
2.13 Statistical analysis
Every experiment was repeated. Data presented are the means ± standard errors (SE) of at least three replicates from three independent experiments (see figure legends).
3 Result
3.1 Aluminium causes respiratory inhibition in tobacco cell's mitochondria
In order to determine the mechanism of growth inhibition of tobacco cells under Al stress, respiratory characteristics of mitochondria were analysed. The cells were treated with or without (control) 50 μM Al for 18 h in a Ca-medium containing 3 mM of CaCl2 and 3% sucrose. The Al-treated cells showed a growth capability of 35 ± 6% of that of control cells during post-Al treatment culture in nutrient medium. The crude mitochondrial fraction was prepared from tobacco cells using the protocol described in Section 2. Compared to the mitochondria isolated from control cells, the mitochondria isolated from Al-treated cells exhibited a 23% decrease in state-III respiration and an 18% decrease in state-IV respiration (Table 1). Using succinate as the sole electron donor, the respiratory rate decreased by 16% as compared to control mitochondria. Both the capacities of AOX and CYT pathways of the mitochondria from Al-treated cells showed a 9% decrease as compared to control. The outer membrane of the mitochondria from Al-treated cells showed a 25% decrease in intactness as compared to control mitochondria (Table 1). In accordance with the respiratory inhibitions, a 20% decrease in the intramitochondrial ATP content was observed under Al treatment (Table 1). In vitro treatment of mitochondria with Al showed a little decrease in respiratory rate when studied using different electron sources (Table 2).
O2 consumption characteristics and ATP content of mitochondria isolated from control and aluminum treated cells
| Treatment | O2 consumption (nmol min−1 mg protein−1) | Intactness (%) | ATP (nmol min−1 mg protein−1) (%) | ||||
| State-III | State-IV | Succ. | AOX (%) | CYT (%) | |||
| Control | 109.0 ± 28 | 49.0 ± 16.8 | 77.8 × 19.8 | 100 | 100 | 72 | 24.0 ± 0.01 |
| (100) | (100) | (100) | (100) | ||||
| Al (50 μM) | 84.4 ± 21 | 40.0 ± 13.7 | 65.0 ± 16.6 | 91 | 91 | 55 | 19.5 ± 0.01 |
| (77) | (82) | (84) | (80) |
In vitro effects of Al on respiration in isolated mitochondria of tobacco cells
| Substrate | −Al | +Al |
| State-III | 115 ± 21 | 109 ± 15 |
| Pyr + Mal | 79 ± 8 | 67 ± 7 |
| Succ | 74 ± 3 | 70 ± 2 |
| NADH | 73 ± 2 | 68 ± 2 |
3.2 Aluminium causes ROS ( and H2O2) production in mitochondria
It was examined whether the inhibition of respiratory functions in isolated mitochondria from Al-treated cells is associated with the simultaneous production of ROS in mitochondria or not. This was substantiated quantitatively by measuring superoxide production in percoll-purified mitochondria. A substrate-dependent increase in superoxide production was observed and it was found to be enhanced in mitochondria isolated from Al-treated cells as compared to control (Fig. 1A). The SOD-sensitive production was found to decrease slightly in the presence of complex-III inhibitor, antimycin. To check the production of H2O2 in isolated mitochondria, quantitation of H2O2 content in isolated mitochondria in the presence or absence of substrates for state-III with or without antimycin was performed (Fig. 1B). The Al-induced increase in peroxide content was visible in the mitochondria isolated from Al-treated cells, especially in the presence of state-III substrates. However, the effect of antimycin was not clearly observed quantitatively. These results indicate strongly the ROS production and its dependency to mitochondrial electron transport in tobacco cells under Al stress.
