

1 Introduction
From the time chromosomes were shown to be the physical basis of heredity, scientists have expressed interest in the consequences of physical aberrations resulting in loss and gain of genetic material. Most of these early remarkable achievements and hypotheses originated from members of the group of Thomas H. Morgan and were developed through genetic analysis of Drosophila melanogaster mutants. A notable contribution came from Calvin B. Bridges who, soon after publishing his 1916 key PhD article on chromosomes, support of heredity [1], emphasized the interest duplicating of chromosomal regions as a mean to generate copies of identical genes as early as 1918. Such copies would be prone to accumulate independent mutations and thus acquire new functions, eventually contributing to speciation events [2]. Bridges' research on mutant flies eventually converged with his early interest in duplications to a remarkable climax, providing the basis for the discovery of HOX gene clusters [3]. This cluster appeared, indeed, to result from a series of gene and genome duplications [4]. A very early contribution to the study of segmental duplications (SDs) was made by another member of Morgan's group, Hermann J. Muller, who described X-ray irradiated flies in which a segment from the X chromosome had been duplicated and translocated onto chromosome II [5]. By observing apparent innocuousness from the presence in the same genome of these rearranged chromosomes II with intact X chromosomes, he elegantly proposed that such small duplications, eventually associated with translocations, were less likely to be deleterious than full aneuploidies, and therefore were probably an important source of genetic diversity. As we will see, his hypothesis appears amazingly accurate 70 years later. Early genetic works on filamentous fungi also revealed large duplications of chromosome segments resulting from non-reciprocal terminal translocations [6–8].
The importance of redundancy in eukaryotic genomes was nonetheless dramatically underestimated until the advent of genome sequencing and the revolution of the genomic era. Comparative genomic studies have described a diversity of genomic structures reminiscent of a variety of gene duplication mechanisms. Interestingly, whereas most eukaryotic genomes present a relatively homogenous level of genome redundancy (between 40% and 60% of genes in a given genome belong to gene families), these mechanisms appear somehow lineage-specific among species. Whole genome duplication (WGD), or polyploidy, is a phenomena originally proposed to account for the presence of two sets of identical chromosomes in maize [9] and commonly observed in plants lineages [10]. Such events have since been detected in the evolutionary history of many others species (see Jaillon, Aury and Wincker [11], this issue). However, in addition to these dramatic, relatively rare events, other mechanisms have contributed to generate the genetic redundancy observed in most eukaryotic genomes. Tandem gene duplications are easily recognizable by the physical proximity between the repeated gene copies. On the other hand, dispersed duplicates generally result from the retroposition of mRNA with the limitation that this mechanism often results in the formation of pseudogenes. Finally, a large proportion of eukaryotic genomes appear to have been extensively reshaped by duplications of large DNA segments. Initially considered as peculiar features of genome architecture, interest in SDs intensified with the release of complete genome sequences. A remarkable breakthrough occurred when it was established that recent duplications appear to constitute more than 5% of the human genome [12,13].
2 Segmental duplications among eukaryotic genomes
2.1 Segmental duplications and copy number variation regions in the human genome
The recent interest in segmental duplications as structural elements of eukaryotic genomes has without doubt been magnified by the discovery of their abundance in the human genome sequence. By considering the overrepresentation of some regions among whole-genome shotgun sequence reads, Eichler and collaborators identified potential duplicated regions and were thus able to generate the first genomic map of SDs [12]. With a later refinements, the original predictions proved to be close to the actual map, with approximately 5% of the genome consisting in duplications of DNA segments >1–5 kb sharing >90% identity, usually <300 kb although some of them cover hundreds of kb [14,15]. The nature and distribution of these SDs within the human genome present various levels of heterogeneity. Current analyses estimate that SDs cover approximately 150 Mb of the genome, and are distributed between pericentromeric (31%), subtelomeric (2%) and interstitial (e.g. between pericentromeric and subtelomeric regions, 67%; Fig. 1) regions. In addition, both pericentromeric and subtelomeric regions are enriched in inter- versus intra-chromosomal SDs. Also, the global chromosomal distribution is not uniform, with enriched and impoverished whole chromosomes or whole chromosomal arms. Furthermore, SDs in interstitial regions tend to be interspersed, either as interchromosomal or as intrachromosomal but separated by large distances. The tendency towards SD scattering seem to be specific to the hominoid genomes (Human and chimpanzee) given that in most other sequenced species SDs mainly appear as clusters of tandemly duplicated regions.
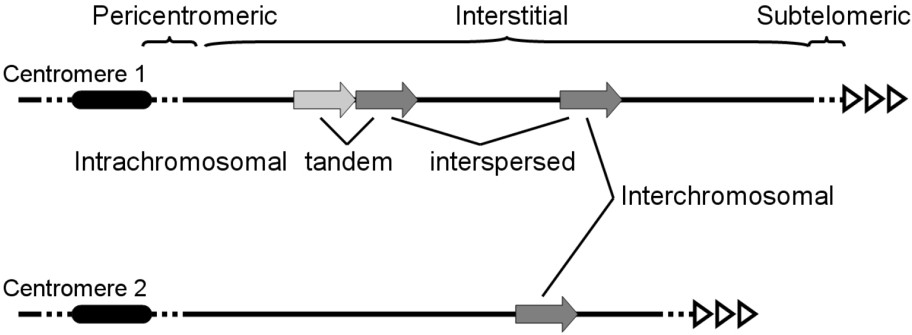
Different classes of SDs, as determined from their chromosomal localization and intra- and interchromosomal positions. The original DNA segment is schematized as a large light-grey arrow and the duplicated copies as repeated dark-grey arrows. Centromeres and telomeres are represented by black ovals and series of open triangles, respectively.