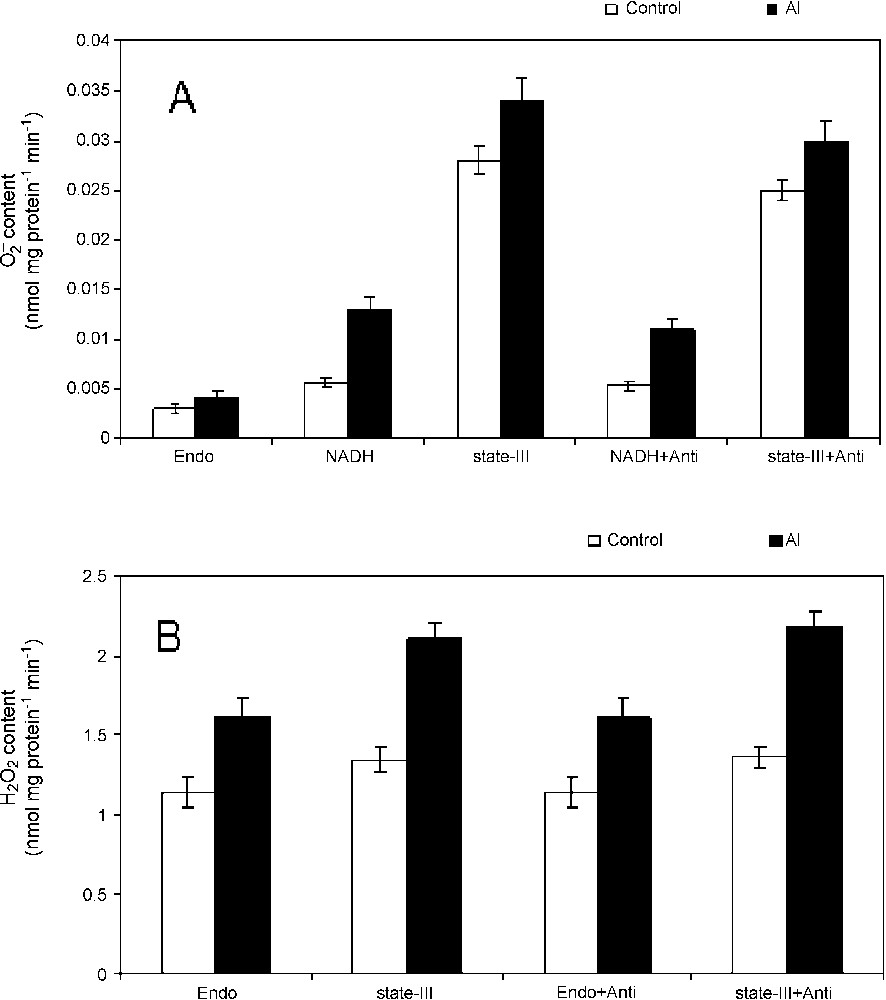
Superoxide radicals and hydrogen peroxide production in isolated mitochondria. Mitochondria were isolated from tobacco cells treated with Al for 18 h. Superoxide radical (A) and hydrogen peroxide (B) were quantified in mitochondria isolated from control (open bar) and Al treated cells (closed bar) directly or under state-III conditions in the presence or absence of antimycin. Others were same as materials and methods.
3.3 Aluminium causes opening of Mitochondrial Permeability Transition (MPT) pore, collapse of inner membrane potential, activation of protease and imbalance in calcium uptake in mitochondria
Addition of CaCl2 (10 μM) to the mitochondria isolated from control cells and Al-treated cells showed a high-amplitude swelling, suggesting the opening of mitochondrial MPT pore. Though there was little opening of MPT pore in the control mitochondria, a pronounced increase in the opening of MPT pore was noticed immediately after the start of assay, and no lag time of opening was observed in the mitochondria isolated from Al-treated cells (Fig. 2). The opening of MPT pore remained almost consistent throughout the assay (Fig. 2A). Both dithioerythritol (DTE) and Pi were found to modulate the pore opening in the absence and presence of Al treatments (Fig. 2B and C). However, the maximum opening of MPT pore was noticed in the presence of DTE and Pi together and in mitochondria isolated from Al-treated cells. Cyclosporine A (CsA), a diagnostic drug known to regulate the MPT pore by closing it [24], was found to be insensitive in tobacco cell mitochondria assayed for MPT pore opening in the presence of Ca2+ and also with Ca2+ and DTE. Rather than that, a normal swelling was observed in CsA-treated mitochondria. Generally, in animal cells undergoing programmed cell death follows the opening of the MPT pore that results in leaking out of cytochrome c (Cyt. c) and other matrix related proteins that help in executing the programmed cell death (PCD). In order to determine whether the tobacco cells undergoing programmed cell death during Al stress follow the similar mitochondrial route via MPT pore, we performed the immunodetection of cytochrome c in mitochondria. As revealed in western blots, the release of cytochrome c was marked in mitochondria isolated from Al-treated cells with a significant MPT pore opening (Fig. 2D).
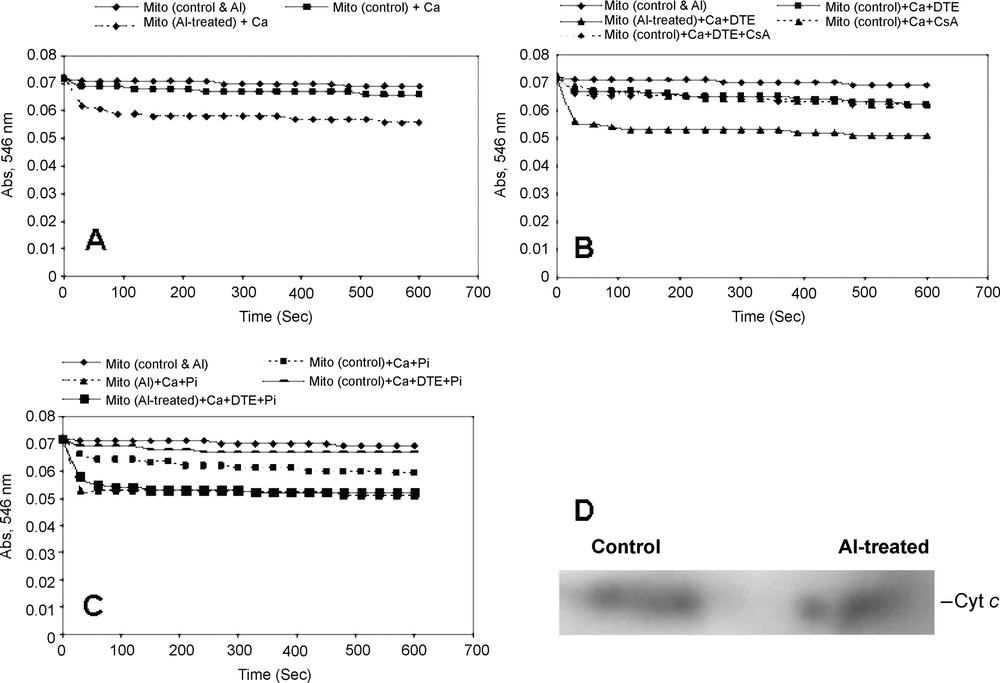
Calcium-dependent opening of mitochondrial permeability transition pore (MPTP) and immunodetection of cytochrome c of the mitochondria isolated from control and Al-treated tobacco cells. Mitochondria were isolated from tobacco cells treated with or without Al (50 μM) for 18 h, as described in Table 1. (A) Ca2+-dependent swelling of isolated mitochondria was observed in control and Al treatment conditions. (B–C) Regulation of opening of MPTP by the addition of Ca2+, dithioerythritol (DTE) and Pi separately or together. 1 μM CsA was added to test the sensitivity of tobacco cell's MPTP opening. (D) Isolated mitochondrial proteins were separated in 10% SDS-PAGE, transferred to PVDF membrane by Western blotting followed by immunodetection of cytochrome c and development by chromogenic reagent.
Rhodamine 123 (Rh-123) was used for monitoring the mitochondrial membrane potential shown by Emaus et al. [25] in isolated mitochondria. It was shown that energization induced a red shift and quenching of Rh-123 fluorescence, suggesting dye accumulation to be a sensitive probe for the mitochondrial inner membrane potential (). As an indication of highly functional mitochondria, a decrease in Rh-123 fluorescence under control condition reflects an intact inner membrane, maintaining its . We observed well maintenance of in control cell's energized mitochondria using succinate as the respiratory substrate, with an increased fluorescence quenching of Rh-123 into the matrix and this quenching maintained a steady state (Fig. 3). However, in the case of mitochondria isolated from Al-treated cells, a decrease in Rh-123 quenching from the beginning of the assay was observed, suggesting a collapse of inner mitochondrial under Al toxicity (Fig. 3).
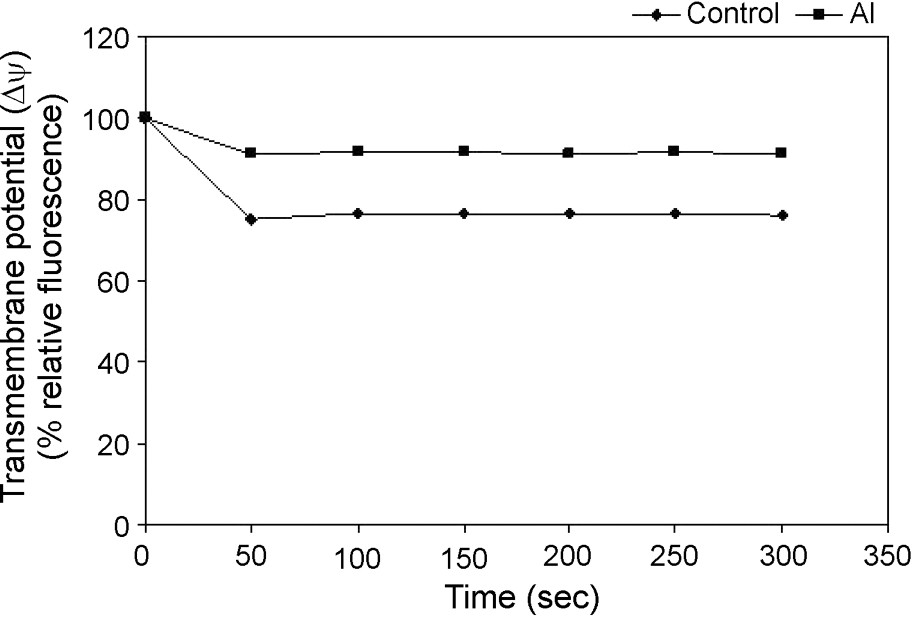
Changes in mitochondrial inner membrane potentials in isolated mitochondria from control and Al-treated cells. Mitochondria were isolated from tobacco cells treated with or without Al (50 μM) for 18 h as described in Table 1. Mitochondria were energized by the complex-ll substrate and the Rh123 quenching was monitored spectrofluorometrically. Others same as in §Materials and Methods.