The suspected dramatically fluidic nature of the human genome has been recently confirmed by the analysis of deletion/insertion and duplication polymorphism between human individuals. Large changes in stretches of genomic DNA segments, generically labeled copy number variations (CNVs), have been detected through SNP genotyping [16–18] and comparative genomic hybridization (CGH) assays [19–24]. The two latest studies were performed between hundreds of individuals, and led to the first CNV maps in the human genome: polymorphic variations in DNA segments larger than 1 kb are estimated to cover from 2% (conservative estimation [25]) to 12% [20] of the genome. Not surprisingly, there is a strong bias for CNVs to be associated with the SDs characterized in the reference genome (∼25%; [20,24]). A specific mapping of CNVs on human chromosome 22 revealed that of the breakpoints intersect with SDs [26]. Although recent SDs, i.e. sharing very similar sequences, are more likely to correspond to CNV regions, this is not always the case: regions of ancient segmental duplications are still prone to exhibit polymorphism within actual populations (for mechanisms of formation, see below). The importance of CNVs, and their association with SDs in human population suggest common mechanisms of formation that still actively reshape our genome.
Both CNVs and SDs are increasingly recognized as a source of genomic diseases. SDs are responsible for numerous gene-dosage imbalances, gene fusions and disruptions events. Also, there is a strong association between SDs and sites of recurrent pathogenic rearrangements such as mental retardation related deletions [27,28]. CNVs are associated with the development of various human cancers [29–32] and the evolution of genetically complex phenotypes including predisposition to autism [33], epilepsy [34], Alzheimer disease [35], glomerulonephritis [36], systemic autoimmunity [37] and susceptibility to HIV/AIDS infections [38]. This strong association between SDs/CNVs and diseases could be the consequence of the significant gene enrichment that is observed in these regions. It is interesting to note that this enrichment is restricted to a small subset of the core segmental duplications. Sequence divergence analysis revealed that the most recent duplications, those that have emerged since the divergence of human and chimpanzee, are centered on this small portion of core SDs [39]. These recent SDs have been fixed despite their association with a large and increasing number of diseases and chromosomal rearrangements. Although some of these SDs may be selectively neutral and be fixed in genomes through genetic drift, it is unlikely that this is the case for all of them: indeed, in some cases, the creation of gene novelties that accompanies their formation may outweighs the negative effects of both chromosomal instabilities and disease appearance.
2.2 SDs in non-human primate species
A FISH analysis and an evolutionary age estimate of SDs from three non-human primates genomes (chimpanzee, baboon and marmoset) supported that bursts in interspersed intra- and interchromosomal duplications occurred during and after the separation of great apes and human lineages from other primates [40]. Old World monkeys genomes nevertheless contain high proportions of SDs, as seen in the sequence of the Rhesus macaque (Macaca mulatta) (SD content of ∼2.5%) or of Macaca fuscata [40–42]. Human, chimpanzee and macaques shared a common ancestor approximately 25 Mya, revealing the important and rapid genomic changes that marked recent primate genome structure evolution. The analysis of the draft sequence of the chimpanzee genome underlined the importance of SDs as a source of genomic diversity between them. Whereas the genomic divergence due to single-nucleotide substitutions between the human and chimp genomes is approximately 1.2% [43], large SDs have had a greater impact (2.7%) in altering genome sequences between the two species [44]. Human and chimp share ∼66% of their duplication and carry a similar amount of sequences duplicated in one but not in the other lineage (32 Mb in human vs. 36 Mb in chimpanzee). However, a significant increase in copy number for shared duplications was identified in the chimpanzee genome (23 Mb vs. 7 Mb in human), mostly representing chimpanzee-specific hyperexpansion of a small number of chromosomal regions. The lineage-specific duplications and expansions induce changes in gene expression that may contribute to the phenotypic differences observed between human and chimpanzee [45,46]. Overviews of duplication polymorphism, corresponding to gain and loss of DNA segments between the genomes of great apes (chimpanzee (Pan troglodytes), bonobo (Pan paniscus), gorilla (Gorilla gorilla) and orangutan (Pongo pygmaeus)) were obtained from CGH onto on human BAC arrays [42,47]. Overall, more than 300 sites of copy number variations, ranging in size from 40 to at least 175 kb, were observed in all chromosomal regions (centromeric, telomeric and interstitial). Most of these CNVs were lineage-specific, the gorilla genome showing the highest frequency of variations and the orangutan, the lowest. Again, human specific CNVs mostly intersect with SDs. Some of these regions also correspond to karyotypic breakpoints, illustrating their importance in promoting chromosomal evolution.