The kinetics of energy-dependent Ca uptake of the isolated mitochondria was examined. The addition of 100 μM Ca2+ triggered a rapid uptake of Ca2+ into the mitochondrial matrix, as shown by a decrease in absorbance of Ca-specific metallochromic dye. This was further checked with the addition of calcium ionophore A23187. However, the uptake of Ca2+ was severely reduced in isolated mitochondria from Al-treated cells, suggesting an increase in matrix volume because of depolarization of the inner mitochondrial membrane (Fig. 4). This result showed a strong correlation between Ca-uptake and . To investigate a possible activation of specific mitochondrial proteases that affect mitochondrial respiratory chain components and other matrix proteins under Al toxicity, we studied the ATP-dependent protease activity of mitochondria isolated from control cells and Al treated cells. A slight increase (8%) in the ATP-dependent mitochondrial protease activity was observed in mitochondria isolated from Al-treated cells as compared to that of control cells (Fig. 5). The ATP-dependent activation of protease activity observed in the mitochondria from Al-treated cells was found to decrease in the presence of a serine protease inhibitor (AEBSF) addition, but not by another inhibitor, i.e. aspartate protease (pepstatin).
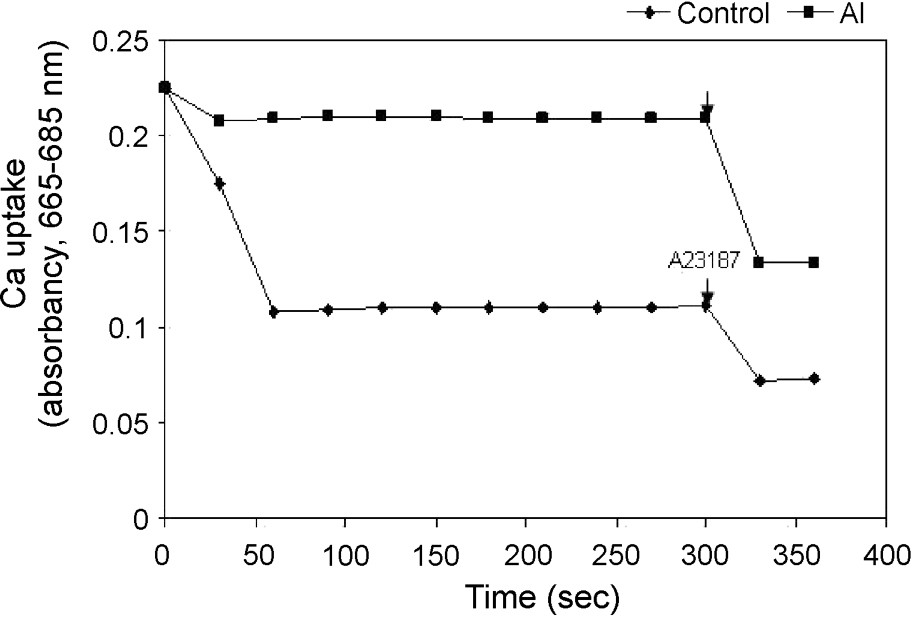
Uptake of Ca2+ by isolated mitochondria of control and Al-treated cells and the effect of Ca-ionophore A23187 on the release of Ca2+ from mitochondria as seen by Ca2+-dependent changes in absorbance. Others same as in §Materials and Methods.
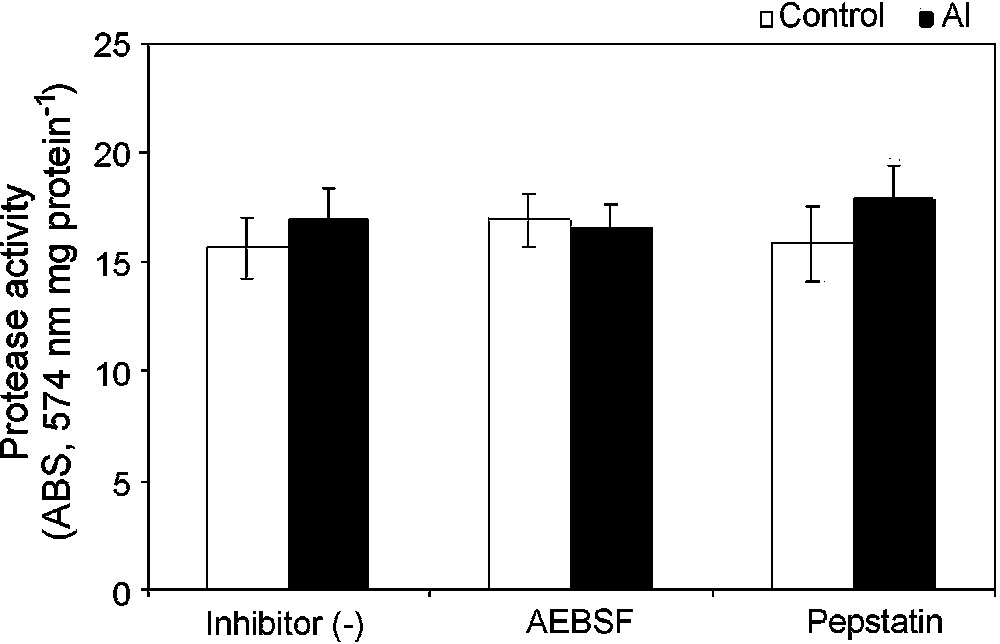
Mitochondrial protease activity in isolated mitochondria from control (open bar) and Al-treated (closed bar) cells. Isolated mitochondria were ruptured with Triton-X 100 and incubated with resourfin-labelled casein in the presence of ATP at 25 °C for 16 h. Proteolytic degradation of the casein released resourfin was detected at 574 nm. Protease inhibitors (AEBSF and pepstatin) were added as described in §Materials and Methods.