2.3 SD in other organisms
The availability of genome sequences for an expanding number of species allows the depiction of a more precise picture of the role played by segmental duplications in structuring eukaryotic genomes. Compared to the particularly rich and dynamic history among primate species, SD formation and content in other vertebrate genomes is somewhat lower, although can remain important. Rat and mouse genomes comprise ∼2–3% of SDs [14,48,49], whereas the SD content for the dog and cat genomes, whose sequences have been released, has not yet been clearly established [50,51]. In each lineage, SD genealogy is reminiscent of the history of primate's SDs, as it also appears as the combination of more or less steady levels of duplications, eventually associated with periods of sudden activity resulting in a burst of duplicated sequences within a genome [49]. When compared to the human and chimpanzee, SDs in other mammals are similar in size and present the same enrichment at pericentromeric and, to a lesser extent, at subtelomeric regions, thus generating a pattern which seems characteristic of mammalian genome architecture [49,52]. However, some notable differences exist: for instance, the duplications in interstitial regions tend to occur as intrachromosomal tandems in rat and mouse, unlike in humanoids (above).
Other vertebrate genomes sequenced so far are relatively compact compared to mammalian genomes and maybe because of this present a significant lower extent of SDs that appear significantly smaller in size. For instance, segmental duplications in the chicken (Gallus gallus) genome are usually smaller than 10 kb and appear mostly as intrachromosomal tandems [53].
In other species, the proportion and arrangements of detectable SDs present significant variations. For the worm Caenorhabditis elegans and the fruit fly Drosophila melanogaster, despite a significant SD content of 4.3% and 1.4%, respectively, the proportion of SDs larger than 10 kb is very low (0.7% and 0.1%) [52,54]. In addition, the contribution of recent segmental duplication is relatively limited. For instance, only 10 interspersed and 4 tandem new gene duplications were detectable in the Caenorhabditis briggsae partial genome sequence when compare to C. elegans, whereas 100 My separate the too species [55]. Few interstitial SDs have been detected in the genomes of different yeast species [56] while the vast majority of the duplicated segment characterized in S. cerevisiae relate to the whole genome duplication event that occurred in this lineage [57]. Paradoxically, yeast remains an important tool to analyze SD formation (below), although little recent duplication has been found in genomes sequenced so far.
The global picture emerging from these studies is that, although the proportion of duplicated genes in most genomes remains high, the contribution of SDs to this feature varies among lineages and periods. It is very likely, however, that bursts similar to that observed within the primate lineage will be characterized in other lineages. These studies will probably help understanding the contribution of SDs polymorphism to speciation and phenotypic variations among closely related species.
3 Mechanisms of SD formation
Numerous studies have approached some of the mechanisms by which large DNA segments can get duplicated in eukaryotic genomes. Important contributions result from both genome sequence analysis (e.g. extent of the duplicated segments, genomic localizations and nature of the breakpoints, Fig. 2), that helped to formulate hypotheses about the molecular mechanisms involved, and experimental studies, where the validity of these hypotheses can be actually tested. From these approaches, it appears that the structures pooled under the generic designation of SDs result from a large variety of molecular mechanisms. In addition, there is an important distinction that can be made between SDs according to their respective origins. On the one hand, duplicated segments can result from the unequal re-assortment of the genetic material of a progenitor cell among its offspring, and thus emerges without any effective DNA duplication. On the other hand, large duplicated segments can result from a round of untimely DNA synthesis, leading to a net increase of genetic material. In the latter scenario, an unscheduled DNA replication step must occur. Although conceptually and mechanistically dissociated, these two classes of SD involve multiple and often share similar genetic pathways. Indeed, whatever the class they belong to, an important aspect of duplications is that like for all chromosomal rearrangements, two distant DNA loci have to be rejoined. This result in the formation of a “breakpoint” sequence, whose nature provides important clues about the genetic pathway involved in its generation (Fig. 2A). Classical hypotheses about breakpoint formation imply a DNA double-strand break (DSB) as the initiating event. However, some recent findings suggest that this might not be an absolute pre-requisite ([58]; below).