3.4 Aluminium affects the mitochondrial structure as revealed by Transmission Electron Microscopy and causes PCD assayed by in-situ TUNEL
Ultrastructural analysis was done with tobacco cells treated for 18 h with or without 50 μM AlCl3 (Fig. 6). Most of the cells maintained normal structures under Al stress. However, some cells (30%) exhibited ultrastructural aberrations induced by Al. In control cells, the nucleus remained normal with an intact nuclear membrane. But in Al-treated cells, outer membrane of the nucleus showed shrinkage and the presence of many electron-dense bodies (Fig. 6A and B). In control cells, the cell wall and cell membrane remained almost intact and it was quite difficult to distinguish between them (Fig. 6C). However, in Al-treated tobacco cells, the shrinkage of the cytoplasmic region was clearly visible, with the formation of plasma membrane blebbings (Fig. 6D). Al-treated cells maintained as many mitochondria as the control cells and almost 30% of the mitochondrial population maintained normal morphology in conformity with the growth inhibition and respiratory inhibition results. However, about 70% of the mitochondria exhibited abnormalities in Al-treated cells, such as swelling of mitochondria with abnormal outer membrane architecture, breakage of cristae, presence of electron-dense particles and sometimes bursting of outer membrane, whereas the mitochondria of control cells maintained normal globular morphology and membrane structures (Fig. 6C–E). These ultrastructural data support strongly our biochemical data, suggesting an Al-induced opening of outer membrane MPT pore with a subsequent high-amplitude swelling. Numerous vacuoles were observed in Al-treated cells as compared to control cells (data not shown). All these abnormalities observed in Al-treated cells fulfill the regular hallmarks of apoptosis, as reported in animal and plant cells [25].
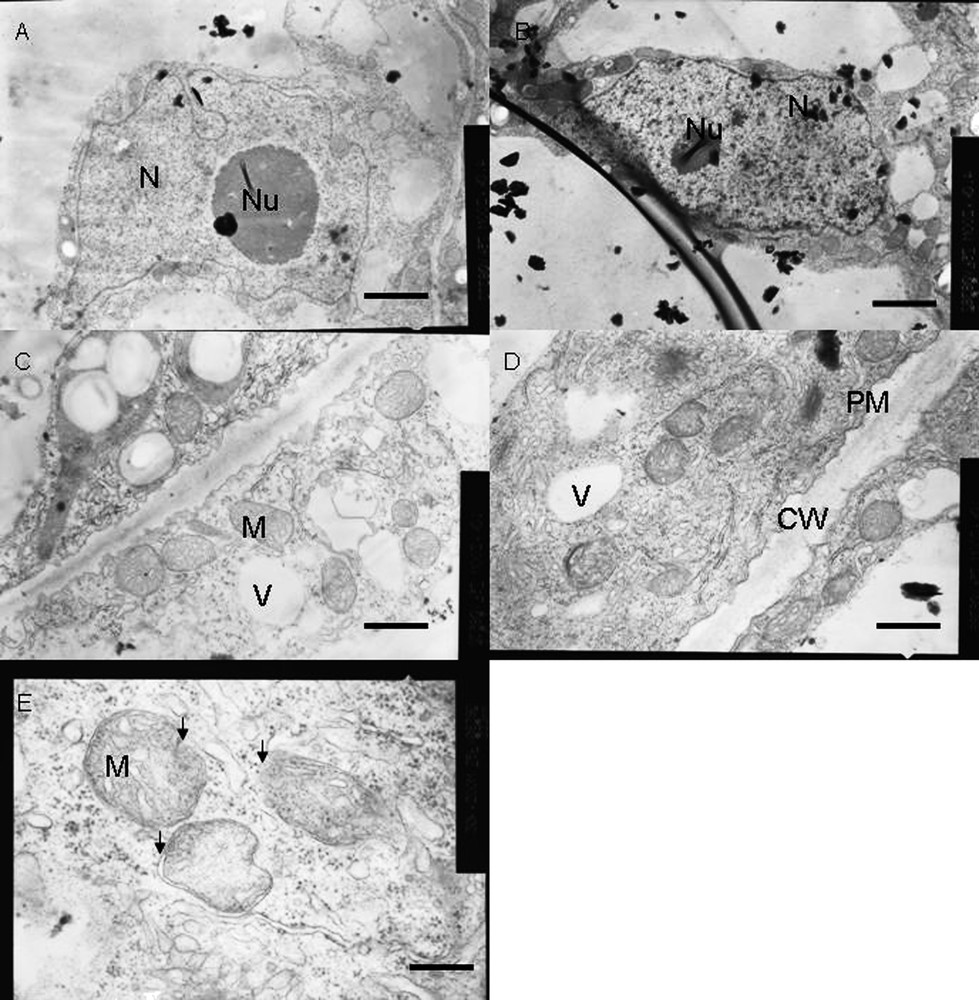
Ultrastructural changes in tobacco cells under control (A, C) and Al-treatment (B, D and E) conditions. Compared to control cells, Al-treated cells (50 μM Al, 18 h) showed, shrinkage of nuclear blebbing with early symptoms of apoptotic nuclear condensation, and plasma membrane blebbing with symplastic shrinkage. Though most mitochondria maintained normal morphology a loss of mitochondrial shape associated with disruption of internal cristae structures with highly dense electron bodies inside them was visible in Al-treated cells. Bars represent 2.5 μm (A, B), 1.0 μm (C, D) and 0.33 μm (E). N – Nucleus, Nu – Nucleolus, CW – Cell wall, PM – Plasma membrane, V – Vacuole and M – Mitochondria respectively. Arrows in (E) indicated structural deformities in mitochondria.