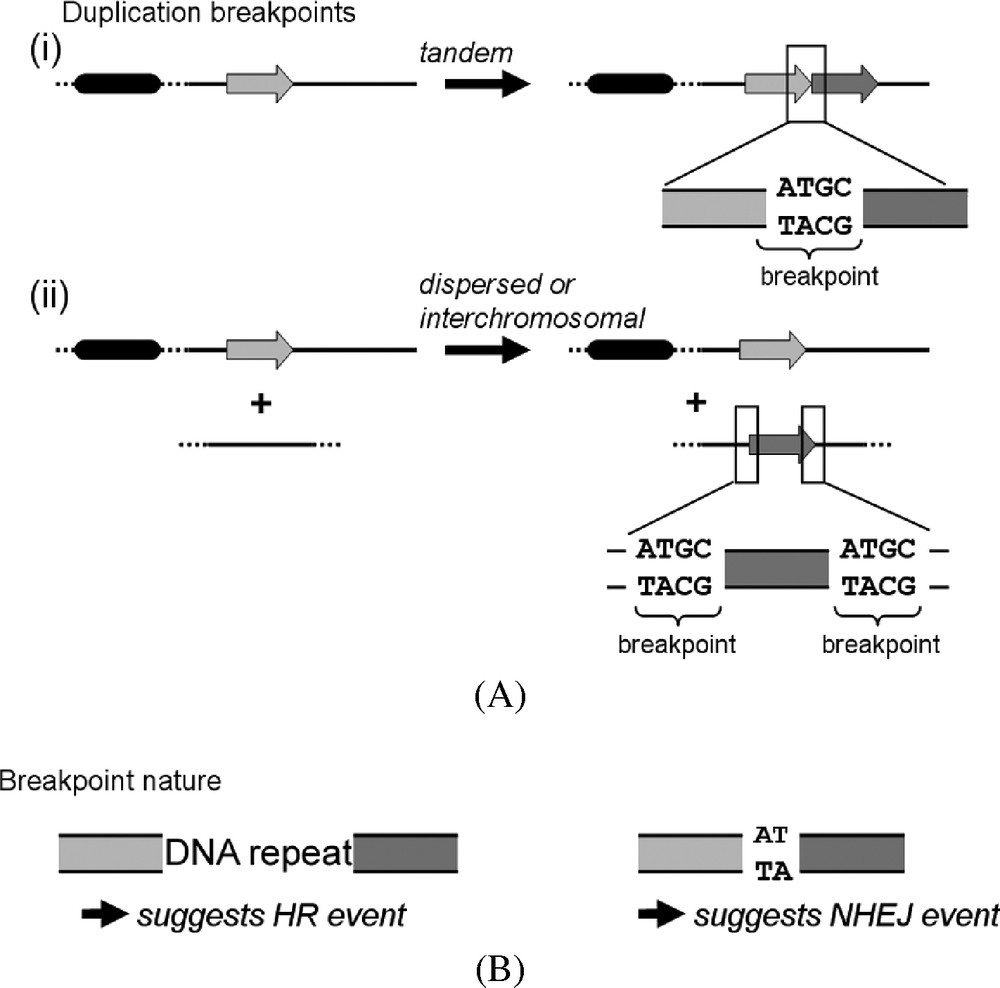
SD breakpoints. Symbols are identical to Fig. 1. (A) Characterization. For direct tandem duplications (i), the breakpoint sequence corresponds to the largest DNA sequence common to both extremities of the duplicated segment that overlaps in-between the two duplicated segments (e.g. the ATGC sequence). (ii) Interspersed intra- or interchromosomal duplications. Breakpoints correspond to the sequence shared by the extremities of the duplication and the insertion locus. In the given example both the extremities of the duplicated and the insertion locus have the same ATGC sequence. (B) Characteristic breakpoints structures of SDs resulting either from HR (left) and NHEJ (right) events.
Briefly, two DSB repair pathways coexist in most eukaryotic cells: non-homologous end joining (NHEJ) and homologous recombination (HR, which consists of different categories, [59]). Maintaining an ability to repair DSBs is an essential function for a cell and both pathways are holders of genome integrity and functionality [60]. The fundamental features of HR consist in the interaction (strand invasion) and exchanges between two homologous DNA sequences. The HR pathway can drive non-allelic homologous recombination events (NAHR), i.e. exchanges between non-allelic sequences showing a high or perfect similarity over dozens of base pairs (bp). Sometimes, a replication fork will be reassembled at the site of strand invasion, which then progresses and replicates dozens of kb in a so-called break-induced replication (BIR) reaction. More than 30 genes are involved in the yeast HR repair pathway, including the group epistatic to the pivotal Rad52 protein [59–61]. The involvement of HR in SD formation can be envisioned when the breakpoint sequence shares a large DNA sequence similarity with the two reunified DNA segments (Fig. 2B). On the other hand, NHEJ allows the ligation of two DNA ends exhibiting virtually no sequence identity. In yeast, the Mre11/Rad50/Xrs2 (MRX) complex binds to the two DNA ends of the break, holding them in the vicinity of each other [62,63]. The Ku70/80 heterodimers are then recruited at these ends, until the ligase Lig4 binds the two DNA ends together. If the two ends are cohesive, then the ligation will be perfect with no trace of the damage remaining. However, in some other cases, small deletions and insertions can eventually occur [60]. Therefore, the breakpoint sequence at the borders of SDs can also serve as a footprint to reveal an origin resulting from a NHEJ event.
3.1 Conservative mechanisms: DNA duplications without duplicating DNA
Large segmental polymorphism in a population can result in the emergence of large duplicated regions in some individual, as a direct consequence of meiosis/mitotic recombination and chromosome segregation. For instance, a translocation-based model could explain how NHEJ-mediated translocations were responsible for the segmental polymorphisms observed in human subtelomeres ([64]; Fig. 3A). This model is reminiscent of the translocation events shown to stabilize SDs in the yeast genome [65]. Dynamic subtelomeric plasticity may allow rapid generation of new alleles combinations or amplifications: for instance, Plasmodium falciparum telomeres carry large gene families involved in the adaptation of the parasite to the host [66,67]. Subsequent rearrangements can eventually disperse these regions all over the genome, ending up in the creation of true “hot-spot” of rearrangements undergoing positive selection [39].
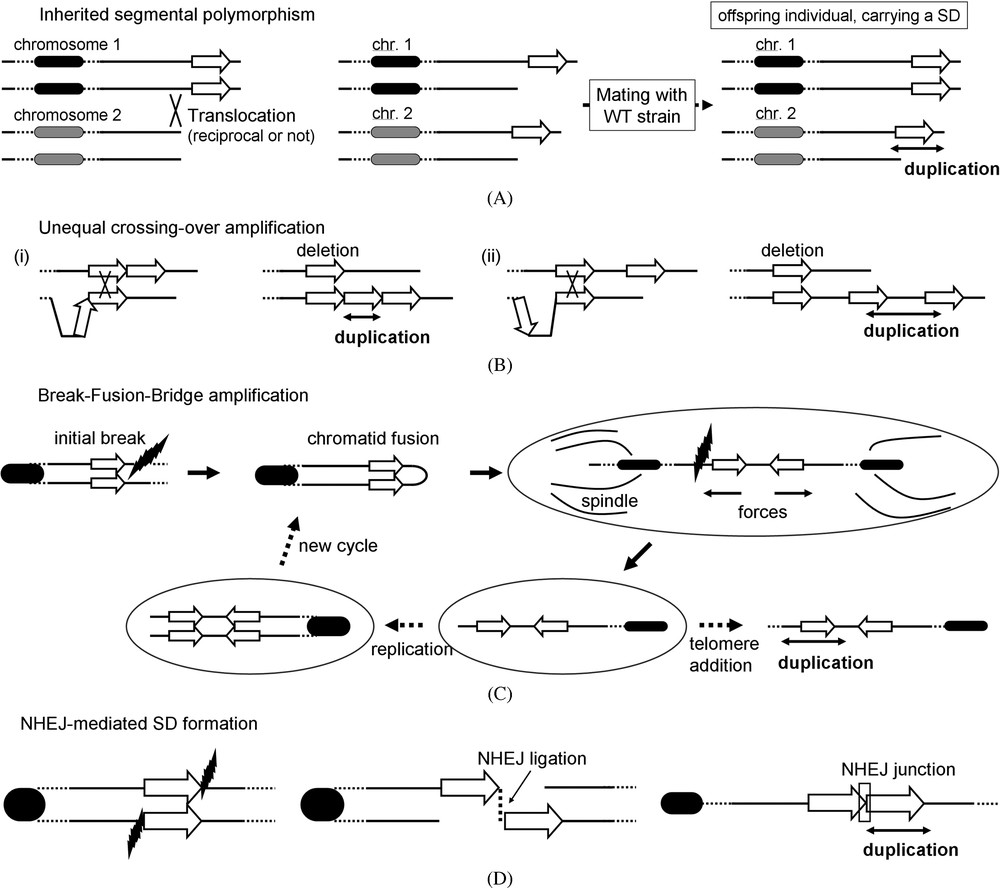
Conservative mechanisms of SD formation. (A) Inherited segmental polymorphism: a translocation of a large DNA segment from chromosome 1 to chromosome 2 can appear as a segmental duplication in offspring. (B) Unequal crossing-over between direct tandem (i) and interspersed (ii) repeats. (C) Break-Fusion-Bridge amplification. Black lightning: DSB formation along a chromatid. (D) NHEJ ligation model: two homologous chromatids each experiencing a DSB at different positions can lead to SD formation if incorrectly repaired by NHEJ.
3.1.1 The unequal crossing-over mantra
In most eukaryotic genomes, SDs consisting of DNA segments repeated as direct tandems are commonly found. Mitotic and meiotic NAHR events, resolved as unequal crossing-over, have been traditionally invoked to account for the presence of these structures within genomes [68,69]. These recombination events occur between identical regions located either within the same chromatid or on sister-chromatids. They can also occur between homologous chromosomes. These regions can be organized as direct tandems (e.g. the clusters of tandem rDNA units [70], Fig. 3B(i)) but also be separated by hundreds of kb (Fig. 3B(ii)). The size of these regions is variable and matters little, as long as they share a high identity. For instance, they can be large SDs generated beforehand, transposable elements or simple DNA regions containing one or more genes.
Larger chromosomal duplications can result from NAHR events [28]. A classical example consists in Type 1A Charcot-Marie-Tooth and Hereditary Neuropathy with liability to Pressure Palsies syndromes that result either from the gain or loss, respectively, of a 1.5 Mb segment from chromosome 17 following a NAHR event between two 24 kb SDs.
Recombination events between the repeated Alu transposable elements (∼300 bp size) have dramatically reshaped the genomic structure of eukaryotic genomes [71]. In humans, NAHR events between dispersed Alu sequences is a source of oncogenic duplications and deletions [72,73]. In the human genome approximately 27% of SDs are bordered by Alu repeats, whereas they constitute 10.6% of the total sequence [74]. As these sequences tend to be fixed within GC rich regions [75], that could explain why SDs have also colonized gene-rich regions of human chromosomes. However, the presence of repeated sequences at duplication breakpoints does not necessary reflect past occurrence of NAHR events: for instance, a study conducted in yeast revealed that the large spontaneous SDs recovered and flanked by dispersed repeated sequences (essentially Long Terminal Repeats (LTRs) from Ty retroposons [76]) actually result from BIR events (see below) [77]. In this assay, NAHR between repeated sequences are probably suppressed by the DNA divergence between LTRs.
3.1.2 Break-fusion-bridge amplification
Break-fusion-bridges (BFB) is a mechanism responsible for dynamic segmental amplification, notably within subtelomeric regions [78]. The amplification results from the fusion of two sister chromatids, after the loss of a telomere by degradation or because of a DSB improperly repaired. Telomeric proteins usually prevent such fusion from occurring, but sometimes a dicentric chromosome will be generated that will undergo a break during segregation because of the forces dragging the two centromeres towards opposite poles. As a consequence, one of the daughter cells will carry a deletion (Fig. 3C). The new break can be repaired through another chromatid fusion, followed by a new cycle, adding another duplicated unit at the end [79]. The resulting structure consists in series of direct/inverted tandems and eventually leads to gene dosage unbalance characterized in numerous tumorigenesis processes [52].