In situ TUNEL assay strongly suggested the presence of nuclear fragmentation in Al-treated cells that were stained fluorescent yellow with fluorescein-dUTP that binds to free 3′-OH groups resulting from nucleosomal fragmentation as compared to control cells (Fig. 7).
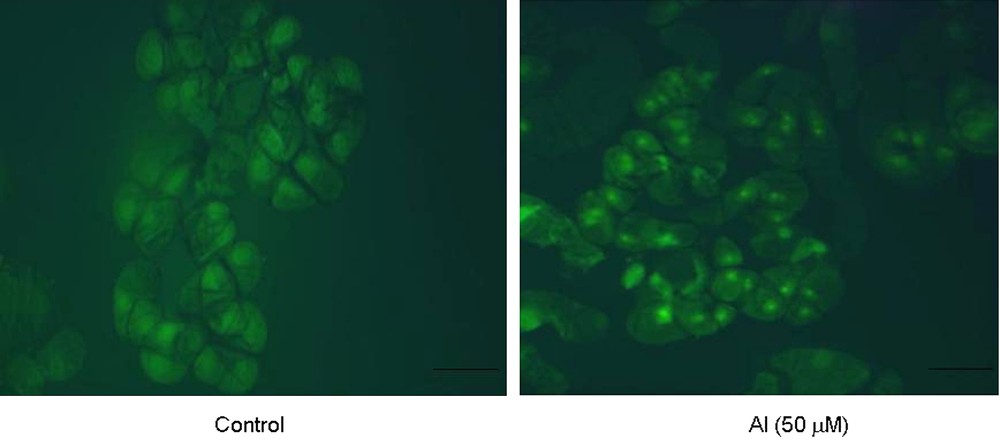
In situ detection of nuclear fragmentation in tobacco cells under control and Al (50 μM) treatment for 18 h by TUNEL assay technique. Bars = 50 μM. (For visualizing this figure in colour, see the web version of this article.)
4 Discussion
Our investigation on mitochondria isolated from control and Al-treated tobacco cells and from mitochondria isolated from root apex of pea under Al treatment clearly showed that treatment with Al affected the mitochondrial functions, namely respiratory inhibitions under both phosphorylating and non-phosphorylating conditions, enhanced production of ROS ( and H2O2), opening of MPT pores, collapsing of inner membrane potential, activation of mitochondrial proteases, regression in Ca2+ uptake, and ultrastructural changes. Our results showed clear inhibition of respiration in both the AOX- and CYT-pathway capacities respectively in isolated mitochondria of tobacco cells and in pea roots under Al stress. Inhibition of electron flow leading to a decreased respiration has earlier been shown indirectly, using whole cells of tobacco under Al treatment [9]. It has not yet been elucidated how Al ion affects mitochondrial functions. Although it seems that most Al remains in apoplasts, it is still possible that the inhibition of respiration may be possibly due to the binding of trace amounts of Al to functional electron transport chain proteins and its subsequent interactions with the Cu and Fe present in the electron carriers of mitochondria. However, in vitro treatment experiment with Al negated this effect of Al, as there was very little decrease in respiration. The in vitro Al effect on respiration was not so much marked as most of the Al gets precipitated out at almost neutral pH (pH 7.5) used for respiratory analysis and Al toxicity is perceived at acidic pH. Perhaps some other signal induced by Al toxicity may be involved in mitochondrial alteration of function, as most Al bind to apoplast, suggesting possibly its absence in the mitochondrial membrane after a cellular uptake or some other signals like ROS which potentially cause mitochondrial alterations. In vitro and in vivo studies with various metal cations in potato, corn and pea mitochondria have showed respiratory inhibitions [26–28]. In animal mitochondria, there were reports of inhibition of cytochrome oxidase by Pb, Cd and Hg and complex-III activity by the Zn ion [5,29]. Our results are well in conformity with Dixit et al. [27], who reported that chromium (Cr6+) is found to affect almost all the electron transport carriers in wheat root mitochondria, inhibiting respiration. AOX, a unique protein in plant mitochondria helps in diverting electrons from not undergoing univalent reductions and thus reduces the generation of ROS. The decrease in the AOX-pathway capacity in Al-treated cells may account for the enhanced ROS production under Al-stress condition, which was substantiated by our results on ROS, showing a trigger of superoxide and hydrogen peroxide production [30]. In intact tobacco cells, Yamamoto et al. [10] reported a high increase in ROS production together with decreases in respiration and cellular ATP level under Al stress. These results at cellular level and the results with isolated mitochondria shown in this paper strongly suggest that the respiratory inhibitions under Al stress decreases the mitochondrial ATP level, which will severely affect the bioenergetic conditions of the cells and may signal for a subsequent programmed cell death under energy deprivation [31].