3.1.3 NHEJ
A subset of tandem SDs either observed in sequenced genomes or experimentally isolated present breakpoints made of few nucleotides, suggesting the involvement of a NHEJ event [76,80]. A conservative hypothesis would be that two sister/homologous chromatids, experiencing a DSB each at a different location and escaping integrity maintenance pathways, would be unified together (Fig. 3D). However, this mechanism appears unlikely in light of a recent study: these events are indeed suppressed in the absence of Pol32, a subunit of the major replicative DNA polymerase δ in S. cerevisiae (see below, [77]).
Although these mechanisms can result in duplications of large DNA regions, the resulting structures are not representative of the diversity of inter- and intrachromosomal SDs. Furthermore, despite the attractiveness of Alu- and other repeat-mediated SDs, such events would only effectively represent a subset of all SD junctions characterized so far [74]. In addition, the prerequisite for any NAHR events to occur consists in the initial presence in the genome of two copies of the region between which the recombination event will occur. As stated by Seymour Fogel in 1983 [81], “It is conceivable that the rate limiting step in tandem gene amplification is the very first event leading to the initial duplication. Once two or more copies of the segment are present, the likelihood of tandem amplification is much greater.” Other mechanisms must therefore play an even, if not more important, role for generating SDs so that a sequence could get duplicated without necessitating pre-existing duplicated sequences.
3.2 De-novo duplication: replication-dependant mechanisms
Invoking replication to account for SD formation may seems straightforward although the multiple roles played by replication are only emerging and remain partially unknown. Interestingly, recent data now favor original new models that are discussed below.
3.2.1 A direct, causative role for replication accidents
Only a subset of SDs breakpoints characterized in eukaryotic genomes are associated with repeated sequences (above). Other sequences, presenting the physicochemical properties of “fragile site”, i.e. regions where DSBs of the DNA molecule are more likely to occur, have been shown to be enriched at breakpoints as well [82]. Fragile sites are actually often associated with hotspots of eukaryotic chromosomal rearrangements as revealed experimentally in several studies, conducted notably in S. cerevisiae. The fragility of some of these loci appears directly linked to replication, the DNA molecule being more prone to break when replication progression is altered chemically and/or physically [83–87]. Improperly repaired, the resulting DSB could induce the formation of a chromosomal rearrangement. Interestingly, spontaneous SDs directly selected within the yeast genome also emphasize a fundamental interplay with DNA replication [76,77]. In these studies, more than 300 SDs (direct tandems as well as inter-chromosomal duplications), strongly reminiscent in size to mammalian SDs (∼40 to 600 kb) and with no constraints exerting on the location of their breakpoints, were isolated. More than 20% of the SDs analyzed were bordered by low complexity DNA sequences (poly A/T and trinucleotide repeats) known to interfere with progression of the replication fork. In addition, the frequency of SDs formation is tremendously enhanced when S-phase DNA damages are induced either by a mutation delaying the firing of replication origins [77,88] or in presence of the Top1 inhibitor, camptothecin [77]. A role for replication accidents in the emergence of SDs is thus supported by recurrent and concordant experimental observations.
3.2.2 Recombination-dependant replication
A DSB originating from replication fork collapse generates a single DNA free-end able to invade the allelic homologous locus on the sister chromatid. This recombination-dependant strand invasion can be followed by reassembly of a replication fork and its progression until the chromosome end through a BIR event [89,90]. However, the DNA free-end can also, and sometimes will, invade a homologous locus located at a non-allelic position, either on the sister-chromatid and upstream the break, or on a different chromosome. Replication fork reassembly will then lead either to a direct tandem SD or to a non-reciprocal translocation, respectively (Fig. 4A). This mechanism is dependent on Pol32, a subunit of the major replicative DNA polymerase δ in S. cerevisiae [91]. Interestingly, all of the recombination-mediated (Rad52-dependant) direct tandem SDs recovered in yeast are actually dependent on the presence of this protein [77], revealing a direct implication of inter sister-chromatid BIR events. In addition, some of the SDs observed in drosophila or recovered using an embryonic stem cell system [54,92] were proposed to result from a mechanisms derived from this classical model, the difference consisting in the possibility for the replication fork to disassemble and the DNA free end to reinvade the original chromosome (Fig. 4B). This model is experimentally supported by evidence of multiple rounds of strand invasion/replication progression in yeast [93].
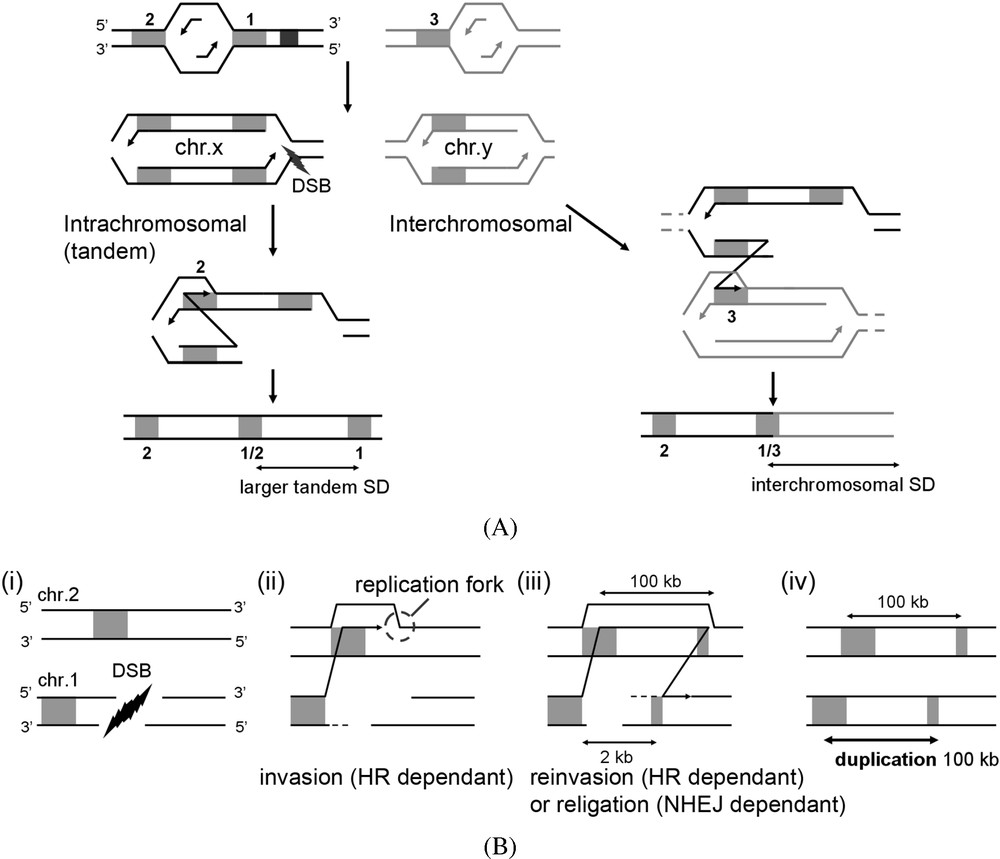
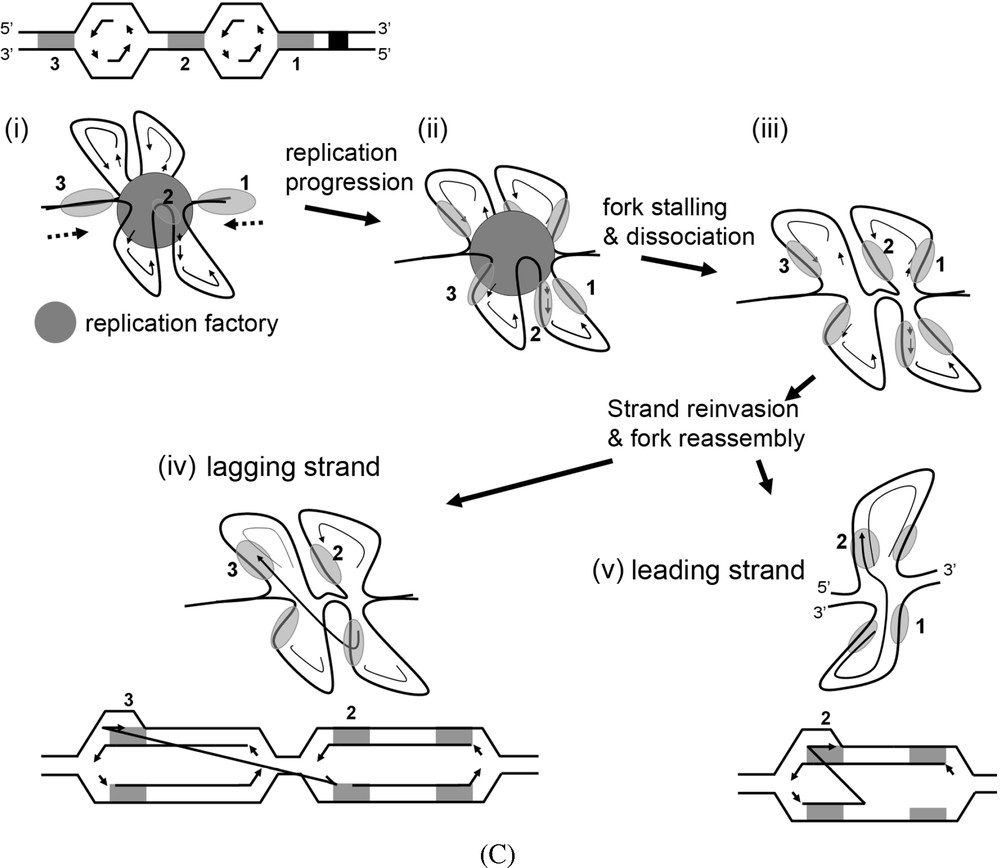
Replication-based mechanisms. Black squares stand for fragile sites slowing down replication forks. Black lightning represents emergence of a DSB at these fragile sites. Grey areas represent non-allelic homologous sequences (NAHS) or microhomologies dispersed within the genome, and are numbered for convenient reasons. (A) Intra- and interchromosomal BIR models. Following DSB, the 3′-end of the DSB (NAHS 1) invades a non-allelic region located either within the same chromosome (NAHS 2) or onto a different one (NAHS 3). Reassembly of a replication fork at these locations, and progression until the end of the chromosome will lead either to direct tandem or interchromosomal duplications. (B) Reinvasion & dissociation model. Following a DSB (i), a classical BIR event is engaged (ii). The replication fork progress, duplicating a large DNA region until a new dissociation occurs (iii). The 3′-end subsequently reinvades or is religated to the original chromosome (iv). (C) Replication factory switch of template and MMIR model. (i) Two replication forks of a same replicon co-localize within a replication factory (grey disk). Eventually, more than one replicon will be present within a factory. We propose that following stalling (ii), the replication fork disassembles (iii), leaving the lagging strand of one of the replicon bubble free to interact and invade, in a homologous-independent manner (through microhomology interactions, see MMIR [77]), a sequence located upstream along the chromosome in another replicon (iv). Reassembly of the fork at this location then allows duplication of a large region, using the neo-synthesized strand as a template. This mechanism will generate tandem or interchromosomal duplication depending whether the invading and invaded sequences are located on the same or on different chromosomes, respectively. A similar mechanism involving the leading strand can also be invoked (v) but appear less likely in regards to recently unveiled role for the DNA polymerase δ in replicating the lagging strand template [96].