Aluminium toxicity has been shown to be connected with ROS production and oxidative stress in tobacco cell and pea root system [10]. The relationship between ROS production and oxidative stress has been also reported with various other abiotic stress factors, resulting in necrotic lesions and PCD [8,32–34]. In non-chlorophyllous cells and roots, mitochondrion was suggested to be the chief organelle which produces superoxide anion [10]. In isolated mitochondria from Arabidopsis cells under oxidative stress, high ROS levels have been observed [5,35]. The important function of mitochondrial electron transport chain is to produce ATP coupled to the electron transport. With a limitation on ATP production or inhibition of electron transport, a leakage of electron is possible, resulting in the production of superoxide radicals. Though there was a clear increase in NADH-dependent production, with the addition of substrates for state-III, an enhancement in was observed in both control and Al-treated conditions, suggesting a threshold production of associated with simultaneous conversion of to H2O2. This ROS enhancement may be due to the damage that occurs to the respiratory chain, causing an impairment of respiration and diverting more electrons to oxygen, especially in a repressed AOX pathway capacity condition under Al stress.
Programmed cell death (PCD) is a biochemically and genetically programmed process that occurs throughout the life cycle of plants. Plant cells show many similarities of apoptosis like PCD of animal cells, especially in the early and late phases of PCD. Very little is known about the executioning mechanism of PCD in plant cells, which varies from animal cells and also among different species of plants. Mitochondria have been attributed well as the chief programmer of animal apoptosis where it undergoes a membrane permeability transition in response to apoptogenic signals followed by intramitochondrial imbalances in redox and calcium homeostasis that opens up the MPT pore, via which cytochrome c goes outside the mitochondria. In the cytosol, Cyt. c binds to Apaf1 protein that in turn activates caspase-9, forming an apoptosome. This apoptosome activates a series of other proteases, including caspase-3, which results in nuclear apoptosis and cell death. Many similarities have been found with the mechanisms seen in animal cell apoptosis in plant cell PCD. However, plant cells being different from animal cells structurally and also biochemically to some extent, they are expected to follow differences in mechanisms of PCD especially induced by abiotic and biotic stress. Cyt. c release has been seen also in plant cells undergoing opening of MPT pore and subsequent PCD under the influence of biotic and abiotic stress conditions [5,36–38]. These results emphasized a similar PCD execution step associated with Cyt. c release, which was found essential in their experimental systems. However, no cytochrome c release was observed in PCD of pollination induced petunia petals [39]. In our experimental system, the release of cytochrome c from mitochondria undergoing MPT pore opening was also observed under Al toxicity, indicating that the animal model of apoptotic cell death is followed during Al induced PCD in tobacco cells. The activation of mitochondrial proteases, depletion of ATP and the release of cytochrome c by which the mitochondria signal the cytosol for executioning the PCD. Oxidative stress has been shown to trigger the opening of MPT pore directly and possibly by elevating calcium levels indirectly [40]. The role of ROS has been well attributed to open MPT pore with studies from blockage of calcium-induced MPT pore by scavengers of ROS [41]. Mitochondria has been thought to integrate various stresses and, once triggered by calcium, increase redox changes, ATP depletion, opening of MPT pores and collapse of the inner membrane potential (), and signals the cytosol for the execution of PCD. Our results also showed insensitivity to CsA, a diagnostic drug that closes the opening of MPT pores. In some animal cells, isolated mitochondria treated with butylated hydroxytoluene and palmitic acid, and in yeast and wheat mitochondria a similar insensitivity of opening of MPT pore to CsA was noticed [15,42,43], suggesting the existence and operation of different mechanisms for the regulation of MPT pore opening in different organisms. As one of the prerequisites of PCD, accompanied with the collapse of , the mitochondrial matrix volume is affected, which releases calcium out of the matrix [15]. Our results indicated a collapse of the inner mitochondrial membrane potential () with the opening of MPT pores, as reported earlier by Yamamoto et al. [10], suggesting this to be an early event in Al toxicity. The matrix location of Ca2+ was verified by Ca-ionophore treatment and the collapse of also showed clearly the variation of Ca2+ uptake under Al treatment in mitochondria of tobacco cells, as reported earlier for wheat root mitochondria [15].