3.2.3 Replication-based duplications: microhomology/microsatellite-induced replication (MMIR)
Interestingly, in yeast, the absence of all known DNA repair pathways (i.e. when HR () and NHEJ/MMEJ () are suppressed) does not alter the rate of SD formation as compared to WT [77]. Whereas LTR sequences are not involved at the breakpoints, low-complexity as well as short identical sequences are commonly found. Thus, HR homology search is not necessary for distant sequences to interact. Furthermore, when recombination is impaired SDs appear both Pol32-dependant, and smaller and of a size reminiscent of the distance separating two replication origins [77]. These observations may be fundamental. Indeed, in budding yeast, two replication forks originating from the same origin are processed simultaneously at a single position defined as a replication factory [94]. Combined with evidence in fission yeast for a repeated replication factory “pattern” [95], it is possible that transient colocalization of two regions within a factory increases the chance of physical interactions between them without involvement of a HR step. A DNA free-end resulting from a collapsed replication fork will indeed be located in the vicinity of the symmetrical fork originating from the same origin. The DNA strand may be able to spontaneously invade a sequence in its vicinity sharing a low complexity structure (such as polyA/T repeats), followed by the reassembly of a new replication fork (MMIR mechanism described in [77], Fig. 4C). Such replication factory switches of template may occur spontaneously and rather frequently (in yeast SD rate formation for a 1.6 kb locus is 10−7 event/cell/division). A recent report by Lupski and collaborators propose that the large PLP1 encompassing SDs that cause the dysmyelinating PMD disease could result from a DNA replication mechanism relying on template switching between microhomologous sequences [58]. The analysis of the junction sequences leads to the proposal that after the replication fork stalls or pauses, the lagging (or, but less likely, leading [96]) strand serially disengages and switches to another nearby replicating template without involving any DSB as the initiating event. This is consistent with results suggesting that SDs might not necessitate the presence of a DSB DNA free-end to occur: in the absence of all known DSB repair pathways in yeast, SD still form at a level close to the WT [77].
4 Conclusion
The histories of the duplicated structures found in eukaryotic genomes probably result from complex interplays between all these mechanisms. We distinguish replication fork switching of template as a key player leading to the primary amplification of any sequence in the genome. SDs emerge as natural events likely to occur at high frequencies anywhere in the genome, having a dramatic impact both onto eukaryotic genome evolution and chromosome dynamic.
First of all, duplicated regions containing entire genes will eventually become fixed into genomes [64,76]. Duplicated genes are thought to be the main source of new genes (neofunctionalization, subfunctionalization or degeneration-complementation, etc.) [97–100], and to promote genetic robustness [101].
Interestingly, SDs also lead to gene fusions and domain accretion without necessarily altering the pre-existing genes present in the genome (Fig. 2A, compare tandem to interspersed duplications [76,77]), allowing evolutionary “experiments” currently observed within genomes [13,102,103]. The intragenic sequences serving as SD breakpoints sometimes show specific features such as nucleotide repeats, and local amplifications/contraction that can induce protein changes and have important consequences.
SDs contributions to the evolutionary history of primate and other eukaryotic lineages remain a large field of investigation. Their consequences in terms of genome stability are intensively addressed because of their association with numerous human genomic diseases. Paradoxically, one possibility would be that the duplication of some DNA regions would be favored even though the genome stability is compromised by such expansion. The primate lineage would be dramatically affected by the positive selection of a subset of SDs despite the structural instability they generate [39,52]. Without a doubt, the near future will see more bridges thrown between these fields, which will shed new light onto the roles played by SDs in speciation processes and adaptation.
Acknowledgements
We thank Celia Payen and members from the Unité de Génétique moléculaire des levures (Institut Pasteur) for helpful discussions and Beth Weiner (Harvard University) for constructive reading of the manuscript. This work was supported by grants from the Association pour la Recherche sur le Cancer (3266), the Agence nationale de la recherche (ANR-05-BLAN0331) and by CNRS (GDR2354 “Génolevures II”).