In animal systems, a specific protease system has been found in mitochondria that degrade oxidatively denatured proteins [44]. In Arabidopsis cells under oxidative stress, mitochondria have shown to induce ATP-dependent proteases that may help in degradation of oxidatively damaged proteins. Al toxicity has been shown in our results to produce ROS in mitochondria that may create an oxidative stress condition, thus damaging many important respiratory chain proteins [35]. Similar activation of ATP-dependent mitochondrial proteases under Al toxicity in tobacco cells may suggest, though to a less extent, a possible role for these proteases in the regulated breakdown of important mitochondrial proteins affected by oxidative stress as imposed by Al.
Ultrastructural analysis of Al-treated cells showed that, though the cell wall was intact, the plasma membrane seemed to have several infoldings and was separated from the cell wall, showing cell shrinkage and plasmolytic features, as reported earlier for red spruce cells and tobacco cells under Al treatment [45,46]. Mitochondria showed significant alterations in morphology with distortion of cristae, outer membrane bursting, and deposition of electron-dense bodies accounting for early impairment of respiratory functions, loss of intactness, opening of MPT pore causing high-amplitude swelling that ultimately affect PCD. Plant cells are unique in a sense that they are endowed with vacuoles that sequester many hydrolases in it. In plants, many gene-encoding hydrolases are shown to be upregulated during PCD [47]. The release of hydrolases, possibly from vacuoles in response to a signal from mitochondria, may result in plant cell PCD, which may be the reason for the appearance of numerous vacuoles under Al treatment preceding growth inhibition and PCD, with release of cytochrome c. Al-induced PCD was further evidenced by nucleosomal fragmentation in Al-treated cells, showing in situ TUNEL-positive cells as compared to control cells. From this quantum of data, a new intracellular mechanism of Al toxicity (Fig. 8) emerges in tobacco cells, which leads, via an imbalance in the mitochondrial redox and bioenergetic homeostasis, to ultrastructural changes that signal the cytosol, resulting in nuclear fragmentation and execution of cell death.
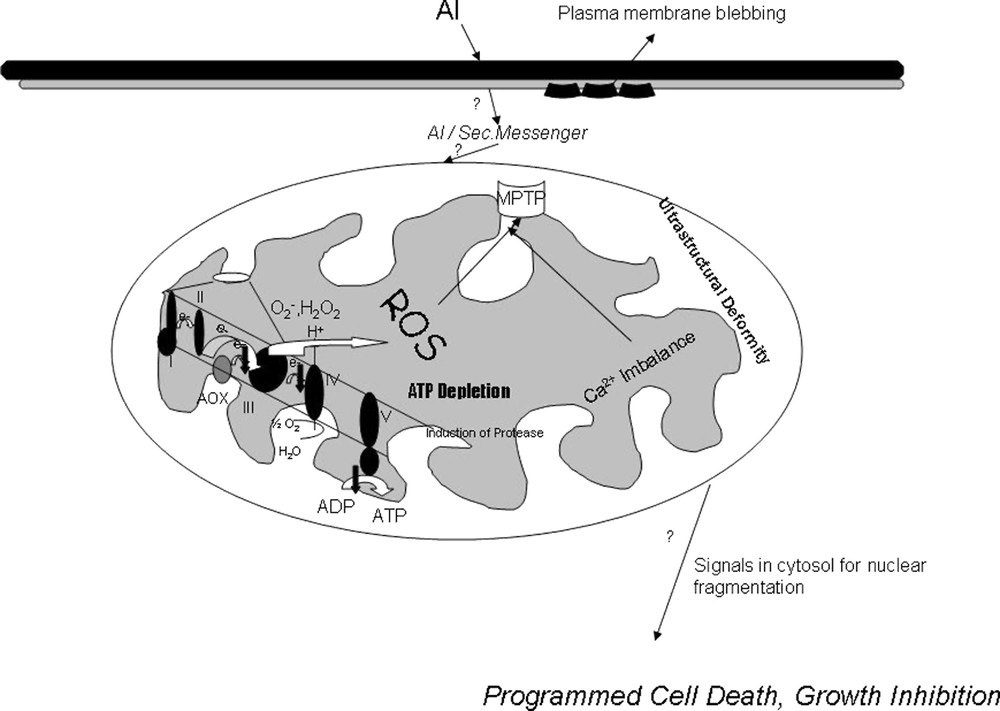
Putative Model for the Intracellular Mechanism of Aluminium Toxicity in Tobacco cells via Mitochondria. Aluminium toxicity is associated with ion pump inhibition, membrane rigidification, lipid peroxidation, membrane blebbing. Possibly some amount of Al enters the cytosol or some second messenger creates respiratory dysfunction, ROS production, Opening of MPTP, loss of inner membrane potential, Imbalance in Ca uptake, induction of proteases in mitochondria which ultimately signals for a nuclear apoptosis and programmed cell death in tobacco cells.
Acknowledgements
This research was supported by the Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan to H.M. (grant No. 14206008) and Y.Y. (grant No. 14540595), the Japan–Australia research cooperative programme (joint research project) from the Japan Society for the Promotion of Science to H.M., and the Postdoctoral Fellowship from the Japan Society for the Promotion of Science (JSPS) to S.K.P.